1. Introduction
AML, the most common type of acute leukemia in adults, is becoming increasingly prominent in terms of incidence and severity against the backdrop of global aging [
1]. By 2050, the global population aged 65 and older is projected to increase from 761 million in 2021 to 1.6 billion, rising from 9% to 16% of the total population [
2]. The growth rate of the population aged 80 and above is even faster, with an estimated 140 million in 2021 and 459 million (4.5% of the total population) by 2050 [
3]. Most AML patients are aged 50 or older, with the highest incidence and mortality observed in those aged 70 and above [
4]. From 2010 to 2017, the overall survival rate for AML patients over 70 was only 5% [
5]. The incidence of AML exhibits a pronounced age-dependent pattern. According to the SEER (Surveillance, Epidemiology, and End Results) database, the median age at AML diagnosis is 67 years, with 50–60% of patients diagnosed at ≥65 years and over 30% at ≥75 years [
6]. As global aging intensifies (the proportion of the population aged 60 and above is expected to reach 22% by 2050), the absolute number of elderly AML patients will continue to rise [
7]. Older AML patients typically present with higher genetic risk (e.g.,
TP53 mutations and complex karyotype) and poorer performance status (PS ≥ 2 in over 40% of cases) [
8], leading to lower treatment tolerance, reduced remission rates, and shorter survival. The complete remission (CR) rate for standard induction chemotherapy in patients ≥65 years is only 30–50%, with a 5-year overall survival (OS) rate of less than 10% [
9], significantly lower than the 40–60% observed in younger patients.
CircRNA, a class of highly stable non-coding RNA with multidimensional regulatory functions, exhibits complex regulatory networks in the pathogenesis of age-related diseases and the aging process [
10]. Following ischemic stroke,
circSCMH1 enhances vascular endothelial cell repair and neurological recovery by modulating the ubiquitination of fat and obesity-associated (FTO) protein, reducing m6A modification of the lipid phosphatase phospholipid phosphatase 3 (Plpp3) and increasing its mRNA stability. In Alzheimer’s disease (AD) patients [
10],
circPSEN1 is significantly up-regulated, promoting amyloid-β (Aβ) production by inhibiting
miR-137 expression and activating the nuclear factor of activated T cells 1 (NFATC1) and epidermal growth factor receptor (EGFR) pathways [
11].
circSTX12 competitively binds to Casitas B-lineage lymphoma (CBL) protein, inhibiting the Hippo/YAP (Yes-associated protein) pathway, promoting adipogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) and suppressing osteogenic differentiation, thereby contributing to bone loss in senile osteoporosis (OP). Antisense oligonucleotides (ASOs) targeting
circSTX12 significantly improve bone microstructure in mouse models [
12]. These studies highlight the central regulatory role of circRNA in age-related and aging-associated diseases, providing novel insights for targeted interventions.
Cellular senescence, first proposed by Hayflick in 1961, describes a state of irreversible growth arrest in actively proliferating cells [
13]. Cellular senescence represents the third major tumor-suppressive mechanism following apoptosis and DNA damage repair. Senescent cells exhibit irreversible cell cycle arrest while retaining metabolic activity and secrete a variety of factors, including cytokines, chemokines, and extracellular matrix proteases, which collectively constitute the senescence-associated secretory phenotype (SASP). Therapy-induced senescence (TIS), characterized by irreversible cell cycle arrest and activation of immune clearance mechanisms, has emerged as a cutting-edge strategy in cancer treatment, particularly for elderly patients intolerant to chemotherapy or radiotherapy. Senescence-like phenotype mediated by IFI16 in glioblastoma (GBM) suppresses ferroptosis via Heme Oxygenase-1 (HMOX1) activation, conferring radiation resistance. The classic antidiabetic drug glyburide disrupts the binding of Interferon Gamma Inducible Protein 16 (IFI16) to transcription factors, restoring radio sensitivity [
12]. Additionally, circRNA-induced tumor cell senescence is gaining attention. Inhibition of
circPETH-147aa significantly increases Reactive Oxygen Species (ROS) accumulation, inducing DNA damage and cellular senescence [
14]. ASOs targeting
circITGB6 markedly suppress tumor progression in colorectal cancer liver metastasis models and induce a senescent phenotype [
12]. However, research on circRNA-induced cellular senescence in AML remains limited.
In this study, we identified circSATB1 as a significantly overexpressed circRNA in AML through whole-transcriptome sequencing and bioinformatics analysis. We validated its circular nature and confirmed its predominant cytoplasmic localization. Further siRNA knockdown experiments demonstrated that circSATB1 inhibition suppresses cell proliferation and promotes cellular senescence, with no significant effects on apoptosis or DNA damage. These findings suggest that circSATB1 holds considerable therapeutic potential in AML, possibly serving as a critical therapeutic target for inducing AML cell senescence.
2. Materials and Methods
2.1. Sample Collection and Transcriptome Sequencing
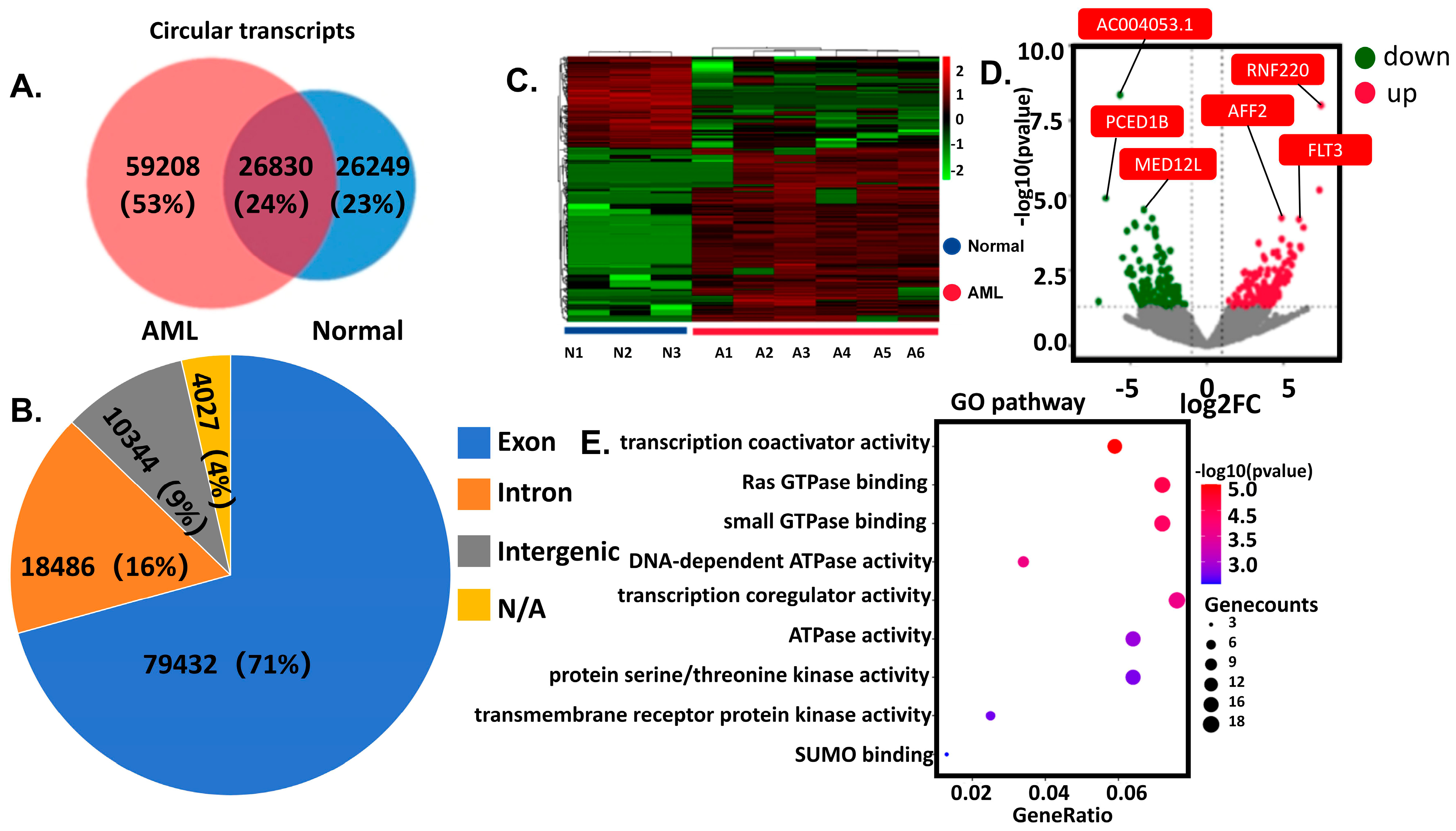
Discovery Cohort (RNA sequencing): Peripheral blood was collected from 6 AML patients and 3 healthy controls. Demographic and clinical characteristics have been comprehensively documented in
Supplementary Material Table S1. Nuclear cells isolated via density gradient centrifugation underwent RNA sequencing (Illumina platform, Novogene, Sacramento, CA, USA). Differentially expressed circRNAs were identified through integrated computational analysis (CIRCexplorer2, CircFinder) with cross-database annotation. Differentially expressed genes (DEGs) were defined as |logFC| > 1 and
p-value < 0.05. Validation Cohort (Clinical validation):
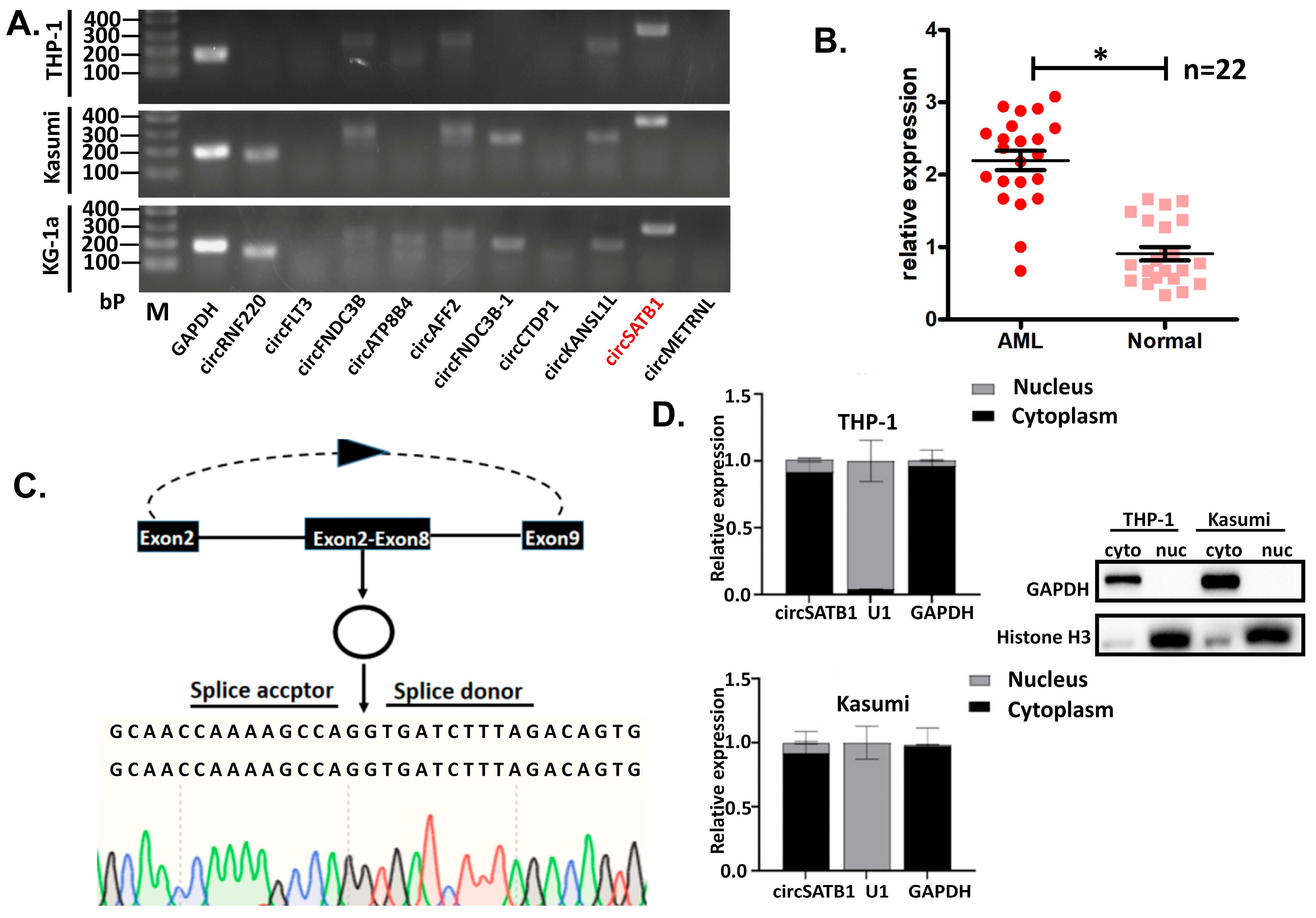
circSATB1 expression was assessed in 44 independent anticoagulated blood samples (22 AML vs. 22 controls). All procedures complied with institutional ethics guidelines, with written informed consent obtained from participants.
2.2. Cell Culture and Treatment
KG-1a, THP-1, and Kasumi cells were purchased from the cell bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were maintained in RPMI-1640 medium (HyClone, South Logan, UT, USA) supplemented with fetal bovine serum (FBS; Gibco, Carlabad, CA, USA, 10% for KG-1a and THP-1, 20% for Kasumi) and 1% penicillin/streptomycin (Gibco, Carlabad, CA, USA) under standard conditions (37 °C/5% CO2) with 2–3 times weekly passaging. siRNA transfection utilized Zlipo2000 (ZomanBio, Beijing, China) in 6-well plates (4–6 h incubation), followed by 24–72 h culture post-medium replacement. For siRNA transfection, Pepmute (ZomanBio, Beijing, China) complexes (40 pmol siRNA, RT incubation: 5 + 15 min) were applied to plates, with cells harvested after 24–72 h.
Nuclear-cytoplasmic fractionation was performed in THP-1 and Kasumi cells using pre-chilled Cell Fractionation Buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with PMSF (Beyotime, Shanghai, China, 1 mM final concentration). Sequential centrifugations (500× g, 4 °C, 5 min) isolated cellular components, followed by lysis in 2× Lysis Binding Solution (Thermo Fisher Scientific, Waltham, MA, USA; β-mercaptoethanol-supplemented to 1% (v/v)) and ethanol precipitation. RNA was purified via filter cartridge washes (Wash Solutions 1–3) and eluted in 95 °C Elution Buffer. Nuclear/cytoplasmic proteins were reserved for immunoblotting, while RNA underwent reverse transcription for qPCR analysis.
2.3. PCR and qPCR
Total RNA was reverse-transcribed using a Hifair® II 1st Strand cDNA Synthesis Kit (gDNA digester plus) (Yeasen, Shanghai, China) according to the manufacturer’s instructions. Briefly, 1 μg total RNA was mixed with gDNA digester and incubated at 42 °C for 30 min, and 85 °C for 5 min. For PCR, 2× HieffTM PCR Master Mix (With Dye) (Yeasen, Shanghai, China) was used. The cDNA and gDNA PCR products were evaluated using 2% agarose gel electrophoresis. The qPCR was conducted using 2× SYBR Green qPCR Master Mix (Bimake, Houston, TX, USA). GAPDH, β-actin, and U1 were used as controls. For quantification of gene expression, the 2−∆∆CT method was used, and the data were normalized to an endogenous control.
2.4. Western Blot Analysis
Cells were lysed in RIPA buffer (Beyotime, Shanghai, China), denatured in 5× loading buffer (100 °C, 10 min), and resolved via SDS-PAGE (10% or 15%). Proteins were transferred to PVDF membranes, blocked with 5% BSA (Biosharp, Hefei, China), and incubated overnight at 4 °C with primary antibodies: anti-GAPDH, Histone H3, CDK2, hTERT, BAX, PCNA and BCL-2 (Abclonal, Wuhan, China). Membranes were washed (TBST × 3), incubated with HRP-conjugated secondary antibodies (1:5000, 1 h, RT), and visualized using a ChemiDoc system (Bio-Rad, Hercules, CA, USA) with ECL (Beyotime Biotechnology, Shanghai, China). Band intensity was quantified via ImageJ (NIH, ImageJ 1.x).
2.5. Cell Proliferation
THP-1 cells were incubated with EdU (RiboBio, Guangzhou, China) in complete medium (1:1000 dilution) for 2 h post-transfection 48 h. Cells (1 × 104/well, 96-well plate, n = 3) were fixed with 4% paraformaldehyde (Biosharp, Hefei, China), permeabilized (0.5% Triton X-100), and stained with Apollo reaction mix and Hoechst 33,342 (Beyotime, Shanghai, China; RT, dark, 30 min). Fluorescence imaging was performed using Optoelectronic Technology microscope (Mingmei, Guangzhou, China). Additionally, cell viability was assessed by a CCK-8 assay kit (Biosharp, Hefei, China) in Kasumi cells.
2.6. Cell Apoptosis
THP-1 cells were harvested by centrifugation (500× g, 5 min) post-transfection 48 h, washed twice with ice-cold PBS, and stained with Annexin V-FITC (Beyotime, Shanghai, China) in binding buffer (4 °C, dark, 15 min), followed by PI addition (4 °C, dark, 5 min). Unstained, FITC-only, and PI-only controls were included. Apoptotic subpopulations (Annexin V-FITC+/PI−: early apoptosis; Annexin V-FITC+/PI+: late apoptosis/necrosis) were quantified via flow cytometry (BD Biosciense, San Jose, CA, USA).
2.7. Cell Cycle
THP-1 cells were harvested by centrifugation (500× g, 5 min, RT), washed twice with ice-cold PBS, and fixed in 70% ethanol (−20 °C, ≥2 h). After rehydration, pellets were stained with PI (BD Biosciences, San Jose, CA, USA, 50 μg/mL), RNase A (Sigma-Aldrich, St. Louis, MO, USA, 100 μg/mL), and 0.1% Triton X-100 in PBS (Servicebio, Wuhan, China, 37 °C, 30 min, dark). Cell suspensions were filtered through 40 μm mesh and analyzed on a flow cytometer (488 nm laser; PI detection: bandpass filter centered at 585 nm with 40 nm bandwidth [585/40 nm]), with ≥10,000 events recorded per sample. Instrument calibration utilized unstained controls and fluorescent beads.
2.8. Comet Assay
The alkaline single-cell gel electrophoresis technique (comet assay) was used to measure the DNA damage in THP-1 cells using an OxiSelect Comet Assay Kit (Cell Biolabs, Chicago, IL, USA). Briefly, liquid agarose was pipetted onto a comet slide and chilled. Next, cell samples were combined with comet agarose, added on top of the base layer, lysed to form nucleoids containing supercoiled loops, and then immersed in alkaline solution. Then, the samples were electrophoresed for 30 min under voltage 25 V and current 300 mA to separate intact DNA from damaged fragments, stained with a diluent DNA dye (Cell Biolabs, Chicago, IL, USA), and visualized by epifluorescence microscopy using a FITC filter (MshOt, Guangzhou, China, 40×). Comet Assay Software Project (CaspLab-1.2.2) was used to quantify the tail parameter.
2.9. β-Galactosidase Staining
β-Galactosidase Staining was measured in THP-1 using the β-galactosidase staining kit (BeiboBio, Shanghai, China) according to the manufacturer’s protocol. Harvest cells by centrifugation at 300× g for 5 min at room temperature into a 1.5 mL centrifuge tube. Wash once with PBS, add 1 mL of β-galactosidase staining fixative, and fix at room temperature for 15 min. During fixation, slowly shake the tube on a shaker to avoid the cells from clumping together. After centrifugation (300× g, 5 min, RT), remove the cell fixative, wash the cells 3 times with PBS, each time for 3 min. Centrifuge (300× g, 5 min, RT), remove the PBS, and add 0.5–1 mL of staining working solution to each tube. Incubate at 37 °C overnight. Drop some of the stained cells onto a slide or into a 6-well plate, and observe under a common optical microscope.
2.10. Southern Blot
The measurements of terminal restriction fragment (TRF) length were applied using the TeloTAGGG telomere length assay kit (Roche, Basel, Switzerland). Briefly, DNA extraction from KG-1a cells was performed through sequential lysis, and nucleic acid purity was verified by NanoDrop 2000 spectrophotometry (Thermo Fisher, Waltham, MA, USA), confirming the A260/280 ratio (≥1.8). Equal volumes of HinfI and RsaI were combined for concurrent DNA digestion and positive control preparation, while the molecular marker was formulated by mixing 4 μL Bottle 6, 12 μL RNase-free water, and 4 μL Bottle 7. Electrophoresis was conducted in 0.8% agarose gel with 1× TAE running buffer (Servicebio, Wuhan, China) at 50 V for 5 h, followed by sequential post-electrophoretic processing: membrane transfer via capillary action (ambient temperature, overnight), UV cross-linking, prehybridization for 1 h and hybridization (42 °C, 3 h) in freshly prepared buffer, and chemiluminescent detection using Bottle 15 substrate.
2.11. Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (version 7.0). Each experiment was independently repeated three times, and the representative data are shown. All the values are presented as mean ± SD of three biologically independent samples. Statistical analyses were performed using a two-way analysis of variance (ANOVA) when comparing at least three groups. The sample size is indicated in the corresponding figure legends. Statistical significance was defined as * p < 0.05, ** p < 0.01.
4. Discussion
circSATB1, a previously uncharacterized circular RNA, was highly expressed in elderly AML patients and orchestrates a telomere-independent senescence program upon knockdown. Using multi-omics and functional validation, we demonstrate that (1)
circSATB1 is significantly overexpressed in AML clinical samples and cell lines (
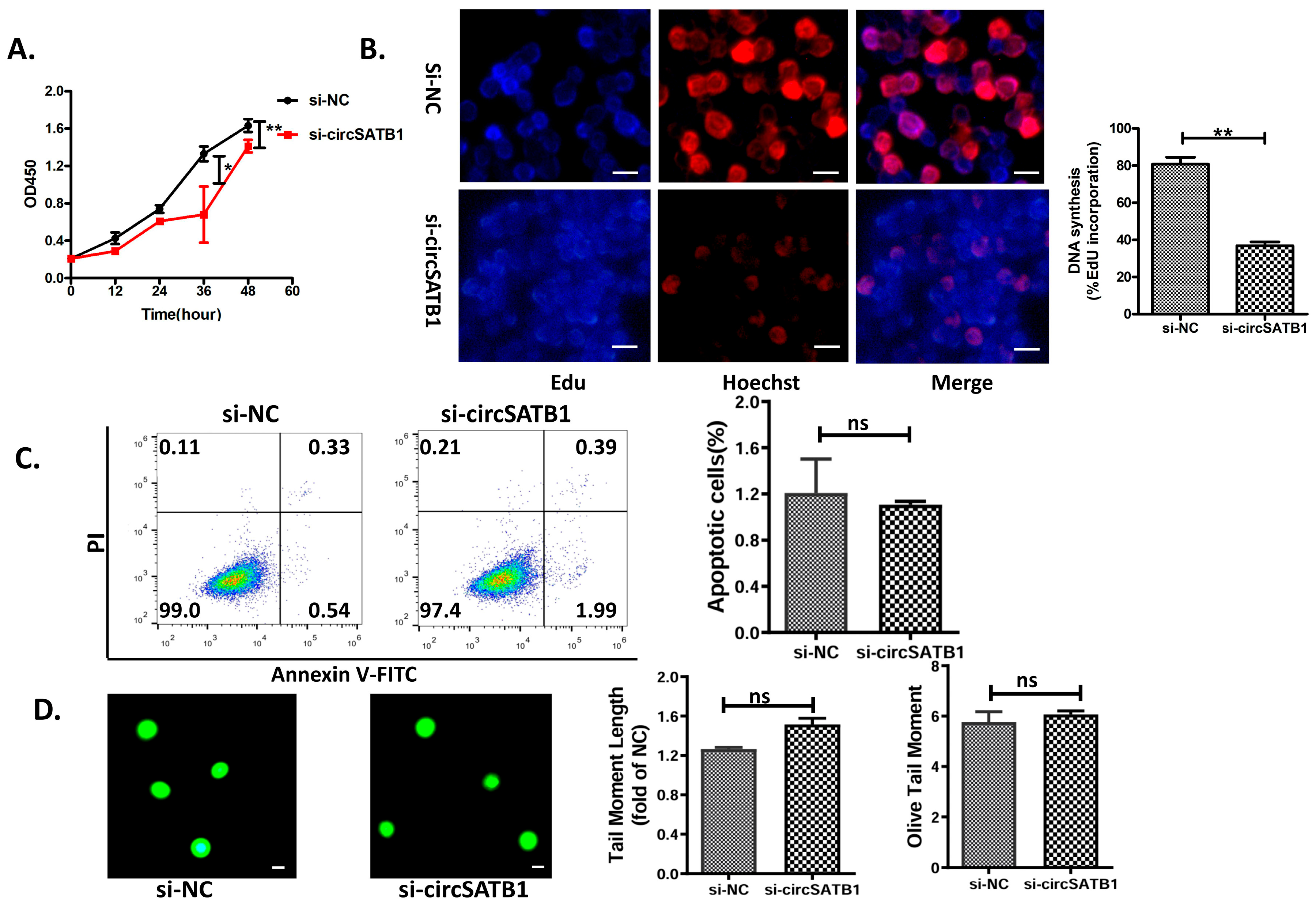
Figure 2A,B); (2) its inhibition induces G0/G1 arrest and premature senescence without triggering apoptosis or DNA damage (
Figure 3 and
Figure 4); (3) this senescence phenotype occurs independently of telomere shortening (
Figure 4E). These results establish
circSATB1 as a novel regulator of cellular senescence in AML.
Population aging is a global public health challenge. Age-related diseases are becoming an increasingly serious public health concern [
15]. It is both imperative and urgent to address these challenges through specific aging biomarkers. Also, their impact on age-related diseases is relatively unexplored, and their potential mechanisms to minimize or alleviate this rapid aging trend remain unclear. Recent research has proposed twelve hallmarks of aging, such as genomic instability, telomere attrition, mitochondrial dysfunction, cellular senescence, and chronic inflammation, which all contribute to understanding the progression of aging. Identifying biomarkers of aging and risks associated with age-related diseases is crucial for supporting public health initiatives for healthy aging, developing a priority control list, and enhancing regulation. Acute myeloid leukemia, the most common adult leukemia, predominantly affects individuals aged 60–70 years [
16]. Elderly patients (≥65 years) exhibit significantly higher mortality rates, with a dismal 5-year relative survival rate of only 29.5% [
17].
circRNAs play important roles in a variety of tumors. Signature circRNAs have been identified in Alzheimer’s disease [
18], brain cell glioma [
19], lung cancer [
20], and other tumors in the elderly. In addition, tissue fluids and body fluids (saliva, blood, etc.) have been found to be rich in circRNAs [
21], which are ideal biomarkers for liquid biopsy and may be a potential target for hematological tumor therapy, but it is not yet clear which circRNAs are involved in the development of AML. Certain circRNAs exhibit spatiotemporal expression patterns correlating with cell cycle phases, while others serve as molecular indicators of cellular senescence. As the third tumor barrier following apoptosis and DNA damage repair, senescence induces irreversible growth arrest while maintaining metabolic activity, thereby limiting cancer proliferation [
22]. The Belgian group’s “one-two punch” strategy “CDC7 inhibitor-induced senescence” + “mTOR blocker-triggered apoptosis” effectively eliminated hepatocellular carcinoma cells with improved patient tolerance compared to conventional cytotoxic therapies [
23]. Achieving cancer eradication while preserving quality of life remains a therapeutic dilemma, particularly for elderly AML patients. Therefore, exploring a similar therapeutic strategy to be adopted for AML is of significant interest. Identifying circRNAs that induce senescence in AML cells is a crucial step in this direction. Our study addresses this gap by identifying
cirSATB1 as a senescence modulator in AML. While our data demonstrate that
circSATB1 knockdown induces senescence (
Figure 3 and
Figure 4), the preliminary nature of the predicted regulatory networks (
Figure 5) must be emphasized. Causal relationships between
circSATB1 and senescence effectors remain to be established.
The ability of cancer cells to evade senescence is contingent on their sustained proliferative capacity. Cellular senescence can be classified into replicative senescence (telomere-dependent) and premature senescence (telomere-independent and stress-induced) [
24,
25,
26,
27,
28]. In this study, we demonstrated that
circSATB1 knockdown induced premature senescence in AML cells, characterized by maintenance of telomere length and obstruction of cell cycle progression. This suppression of uncontrolled AML cell proliferation offers a novel and mild therapeutic approach, providing a framework for identifying more tolerable AML treatment options. Such options include the induction of cellular senescence to halt disease progression, which holds significant prognostic value for elderly patients. Our findings align with emerging interest in senescence-inducing therapies [
12,
14,
22,
23], though the specific role of circRNAs in AML senescence was previously unexplored. The predicted interaction networks involving
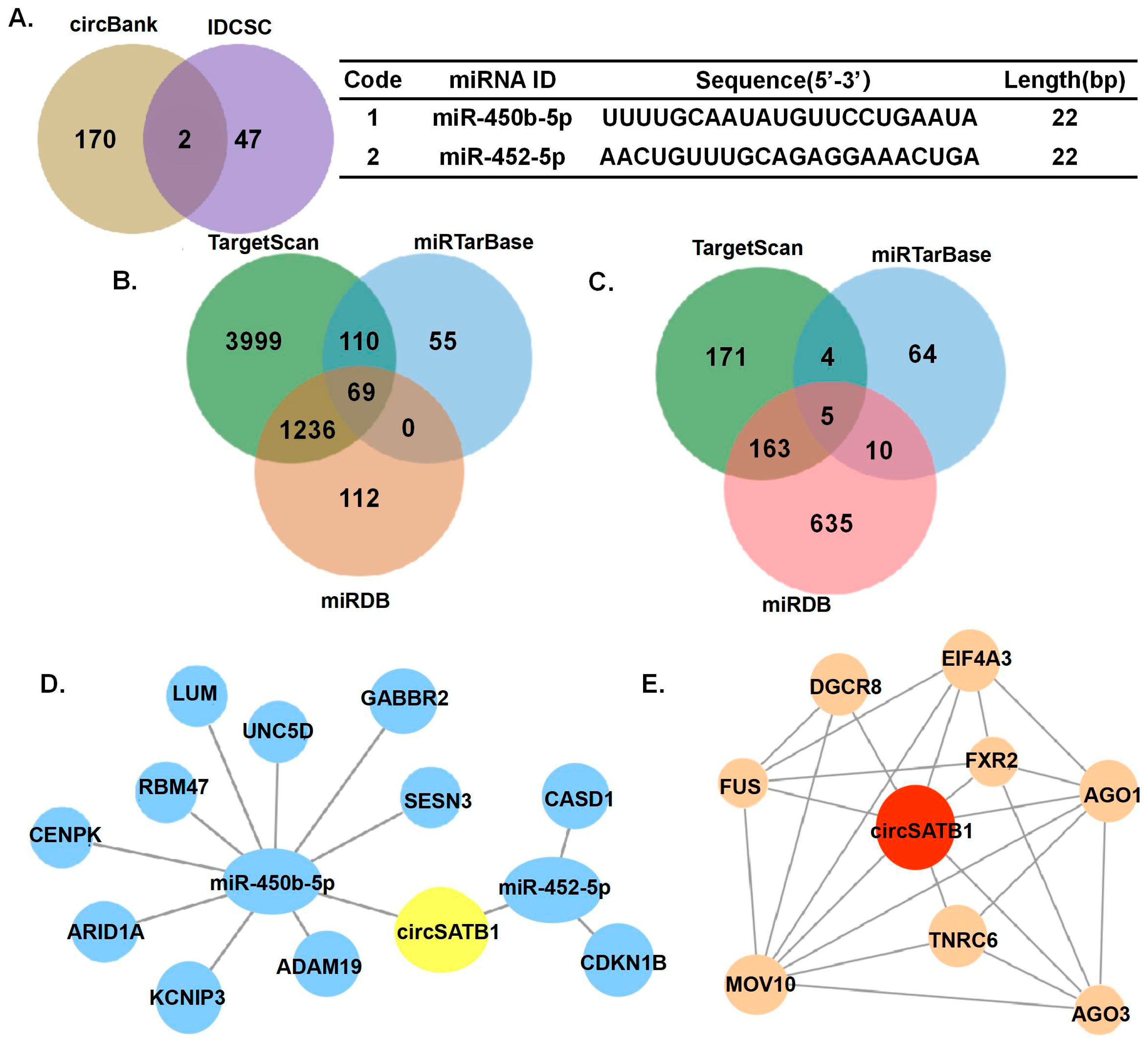
miR-450b-5p,
miR-452-5p, and specific RBPs (
Figure 5) provide testable hypotheses for future mechanistic studies to understand how
circSATB1 exerts its anti-senescence function. While our data provided evidence for identifying
circSATB1 as a novel regulator of senescence in AML and suggest its potential as a therapeutic target, several limitations warrant acknowledgment: (1). Cohort size: Our conclusions are derived from in vitro analyses using AML cells from a limited cohort, which may not fully capture the heterogeneity of AML. Larger-scale validation in diverse populations is needed. (2). Model constraints: The exclusive use of in vitro models precludes assessment of systemic factors. Future work should integrate probing physiological relevance. (3). Mechanistic depth: Although we identified
circSATB1 as a novel regulator of senescence in AML and suggest its potential as a therapeutic target, causal links require further deconvolution. Its therapeutic potential and mechanism of action remains untested.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}