

Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation


Abstract

1. Introduction
1.1. Current Understanding and Clinical Definition of Parkinson’s Disease (PD)
1.2. Overview of Therapeutic Evolution: From Levodopa to Alpha-Synuclein (α-Syn) Therapies
1.3. Rationale and Objectives of This Review
2. Overview of Parkinson’s Disease (PD) Pathogenesis
2.1. Pathological Hallmarks: Alpha-Synuclein (α-Syn) Aggregation and Lewy Bodies
2.1.1. Alpha-Synuclein (α-Syn)’s Role in Parkinson’s Pathology
2.1.2. Formation, Distribution, and Significance of Lewy Bodies
2.2. Genetic Contributions and Risk Factors
2.2.1. Monogenic Forms: Key Genes (LRRK2, SNCA, PARKIN, PINK1)
2.2.2. Polygenic Influences and Genetic Risk Profiling
2.3. Environmental Factors and Gene–Environment Interactions
2.3.1. Epidemiological Evidence of Environmental Influences
2.3.2. Interplay Between Environmental Triggers and Genetic Susceptibility
2.4. Neuroinflammation and Oxidative Stress Mechanisms
2.4.1. Neuroinflammatory Pathways in Parkinson’s Progression
2.4.2. Oxidative Stress and Mitochondrial Dysfunction: Central Drivers in Parkinson’s Pathology
3. Core Research Gaps in Parkinson’s Disease (PD)
3.1. Contradictory Findings in Parkinson’s Disease (PD) Research
3.1.1. Examples of Inconsistent Studies
3.1.2. Reasons Behind Conflicting Data
3.1.3. Impact on Therapeutic and Diagnostic Development
3.2. Knowledge Voids in Pathophysiology
3.2.1. Unresolved Mechanisms of Neurodegeneration
3.2.2. Unknown Functions of Genetic Risk Loci
3.2.3. Areas Needing Deeper Molecular Characterization
3.3. Action–Knowledge Conflict
3.3.1. Discrepancy Between Research Outcomes and Clinical Application
3.3.2. Misalignment of Preclinical Successes and Clinical Failures
3.3.3. Examples: Neuroprotective Treatments
3.4. Methodological Shortcomings
3.4.1. Limitations in Experimental Parkinson’s Disease (PD) Models (Animal vs. Human Rele-Vance)
3.4.2. Biomarker Discovery and Validation Challenges
3.4.3. Technological Barriers in Longitudinal and Predictive Studies
3.5. Evaluation Voids
3.5.1. Absence of Standardized Evaluation Criteria for Early Detection
3.5.2. Gaps in Clinical Trial Endpoint Definitions
3.5.3. Insufficient Use of Patient-Reported Outcomes
3.6. Theory Application Gaps
3.6.1. Insufficient Theoretical Integration
3.6.2. Over-Reliance on Reductionist Models
3.6.3. Limited Cross-Disciplinary Collaboration
3.7. Underrepresented Cohorts
3.7.1. Lack of Diversity in Genetic Studies
3.7.2. Underrepresentation in Clinical Trials
3.7.3. Consequences of Biases for Therapeutic Efficacy and Generalizability
4. Beyond Alpha-Synuclein (α-Syn): Emerging Therapeutic Targets and Approaches
4.1. Novel Targets: Lysosomal Pathways, Mitochondrial Dynamics, Neuroinflammation Modulation
4.2. Personalized Medicine and Precision Neurology Potentials
4.3. Integration of Multi-Modal Therapies: Pharmacological, Genetic, Lifestyle Interventions
5. Bridging Research Gaps: Strategic Recommendations
5.1. Enhancing Methodological Rigor and Reproducibility
5.2. Standardizing Clinical Outcomes and Biomarker Criteria
5.3. Encouraging Interdisciplinary Collaboration and Global Representation
5.4. Developing Frameworks for Translating Bench Findings to Bedside Interventions
6. Conclusions and Future Perspectives
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 18F-FDG | 18-fluorodeoxyglucose |
| AI | artificial intelligence |
| α-syn | alpha-synuclein |
| COMT | catechol-O-methyltransferase |
| CSF | cerebrospinal fluid |
| DAT | dopamine transporter |
| FDG | fluorodeoxyglucose |
| GBA1 | glucocerebrosidase 1 |
| GIP | glucose-dependent insulinotropic polypeptide |
| GLP-1 | glucagon-like peptide-1 |
| GP2 | Global Parkinson’s Genetics Program |
| GWAS | genome-wide association studies |
| IoT | Internet-of-Things |
| LRRK2 | leucine-rich repeat kinase 2 |
| MDS | Movement Disorder Society |
| MDS-UPDRS | Movement Disorder Society–Unified Parkinson’s Disease Rating Scale |
| MRI | magnetic resonance imaging |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| PD | Parkinson’s disease |
| PET | positron emission tomography |
| PINK1 | PTEN-induced kinase 1 |
| PRKN | Parkin RBR E3 ubiquitin-protein ligase |
| RBD | REM-sleep behavior disorder |
| REM | rapid eye movement |
| SNCA | synuclein alpha gene |
| SNP | single-nucleotide polymorphism |
References
- Lamotte, G.; Becker, B. Toward biomarker-based clinical subtyping of Parkinson disease. Neurology 2020, 95, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Le, W. Milestones of Parkinson’s Disease Research: 200 Years of History and Beyond. Neurosci. Bull. 2017, 33, 598–602. [Google Scholar] [CrossRef]
- Waleed, M.; Sadiq, W.; Subhan, S. Parkinsonism and D-512, dopamine D2/3 receptor agonist; A review of literature. Inter-Natl. J. Clin. Case Rep. Rev. 2020, 4. [Google Scholar] [CrossRef]
- Scorza, F.A.; de Almeida, A.C.G.; Scorza, C.A.; Finsterer, J. Prevention of Parkinson’s disease-related sudden death. Clinics 2021, 76, e3266. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.A.; Menezes-Rodrigues, F.S.; Olszewer, E.; Errante, P.R.; Tavares, J.G.P.; Scorza, C.A.; Ferraz, H.B.; Finsterer, J.; Caricati-Neto, A. The mitochondrial calcium uniporter: A new therapeutic target for Parkinson’s disease-related cardiac dysfunctions? Clinics 2020, 75, e1299. [Google Scholar] [CrossRef]
- Pearce, J.M. Aspects of the history of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1989, 52, 6–10. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Jimenez-Del-Rio, M. Oxidative stress signaling and regulated cell death in Parkinson’s disease. In Genetics, Neurology, Behavior, and Diet in Parkinson’s Disease; Elsevier: Amsterdam, The Netherlands, 2020; pp. 157–173. [Google Scholar]
- Jreige, M.; Kurian, G.K.; Perriraz, J.; Potheegadoo, J.; Bernasconi, F.; Stampacchia, S.; Blanke, O.; Alessandra, G.; Lejay, N.; Chiabotti, P.S.; et al. The diagnostic performance of functional dopaminergic scintigraphic imaging in the diagnosis of dementia with Lewy bodies: An updated systematic review. Eur. J. Nucl. Med. 2023, 50, 1988–2035. [Google Scholar] [CrossRef]
- Walker, R.W.; Walker, Z. Dopamine transporter single photon emission computerized tomography in the diagnosis of dementia with Lewy bodies. Mov. Disord. 2009, 24, S754–S759. [Google Scholar] [CrossRef]
- Piggott, M.A.; Marshall, E.F.; Thomas, N.; Lloyd, S.; Court, J.A.; Jaros, E.; Burn, D.; Johnson, M.; Perry, R.H.; McKeith, I.G.; et al. Striatal dopaminergic markers in dementia with Lewy bodies, Alzheimer’s and Parkinson’s diseases: Rostrocaudal distribution. Brain 1999, 122, 1449–1468. [Google Scholar] [CrossRef]
- Duda, J.E. Pathology and Neurotransmitter Abnormalities of Dementia with Lewy Bodies. Dement. Geriatr. Cogn. Disord. 2003, 17, 3–14. [Google Scholar] [CrossRef]
- Yoo, H.S.; Jeong, S.H.; Oh, K.T.; Lee, S.; Sohn, Y.H.; Ye, B.S.; Yun, M.; Lee, P.H. Interrelation of striatal dopamine, brain metabolism and cognition in dementia with Lewy bodies. Brain 2022, 145, 4448–4458. [Google Scholar] [CrossRef] [PubMed]
- Patterson, L.; Rushton, S.P.; Attems, J.; Thomas, A.J.; Morris, C.M. Degeneration of dopaminergic circuitry influences depressive symptoms in Lewy body disorders. Brain Pathol. 2018, 29, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Holmes, C.; Sullivan, P.; Lopez, G.; Gelsomino, J.; Moore, S.; Isonaka, R.; Wu, T.; Sharabi, Y. Cardiac noradrenergic deficiency revealed by 18F-dopamine positron emission tomography identifies preclinical central Lewy body diseases. J. Clin. Investig. 2024, 134. [Google Scholar] [CrossRef]
- Pifl, C.; Reither, H.; Attems, J.; Zecca, L. Dopamine and vesicular monoamine transport loss supports incidental Lewy body disease as preclinical idiopathic Parkinson. npj Park. Dis. 2023, 9, 89. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Beyer, L.; Prix, C.; Schönecker, S.; Palleis, C.; Rauchmann, B.; Morbelli, S.; Chincarini, A.; Bruffaerts, R.; Vandenberghe, R.; et al. Metabolic Correlates of Dopaminergic Loss in Dementia with Lewy Bodies. Mov. Disord. 2019, 35, 595–605. [Google Scholar] [CrossRef]
- Moreira, E.G.; Boll, K.M.; Correia, D.G.; Soares, J.F.; Rigobello, C.; Maes, M. Why Should Psychiatrists and Neuroscientists Worry about Paraoxonase 1? Curr. Neuropharmacol. 2019, 17, 1004–1020. [Google Scholar] [CrossRef]
- LeWitt, P.A. Levodopa therapy for Parkinson’s disease: Pharmacokinetics and pharmacodynamics. Mov. Disord. 2015, 30, 64–72. [Google Scholar] [CrossRef]
- Lees, A.; Tolosa, E.; Stocchi, F.; Ferreira, J.J.; Rascol, O.; Antonini, A.; Poewe, W. Optimizing levodopa therapy, when and how? Perspectives on the importance of delivery and the potential for an early combination approach. Expert Rev. Neurother. 2023, 23, 15–24. [Google Scholar] [CrossRef]
- Beaulieu-Boire, I.; Lang, A.E. Behavioral effects of levodopa. Mov. Disord. 2014, 30, 90–102. [Google Scholar] [CrossRef]
- Hefter, H.; Hueber, R.; Jost, W.H.; Leenders, K.L.; Odin, P.; Schwarz, J.; Müller, T. Is levodopa toxic? J. Neurol. 2004, 251, vi44–vi46. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. Is levodopa toxic? Neurology 1996, 47, S184–S195. [Google Scholar] [CrossRef] [PubMed]
- Verschuur, C.V.; Suwijn, S.R.; Boel, J.A.; Post, B.; Bloem, B.R.; van Hilten, J.J.; van Laar, T.; Tissingh, G.; Munts, A.G.; Deuschl, G.; et al. Randomized Delayed-Start Trial of Levodopa in Parkinson’s Disease. N. Engl. J. Med. 2019, 380, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Odin, P.; Pahwa, R.; Aldred, J.; Alobaidi, A.; Jalundhwala, Y.J.; Kukreja, P.; Bergmann, L.; Inguva, S.; Bao, Y.; et al. The Long-Term Impact of Levodopa/Carbidopa Intestinal Gel on ‘Off’-time in Patients with Advanced Parkinson’s Disease: A Systematic Review. Adv. Ther. 2021, 38, 2854–2890. [Google Scholar] [CrossRef]
- Standaert, D.G.; Patel, V.; Snedecor, S.J.; Thakkar, S.; Jalundhwala, Y.J.; Kukreja, P.; Kratochvil, D.; Bao, Y.; Pahwa, R. Impact of carbidopa-levodopa enteral suspension on quality of life and activities of daily living in patients with advanced Parkinson’s disease: Results from a pooled meta-analysis. Park. Relat. Disord. 2021, 86, 52–57. [Google Scholar] [CrossRef]
- Monteiro, L.; Souza-Machado, A.; Valderramas, S.; Melo, A. The Effect of Levodopa on Pulmonary Function in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Clin. Ther. 2012, 34, 1049–1055. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic Criteria for Parkinson Disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef]
- Marsili, L.; Rizzo, G.; Colosimo, C. Diagnostic Criteria for Parkinson’s Disease: From James Parkinson to the Concept of Prodromal Disease. Front. Neurol. 2018, 9, 156. [Google Scholar] [CrossRef]
- Postuma, R.; Berg, D. MDS Clinical Diagnostic Criteria for Parkinson’s Disease (S19.001). Neurology 2016, 86, S19-001. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Poewe, W.; Litvan, I.; Lewis, S.; Lang, A.E.; Halliday, G.; Goetz, C.G.; Chan, P.; Slow, E.; Seppi, K.; et al. Validation of the MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2018, 33, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Schrag, A. Testing the MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2018, 33, 1518–1520. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Adler, C.H.; Bloem, B.R.; Chan, P.; Gasser, T.; Goetz, C.G.; Halliday, G.; Lang, A.E.; Lewis, S.; Li, Y.; et al. Movement disorder society criteria for clinically established early Parkinson’s disease. Mov. Disord. 2018, 33, 1643–1646. [Google Scholar] [CrossRef]
- Deutschländer, A.B.; Ross, O.A.; Dickson, D.W.; Wszolek, Z.K. Atypical parkinsonian syndromes: A general neurologist’s perspective. Eur. J. Neurol. 2017, 25, 41–58. [Google Scholar] [CrossRef]
- Caproni, S.; Colosimo, C. Diagnosis and Differential Diagnosis of Parkinson Disease. Clin. Geriatr. Med. 2020, 36, 13–24. [Google Scholar] [CrossRef]
- Rus, T.; Tomše, P.; Jensterle, L.; Grmek, M.; Pirtošek, Z.; Eidelberg, D.; Tang, C.; Trošt, M. Differential diagnosis of parkinsonian syndromes: A comparison of clinical and automated—Metabolic brain patterns’ based approach. Eur. J. Nucl. Med. 2020, 47, 2901–2910. [Google Scholar] [CrossRef]
- Schindlbeck, K.A.; Eidelberg, D. Network imaging biomarkers: Insights and clinical applications in Parkinson’s disease. Lancet Neurol. 2018, 17, 629–640. [Google Scholar] [CrossRef]
- Meyer, P.T.; Frings, L.; Rücker, G.; Hellwig, S. 18F-FDG PET in Parkinsonism: Differential Diagnosis and Evaluation of Cognitive Impairment. J. Nucl. Med. 2017, 58, 1888–1898. [Google Scholar] [CrossRef]
- Heim, B.; Krismer, F.; De Marzi, R.; Seppi, K. Magnetic resonance imaging for the diagnosis of Parkinson’s disease. J. Neural Transm. 2017, 124, 915–964. [Google Scholar] [CrossRef] [PubMed]
- Holtbernd, F.; Eidelberg, D. The Utility of Neuroimaging in the Differential Diagnosis of Parkinsonian Syndromes. Semin. Neurol. 2014, 34, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Papathoma, P.-E.; Markaki, I.; Tang, C.; Lindström, M.L.; Savitcheva, I.; Eidelberg, D.; Svenningsson, P. A replication study, systematic review and meta-analysis of automated image-based diagnosis in parkinsonism. Sci. Rep. 2022, 12, 2763. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhao, Y.; Wu, J.; Brendel, M.; Lu, J.; Ge, J.; Bernhardt, A.; Li, L.; Alberts, I.; Katzdobler, S.; et al. Differential diagnosis of parkinsonism based on deep metabolic imaging indices. J. Nucl. Med. 2022, 63, 1741–1747. [Google Scholar] [CrossRef]
- Vidović, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef]
- Nwabufo, C.K.; Aigbogun, O.P. Diagnostic and therapeutic agents that target alpha-synuclein in Parkinson’s disease. J. Neurol. 2022, 269, 5762–5786. [Google Scholar] [CrossRef]
- Stocchi, F.; Bravi, D.; Emmi, A.; Antonini, A. Parkinson disease therapy: Current strategies and future research priorities. Nat. Rev. Neurol. 2024, 20, 695–707. [Google Scholar] [CrossRef]
- Fahn, S. The 200-year journey of Parkinson disease: Reflecting on the past and looking towards the future. Park. Relat. Disord. 2018, 46, S1–S5. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Jasutkar, H.G.; Oh, S.E.; Mouradian, M.M. Therapeutics in the Pipeline Targeting α-Synuclein for Parkinson’s Disease. Pharmacol. Rev. 2022, 74, 207–237. [Google Scholar] [CrossRef]
- Smith, Y.; Wichmann, T.; Factor, S.A.; DeLong, M.R. Parkinson’s Disease Therapeutics: New Developments and Challenges Since the Introduction of Levodopa. Neuropsychopharmacology 2011, 37, 213–246. [Google Scholar] [CrossRef]
- Prasad, E.M.; Hung, S.-Y. Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update. Pharmaceuticals 2021, 14, 717. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tuka, B.; Vécsei, L. Navigating the Neurobiology of Migraine: From Pathways to Potential Therapies. Cells 2024, 13, 1098. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A.; Surguchev, A. Synucleins: New Data on Misfolding, Aggregation and Role in Diseases. Biomedicines 2022, 10, 3241. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Emre, M.; Jenner, P.; Poewe, W. Levodopa in the treatment of Parkinson’s disease. Eur. J. Neurol. 2009, 16, 982–989. [Google Scholar] [CrossRef]
- Olanow, C.W.; Agid, Y.; Mizuno, Y.; Albanese, A.; Bonucelli, U.; Damier, P.; De Yebenes, J.; Gershanik, O.; Guttman, M.; Grandas, F.; et al. Levodopa in the treatment of Parkinson’s disease: Current controversies. Mov. Disord. 2004, 19, 997–1005. [Google Scholar] [CrossRef]
- Salat, D.; Tolosa, E. Levodopa in the Treatment of Parkinson’s Disease: Current Status and New Developments. J. Park. Dis. 2013, 3, 255–269. [Google Scholar] [CrossRef]
- Jankovic, J.; Stacy, M. Medical Management of Levodopa-Associated Motor Complications in Patients with Parkinson’s Disease. CNS Drugs 2007, 21, 677–692. [Google Scholar] [CrossRef]
- Tanaka, M.; Battaglia, S. Dualistic Dynamics in Neuropsychiatry: From Monoaminergic Modulators to Multiscale Biomarker Maps. Biomedicines 2025, 13, 1456. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, X.; Chen, F.; Wen, S.; Zhou, C. Dopamine agonists versus levodopa monotherapy in early Parkinson’s disease for the potential risks of motor complications: A network meta-analysis. Eur. J. Pharmacol. 2023, 954, 175884. [Google Scholar] [CrossRef]
- Wiesen, T.; Atlas, D. Novel anti-apoptotic L-DOPA precursors SuperDopa and SuperDopamide as potential neuroprotective agents for halting/delaying progression of Parkinson’s disease. Cell Death Dis. 2022, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Mariani, L.-L.; Mangone, G.; Nailly, D.L.F.d.; Charbonnier-Beaupel, F.; Corvol, J.-C. Molecular basis of dopamine replacement therapy and its side effects in Parkinson’s disease. Cell Tissue Res. 2018, 373, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Demailly, A.; Moreau, C.; Devos, D. Effectiveness of Continuous Dopaminergic Therapies in Parkinson’s Disease: A Review of L-DOPA Pharmacokinetics/Pharmacodynamics. J. Park. Dis. 2024, 14, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Martos, D.; Lőrinczi, B.; Szatmári, I.; Vécsei, L.; Tanaka, M. Decoupling Behavioral Domains via Kynurenic Acid Analog Optimization: Implications for Schizophrenia and Parkinson’s Disease Therapeutics. Cells 2025, 14, 973. [Google Scholar] [CrossRef]
- Mao, Q.; Qin, W.-Z.; Zhang, A.; Ye, N. Recent advances in dopaminergic strategies for the treatment of Parkinson’s disease. Acta Pharmacol. Sin. 2020, 41, 471–482. [Google Scholar] [CrossRef]
- Liang, K.; Yang, L.; Kang, J.; Liu, B.; Zhang, D.; Wang, L.; Wang, W.; Wang, Q. Improving treatment for Parkinson’s disease: Harnessing photothermal and phagocytosis-driven delivery of levodopa nanocarriers across the blood-brain barrier. Asian J. Pharm. Sci. 2024, 19, 100963. [Google Scholar] [CrossRef]
- Neha; Mazahir, I.; Khan, S.A.; Kaushik, P.; Parvez, S. The Interplay of Mitochondrial Bioenergetics and Dopamine Agonists as an Effective Disease-Modifying Therapy for Parkinson’s Disease. Mol. Neurobiol. 2024, 61, 8086–8103. [Google Scholar] [CrossRef]
- Szabó, Á.; Galla, Z.; Spekker, E.; Szűcs, M.; Martos, D.; Takeda, K.; Ozaki, K.; Inoue, H.; Yamamoto, S.; Toldi, J.; et al. Oxidative and Excitatory Neurotoxic Stresses in CRISPR/Cas9-Induced Kynurenine Aminotransferase Knockout Mice: A Novel Model for Despair-Based Depression and Post-Traumatic Stress Disorder. Front. Biosci. 2025, 30, 25706. [Google Scholar] [CrossRef]
- Barbalho, S.M.; Laurindo, L.F.; Zanuso, B.d.O.; da Silva, R.M.S.; Caglioni, L.G.; de Moraes, V.B.F.N.J.; Laurindo, L.F.; Rodrigues, V.D.; Oliveira, J.d.S.C.; Beluce, M.E.; et al. AdipoRon’s Impact on Alzheimer’s Disease—A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2025, 26, 484. [Google Scholar] [CrossRef]
- Sackner-Bernstein, J. Rethinking Parkinson’s disease: Could dopamine reduction therapy have clinical utility? J. Neurol. 2024, 271, 5687–5695. [Google Scholar] [CrossRef] [PubMed]
- Manza, P.; Amandola, M.; Tatineni, V.; Li, C.-S.R.; Leung, H.-C. Response inhibition in Parkinson’s disease: A meta-analysis of dopaminergic medication and disease duration effects. npj Park. Dis. 2017, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Avenanti, A.; Vécsei, L.; Tanaka, M. Neurodegeneration in Cognitive Impairment and Mood Disorders for Experimental, Clinical and Translational Neuropsychiatry. Biomedicines 2024, 12, 574. [Google Scholar] [CrossRef] [PubMed]
- Charvin, D.; Medori, R.; Hauser, R.A.; Rascol, O. Therapeutic strategies for Parkinson disease: Beyond dopaminergic drugs. Nat. Rev. Drug Discov. 2018, 17, 804–822. [Google Scholar] [CrossRef]
- Barker, R.A.; Drouin-Ouellet, J.; Parmar, M. Cell-based therapies for Parkinson disease—Past insights and future potential. Nat. Rev. Neurol. 2015, 11, 492–503. [Google Scholar] [CrossRef]
- Martos, D.; Lőrinczi, B.; Szatmári, I.; Vécsei, L.; Tanaka, M. The Impact of C-3 Side Chain Modifications on Kynurenic Acid: A Behavioral Analysis of Its Analogs in the Motor Domain. Int. J. Mol. Sci. 2024, 25, 3394. [Google Scholar] [CrossRef]
- Oertel, W.; Schulz, J.B. Current and experimental treatments of Parkinson disease: A guide for neuroscientists. J. Neurochem. 2016, 139, 325–337. [Google Scholar] [CrossRef]
- Pagano, G.; Monnet, A.; Reyes, A.; Ribba, B.; Svoboda, H.; Kustermann, T.; Simuni, T.; Postuma, R.B.; Pavese, N.; Stocchi, F.; et al. Sustained effect of prasinezumab on Parkinson’s disease motor progression in the open-label extension of the PASADENA trial. Nat. Med. 2024, 30, 3669–3675. [Google Scholar] [CrossRef]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Poewe, W.; Volc, D.; Seppi, K.; Medori, R.; Lührs, P.; Kutzelnigg, A.; Djamshidian, A.; Thun-Hohenstein, C.; Meissner, W.G.; Rascol, O.; et al. Safety and Tolerability of Active Immunotherapy Targeting α-Synuclein with PD03A in Patients with Early Parkinson’s Disease: A Randomized, Placebo-Controlled, Phase 1 Study. J. Park. Dis. 2021, 11, 1079–1089. [Google Scholar] [CrossRef]
- Eijsvogel, P.; Misra, P.; Concha-Marambio, L.; Boyd, J.D.; Ding, S.; Fedor, L.; Hsieh, Y.-T.; Sun, Y.S.; Vroom, M.M.; Farris, C.M.; et al. Target engagement and immunogenicity of an active immunotherapeutic targeting pathological α-synuclein: A phase 1 placebo-controlled trial. Nat. Med. 2024, 30, 2631–2640. [Google Scholar] [CrossRef]
- Khrieba, M.O.; Hegazy, S.K.; Mustafa, W.; El-Haggar, S.M. Repurposing celecoxib as adjuvant therapy in patients with Parkinsonian disease: A new therapeutic dawn: Randomized controlled pilot study. Inflammopharmacology 2024, 32, 3729–3738. [Google Scholar] [CrossRef]
- Idowu, O.K.; Oremosu, A.A.; Dosumu, O.O.; Mohammed, A.A. Ribose-cysteine and levodopa abrogate Parkinsonism via the regulation of neurochemical and redox activities in alpha-synuclein transgenic Drosophila melanogaster models. Fly 2024, 18, 2306687. [Google Scholar] [CrossRef] [PubMed]
- Majbour, N.; Aasly, J.; Abdi, I.; Ghanem, S.; Erskine, D.; van de Berg, W.; El-Agnaf, O. Disease-Associated α-Synuclein Aggregates as Biomarkers of Parkinson Disease Clinical Stage. Neurology 2022, 99, e2417–e2427. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Saiki, S.; Shiina, K.; Iseki, T.; Sasazawa, Y.; Ishikawa, K.-I.; Nishikawa, N.; Sako, W.; Oyama, G.; Hatano, T.; et al. Comprehensive data for studying serum exosome microRNA transcriptome in Parkinson’s disease patients. Sci. Data 2024, 11, 1128. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.K.; Farmen, K.; Carstensen, M.; Schulte, C.; Goldeck, D.; Brockmann, K.; Romero-Ramos, M. Changes in CD163+, CD11b+, and CCR2+ peripheral monocytes relate to Parkinson’s disease and cognition. Brain Behav. Immun. 2022, 101, 182–193. [Google Scholar] [CrossRef]
- Simitsi, A.M.; Sfikas, E.; Koros, C.; Papagiannakis, N.; Beratis, I.; Papadimitriou, D.; Antonellou, R.; Fragiadaki, S.; Kontaxopoulou, D.; Picillo, M.; et al. Motor and nonmotor features of p.A53T alpha-synuclein PD vs. idiopathic PD: Longitudinal data from the PPMI study. J. Neurol. 2025, 272, 203. [Google Scholar] [CrossRef]
- Abdi, I.Y.; Bartl, M.; Dakna, M.; Abdesselem, H.; Majbour, N.; Trenkwalder, C.; El-Agnaf, O.; Mollenhauer, B. Cross-sectional proteomic expression in Parkinson’s disease-related proteins in drug-naïve patients vs. healthy controls with longitudinal clinical follow-up. Neurobiol. Dis. 2023, 177, 105997. [Google Scholar] [CrossRef]
- You, J.; Wang, L.; Wang, Y.; Kang, J.; Yu, J.; Cheng, W.; Feng, J. Prediction of Future Parkinson Disease Using Plasma Proteins Combined With Clinical-Demographic Measures. Neurology 2024, 103, e209531. [Google Scholar] [CrossRef]
- Lewis, M.M.; Mailman, R.B.; Cheng, X.V.; Du, G.; Zhang, L.; Li, C.; De Jesus, S.; Tabbal, S.D.; Li, R.; Huang, X. Clinical progression of Parkinson’s disease in the early 21st century: Insights from the accelerating medicine partnership (AMP-PD) data. Park. Relat. Disord. 2024, 130, 107186. [Google Scholar] [CrossRef]
- Bovenzi, R.; Conti, M.; Degoli, G.R.; Cerroni, R.; Simonetta, C.; Liguori, C.; Salimei, C.; Pisani, A.; Pierantozzi, M.; Stefani, A.; et al. Shaping the course of early-onset Parkinson’s disease: Insights from a longitudinal cohort. Neurol. Sci. 2023, 44, 3151–3159. [Google Scholar] [CrossRef] [PubMed]
- Severson, K.A.; Chahine, L.M.; Smolensky, L.A.; Dhuliawala, M.; Frasier, M.; Ng, K.; Ghosh, S.; Hu, J. Discovery of Parkinson’s disease states and disease progression modelling: A longitudinal data study using machine learning. Lancet Digit. Health 2021, 3, e555–e564. [Google Scholar] [CrossRef] [PubMed]
- Almgren, H.; Camacho, M.; Hanganu, A.; Kibreab, M.; Camicioli, R.; Ismail, Z.; Forkert, N.D.; Monchi, O. Machine learning-based prediction of longitudinal cognitive decline in early Parkinson’s disease using multimodal features. Sci. Rep. 2023, 13, 13193. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Basaia, S.; Sarasso, E.; Stojkovic, T.; Stankovic, I.; Fontana, A.; Tomic, A.; Piramide, N.; Stefanova, E.; Markovic, V.; et al. Longitudinal brain connectivity changes and clinical evolution in Parkinson’s disease. Mol. Psychiatry 2020, 26, 5429–5440. [Google Scholar] [CrossRef]
- Pereira, J.B.; Hall, S.; Jalakas, M.; Grothe, M.J.; Strandberg, O.; Stomrud, E.; Westman, E.; van Westen, D.; Hansson, O. Longitudinal degeneration of the basal forebrain predicts subsequent dementia in Parkinson’s disease. Neurobiol. Dis. 2020, 139, 104831. [Google Scholar] [CrossRef]
- Basaia, S.; Agosta, F.; Francia, A.; Cividini, C.; Balestrino, R.; Stojkovic, T.; Stankovic, I.; Markovic, V.; Sarasso, E.; Gardoni, A.; et al. Cerebro-cerebellar motor networks in clinical subtypes of Parkinson’s disease. npj Park. Dis. 2022, 8, 113. [Google Scholar] [CrossRef]
- Schalkamp, A.-K.; Peall, K.J.; Harrison, N.A.; Sandor, C. Wearable movement-tracking data identify Parkinson’s disease years before clinical diagnosis. Nat. Med. 2023, 29, 2048–2056. [Google Scholar] [CrossRef]
- Tanaka, M.; Battaglia, S.; Liloia, D. Navigating Neurodegeneration: Integrating Biomarkers, Neuroinflammation, and Imaging in Parkinson’s, Alzheimer’s, and Motor Neuron Disorders. Biomedicines 2025, 13, 1045. [Google Scholar] [CrossRef]
- Link, T.; Maraghi, S.; Reddy, M.; Sheth, H.; Stark, K.; Vijay, V. Role of Alpha-Synuclein in Parkinson’s Disease. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Hallacli, E.; Kayatekin, C.; Nazeen, S.; Wang, X.H.; Sheinkopf, Z.; Sathyakumar, S.; Sarkar, S.; Jiang, X.; Dong, X.; Di Maio, R.; et al. The Parkinson’s disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability. Cell 2022, 185, 2035–2056.e33. [Google Scholar] [CrossRef]
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The mechanistic role of alpha-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 2020, 29, 3107–3121. [Google Scholar] [CrossRef]
- Cardinale, A.; Calabrese, V.; de Iure, A.; Picconi, B. Alpha-Synuclein as a Prominent Actor in the Inflammatory Synaptopathy of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6517. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: From overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Dettmer, U. Alpha-Synuclein Effects on Mitochondrial Quality Control in Parkinson’s Disease. Biomolecules 2024, 14, 1649. [Google Scholar] [CrossRef]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef]
- Oliveira, L.M.A.; Gasser, T.; Edwards, R.; Zweckstetter, M.; Melki, R.; Stefanis, L.; Lashuel, H.A.; Sulzer, D.; Vekrellis, K.; Halliday, G.M.; et al. Alpha-synuclein research: Defining strategic moves in the battle against Parkinson’s disease. npj Park. Dis. 2021, 7, 65. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Fares, M.B.; Jagannath, S.; Lashuel, H.A. Reverse engineering Lewy bodies: How far have we come and how far can we go? Nat. Rev. Neurosci. 2021, 22, 111–131. [Google Scholar] [CrossRef]
- Moors, T.E.; Maat, C.A.; Niedieker, D.; Mona, D.; Petersen, D.; Timmermans-Huisman, E.; Kole, J.; El-Mashtoly, S.F.; Spycher, L.; Zago, W.; et al. The subcellular arrangement of alpha-synuclein proteoforms in the Parkinson’s disease brain as revealed by multicolor STED microscopy. Acta Neuropathol. 2021, 142, 423–448. [Google Scholar] [CrossRef]
- Lashuel, H.A. Do Lewy bodies contain alpha-synuclein fibrils? and Does it matter? A brief history and critical analysis of recent reports. Neurobiol. Dis. 2020, 141, 104876. [Google Scholar] [CrossRef]
- Petyuk, V.A.; Yu, L.; Olson, H.M.; Yu, F.; Clair, G.; Qian, W.-J.; Shulman, J.M.; Bennett, D.A. Proteomic Profiling of the Substantia Nigra to Identify Determinants of Lewy Body Pathology and Dopaminergic Neuronal Loss. J. Proteome Res. 2021, 20, 2266–2282. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Calixto, A.; Mukherjee, A.; Ramirez, S.; Sepulveda, S.; Sinha, T.; Al-Lahham, R.; De Gregorio, N.; Gherardelli, C.; Soto, C. Lewy Body-like Pathology and Loss of Dopaminergic Neurons in Midbrain Organoids Derived from Familial Parkinson’s Disease Patient. Cells 2023, 12, 625. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.J.; van den Heuvel, L.; Di Fabrizio, M.; Burger, D.; Huisman, E.; Bol, J.G.; van de Berg, W.D.; Stahlberg, H. Ultra-structural diversity of alpha-Synuclein pathology in the post-mortem brain of Parkinson patients: Implications for Lewy Body formation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Borghammer, P.; Horsager, J.; Andersen, K.; Berge, N.V.D.; Raunio, A.; Murayama, S.; Parkkinen, L.; Myllykangas, L. Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol. Dis. 2021, 161, 105557. [Google Scholar] [CrossRef]
- Li, J.; Ng, K.W.; Sung, C.C.; Chung, K.K.K. The role of age-associated alpha-synuclein aggregation in a conditional transgenic mouse model of Parkinson’s disease: Implications for Lewy body formation. J. Neurochem. 2024, 168, 1215–1236. [Google Scholar] [CrossRef]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef]
- Xiao, B.; Zhou, Z.; Chao, Y.; Tan, E.-K. Pathogenesis of Parkinson’s Disease. Neurol. Clin. 2025, 43, 185–207. [Google Scholar] [CrossRef]
- Yao, L.; Wu, J.; Koc, S.; Lu, G. Genetic Imaging of Neuroinflammation in Parkinson’s Disease: Recent Advancements. Front. Cell Dev. Biol. 2021, 9, 655819. [Google Scholar] [CrossRef] [PubMed]
- Over, L.; Brüggemann, N.; Lohmann, K.; Bonne, G. Therapies for Genetic Forms of Parkinson’s Disease: Systematic Literature Review. J. Neuromuscul. Dis. 2021, 8, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Cristina, T.-P.; Pablo, M.; Teresa, P.M.; Lydia, V.-D.; Irene, A.-R.; Araceli, A.-C.; Inmaculada, B.-B.; Marta, B.-T.; Dolores, B.-R.; José, C.-A.M.; et al. A genetic analysis of a Spanish population with early onset Parkinson’s disease. PLoS ONE 2020, 15, e0238098. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chan, S.-W.; Zhao, H.; Miu, K.-K.; Chan, W.-Y. Outlook of PINK1/Parkin signaling in molecular etiology of Parkinson’s disease, with insights into Pink1 knockout models. Zool. Res. 2023, 44, 559–576. [Google Scholar] [CrossRef]
- Planas-Ballvé, A.; Vilas, D.; Pisani, A. Cognitive Impairment in Genetic Parkinson’s Disease. Park. Dis. 2021, 2021, 8610285. [Google Scholar] [CrossRef]
- Landoulsi, Z.; Pachchek, S.; Bobbili, D.R.; Pavelka, L.; May, P.; Krüger, R. The NCER-PD Consortium Genetic landscape of Parkinson’s disease and related diseases in Luxembourg. Front. Aging Neurosci. 2023, 15, 1282174. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Fan, C.C.; Frei, O.; Tan, M.; Karunamuni, R.A.; Blauwendraat, C.; Bandres-Ciga, S.; Gan-Or, Z.; Grosset, D.G.; International Parkinson’s Disease Genomics Consortium (IPDGC); et al. Genetic Stratification of Age-Dependent Parkinson’s Disease Risk by Polygenic Hazard Score. Mov. Disord. 2021, 37, 62–69. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Belete, D.; Bestwick, J.; Blauwendraat, C.; Bandres-Ciga, S.; Heilbron, K.; Dobson, R.; Nalls, M.A.; Singleton, A.; Hardy, J.; et al. Parkinson’s disease determinants, prediction and gene–environment interactions in the UK Biobank. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1046–1054. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Saez-Atienzar, S.; Kim, J.J.; Makarious, M.B.; Faghri, F.; Diez-Fairen, M.; Iwaki, H.; Leonard, H.; Botia, J.; Ryten, M.; et al. Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol. 2020, 140, 341–358. [Google Scholar] [CrossRef]
- Hall, A.; Bandres-Ciga, S.; Diez-Fairen, M.; Quinn, J.P.; Billingsley, K.J. Genetic Risk Profiling in Parkinson’s Disease and Utilizing Genetics to Gain Insight into Disease-Related Biological Pathways. Int. J. Mol. Sci. 2020, 21, 7332. [Google Scholar] [CrossRef]
- Goldstein, O.; Shani, S.; Gana-Weisz, M.; Elkoshi, N.; Casey, F.; Sun, Y.H.; Chandratre, K.; Cedarbaum, J.M.; Blauwendraat, C.; Bar-Shira, A.; et al. The effect of polygenic risk score on PD risk and phenotype in LRRK2 G2019S and GBA1 carriers. J. Park. Dis. 2025, 15, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Kukkle, P.L.; Geetha, T.S.; Chaudhary, R.; Sathirapongsasuti, J.F.; Goyal, V.; Kandadai, R.M.; Kumar, H.; Borgohain, R.; Mukherjee, A.; Oliver, M.; et al. Genome-Wide Polygenic Score Predicts Large Number of High Risk Individuals in Monogenic Undiagnosed Young Onset Parkinson’s Disease Patients from India. Adv. Biol. 2022, 6, e2101326. [Google Scholar] [CrossRef]
- Foo, J.N.; Chew, E.G.Y.; Chung, S.J.; Peng, R.; Blauwendraat, C.; Nalls, M.A.; Mok, K.Y.; Satake, W.; Toda, T.; Chao, Y.; et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. 2020, 77, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, L.; Gaudio, A.; Monfrini, E.; Avanzino, L.; Di Fonzo, A.; Mandich, P. Genetics in Parkinson’s disease, state-of-the-art and future perspectives. Br. Med. Bull. 2024, 149, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, N.; Tremblay, C.; Rajimehr, R.; Yu, E.; Markello, R.D.; Shafiei, G.; Khatibi, N.; ENIGMA-Parkinson’s study; Jahanshad, N.; Thompson, P.M. Neuroanatomical correlates of polygenic risk for Parkinson’s Disease. medRxiv 2022. [Google Scholar] [CrossRef]
- Koch, S.; Laabs, B.-H.; Kasten, M.; Vollstedt, E.-J.; Becktepe, J.; Brüggemann, N.; Franke, A.; Krämer, U.M.; Kuhlenbäumer, G.; Lieb, W.; et al. Validity and Prognostic Value of a Polygenic Risk Score for Parkinson’s Disease. Genes 2021, 12, 1859. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. Parkinson’s Disease Is Predominantly an Environmental Disease. J. Park. Dis. 2024, 14, 451–465. [Google Scholar] [CrossRef]
- Aravindan, A.; Newell, M.E.; Halden, R.U. Literature review and meta-analysis of environmental toxins associated with increased risk of Parkinson’s disease. Sci. Total Environ. 2024, 931, 172838. [Google Scholar] [CrossRef]
- Brolin, K.A.; Schaeffer, E.; Kuri, A.; Rumrich, I.K.; Schuh, A.F.S.; Darweesh, S.K.; Kaasinen, V.; Tolppanen, A.; Chahine, L.M.; Noyce, A.J. Environmental Risk Factors for Parkinson’s Disease: A Critical Review and Policy Implications. Mov. Disord. 2024, 40, 204–221. [Google Scholar] [CrossRef]
- Murata, H.; Barnhill, L.M.; Bronstein, J.M. Air Pollution and the Risk of Parkinson’s Disease: A Review. Mov. Disord. 2022, 37, 894–904. [Google Scholar] [CrossRef]
- Huang, M.; Bargues-Carot, A.; Riaz, Z.; Wickham, H.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Impact of Environmental Risk Factors on Mitochondrial Dysfunction, Neuroinflammation, Protein Misfolding, and Oxidative Stress in the Etiopathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 10808. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Zinchenko, V.P.; Goncharov, N.V. Molecular and Cellular Interactions in Pathogenesis of Sporadic Parkinson Disease. Int. J. Mol. Sci. 2022, 23, 13043. [Google Scholar] [CrossRef]
- Kline, E.M.; Houser, M.C.; Herrick, M.K.; Seibler, P.; Klein, C.; West, A.; Tansey, M.G. Genetic and Environmental Factors in Parkinson’s Disease Converge on Immune Function and Inflammation. Mov. Disord. 2021, 36, 25–36. [Google Scholar] [CrossRef]
- Tsalenchuk, M.; Gentleman, S.M.; Marzi, S.J. Linking environmental risk factors with epigenetic mechanisms in Parkinson’s disease. npj Park. Dis. 2023, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sci. 2022, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, B.R.; Goldman, S.M.; Miller, G.W.; Greenamyre, J.T.; Dorsey, E.R. Preventing Parkinson’s Disease: An Environmental Agenda. J. Park. Dis. 2022, 12, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Periñán, M.T.; Brolin, K.; Bandres-Ciga, S.; Blauwendraat, C.; Klein, C.; Gan-Or, Z.; Singleton, A.; Gomez-Garre, P.; Swanberg, M.; Mir, P.; et al. Effect Modification between Genes and Environment and Parkinson’s Disease Risk. Ann. Neurol. 2022, 92, 715–724. [Google Scholar] [CrossRef]
- Huang, Y.-M.; Ma, Y.-H.; Gao, P.-Y.; Cui, X.-H.; Hou, J.-H.; Chi, H.-C.; Fu, Y.; Wang, Z.-B.; Feng, J.-F.; Cheng, W.; et al. Genetic susceptibility modifies the association of long-term air pollution exposure on Parkinson’s disease. npj Park. Dis. 2024, 10, 23. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Schaffner, S.L.; Kobor, M.S. DNA methylation as a mediator of genetic and environmental influences on Parkinson’s disease susceptibility: Impacts of alpha-Synuclein, physical activity, and pesticide exposure on the epigenome. Front. Genet. 2022, 13, 971298. [Google Scholar] [CrossRef]
- Reynoso, A.; Torricelli, R.; Jacobs, B.M.; Shi, J.; Aslibekyan, S.; Norcliffe-Kaufmann, L.; Noyce, A.J.; Heilbron, K. Gene–Environment Interactions for Parkinson’s Disease. Ann. Neurol. 2024, 95, 677–687. [Google Scholar] [CrossRef]
- Clarimón, J. Genetic-environmental factors finally assessed together in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1030. [Google Scholar] [CrossRef]
- Yang, L.; Mao, K.; Yu, H.; Chen, J. Neuroinflammatory Responses and Parkinson’ Disease: Pathogenic Mechanisms and Therapeutic Targets. J. Neuroimmune Pharmacol. 2020, 15, 830–837. [Google Scholar] [CrossRef]
- Isik, S.; Kiyak, B.Y.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef] [PubMed]
- Barbalho, S.M.; Boaro, B.L.; Oliveira, J.d.S.C.; Patočka, J.; Lamas, C.B.; Tanaka, M.; Laurindo, L.F. Molecular Mechanisms Underlying Neuroinflammation Intervention with Medicinal Plants: A Critical and Narrative Review of the Current Literature. Pharmaceuticals 2025, 18, 133. [Google Scholar] [CrossRef] [PubMed]
- Araújo, B.; Caridade-Silva, R.; Soares-Guedes, C.; Martins-Macedo, J.; Gomes, E.D.; Monteiro, S.; Teixeira, F.G. Neuroinflammation and Parkinson’s Disease—From Neurodegeneration to Therapeutic Opportunities. Cells 2022, 11, 2908. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Janrao, S.; Srivastava, S.; Singh, S.B.; Vora, L.; Khatri, D.K. GSK-3β: An exuberating neuroinflammatory mediator in Parkinson’s disease. Biochem. Pharmacol. 2023, 210, 115496. [Google Scholar] [CrossRef]
- Zhang, P.-F.; Gao, F. Neuroinflammation in Parkinson’s disease: A meta-analysis of PET imaging studies. J. Neurol. 2021, 269, 2304–2314. [Google Scholar] [CrossRef]
- Liu, T.-W.; Chen, C.-M.; Chang, K.-H. Biomarker of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4148. [Google Scholar] [CrossRef]
- Grotemeyer, A.; McFleder, R.L.; Wu, J.; Wischhusen, J.; Ip, C.W. Neuroinflammation in Parkinson’s Disease—Putative Pathomechanisms and Targets for Disease-Modification. Front. Immunol. 2022, 13, 878771. [Google Scholar] [CrossRef]
- Jurcau, A.; Andronie-Cioara, F.L.; Nistor-Cseppento, D.C.; Pascalau, N.; Rus, M.; Vasca, E.; Jurcau, M.C. The Involvement of Neuroinflammation in the Onset and Progression of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 14582. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Ko, E.Y.; Andrews, A.E.; Shin, J.E.; Nance, K.J.; Barman, P.K.; Heeger, P.S.; Freeman, W.M.; Benayoun, B.A.; Goodridge, H.S. Microglia undergo sex-dimorphic transcriptional and metabolic rewiring during aging. J. Neuroinflamm. 2024, 21, 150. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.-V.; Araiz, A.R.; Mela, V.; Gaban, A.S.; O’nEill, E.; Joshi, L.; Chouchani, E.T.; Mills, E.L.; Lynch, M.A. Microglial metabolism is a pivotal factor in sexual dimorphism in Alzheimer’s disease. Commun. Biol. 2021, 4, 711. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Al Mamun, A.; Ngwa, C.; Romana, S.; Ritzel, R.; Arnold, A.P.; McCullough, L.D.; Liu, F. X chromosome escapee genes are involved in ischemic sexual dimorphism through epigenetic modification of inflammatory signals. J. Neuroinflamm. 2021, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, G.L.; Lizama, C.O.; Keown-Lang, A.E.; Niu, A.; Santander, N.; Larpthaveesarp, A.; Chee, E.; Gonzalez, F.F.; Arnold, T.D. A new genetic strategy for targeting microglia in development and disease. eLife 2020, 9, e54590. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Revolutionizing our understanding of Parkinson’s disease: Dr. Heinz Reichmann’s pioneering research and future research direction. J. Neural Transm. 2024, 131, 1367–1387. [Google Scholar] [CrossRef]
- Sharma, S.; Trivedi, S.; Pandey, T.; Ranjan, S.; Trivedi, M.; Pandey, R. Wedelolactone Mitigates Parkinsonism Via Alleviating Oxidative Stress and Mitochondrial Dysfunction Through NRF2/SKN-1. Mol. Neurobiol. 2020, 58, 65–77. [Google Scholar] [CrossRef]
- Rasheed, Z.; Khatoon, R.; Talat, F.; Alam, M.M.; Tabassum, H.; Parvez, S. Melatonin Mitigates Rotenone-Induced Oxidative Stress and Mitochondrial Dysfunction in the Drosophila melanogaster Model of Parkinson’s Disease-like Symptoms. ACS Omega 2023, 8, 7279–7288. [Google Scholar] [CrossRef]
- Ishola, I.; Awogbindin, I.; Olubodun-Obadun, T.; Olajiga, A.; Adeyemi, O. Vinpocetine prevents rotenone-induced Parkinson disease motor and non-motor symptoms through attenuation of oxidative stress, neuroinflammation and α-synuclein expressions in rats. NeuroToxicology 2023, 96, 37–52. [Google Scholar] [CrossRef]
- Godoy, A.C.F.; Frota, F.F.; Araújo, L.P.; Valenti, V.E.; Pereira, E.d.S.B.M.; Detregiachi, C.R.P.; Galhardo, C.M.; Caracio, F.C.; Haber, R.S.; Laurindo, L.F. Phytochemicals and Depression: Plant-Powered Pathways Preventing Neuroinflammation, Ox-idative Overload, and Mitochondrial Malfunction in Major Depressive Disorder. preprints 2025. [Google Scholar] [CrossRef]
- Magaña, J.C.; Deus, C.M.; Baldellou, L.; Avellanet, M.; Gea-Rodríguez, E.; Enriquez-Calzada, S.; Laguna, A.; Martínez-Vicente, M.; Hernández-Vara, J.; Giné-Garriga, M.; et al. Investigating the impact of physical activity on mitochondrial function in Parkinson’s disease (PARKEX): Study protocol for A randomized controlled clinical trial. PLoS ONE 2023, 18, e0293774. [Google Scholar] [CrossRef]
- Hong, C.-T.; Hu, C.-J.; Lin, H.-Y.; Wu, D.; Zhu, Y. Effects of concomitant use of hydrogen water and photobiomodulation on Parkinson disease. Medicine 2021, 100, e24191. [Google Scholar] [CrossRef]
- Jovanovic, M.Z.; Stanojevic, J.; Stevanovic, I.; Ninkovic, M.; Nedeljkovic, N.; Dragic, M. Sustained Systemic Antioxidative Effects of Intermittent Theta Burst Stimulation beyond Neurodegeneration: Implications in Therapy in 6-Hydroxydopamine Model of Parkinson’s Disease. Antioxidants 2024, 13, 218. [Google Scholar] [CrossRef]
- Tanaka, M.; He, Z.; Han, S.; Battaglia, S. Editorial: Noninvasive brain stimulation: A promising approach to study and improve emotion regulation. Front. Behav. Neurosci. 2025, 19, 1633936. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, N.N.; Tadros, M.G.; George, M.Y. Empagliflozin repurposing in Parkinson’s disease; modulation of oxidative stress, neuroinflammation, AMPK/SIRT-1/PGC-1α, and wnt/β-catenin pathways. Inflammopharmacology 2023, 32, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Szabó, Á.; Vécsei, L. Redefining Roles: A Paradigm Shift in Tryptophan–Kynurenine Metabolism for Innovative Clinical Applications. Int. J. Mol. Sci. 2024, 25, 12767. [Google Scholar] [CrossRef] [PubMed]
- Uthman, Y.A.; Ibrahim, K.G.; Abubakar, M.B.; Sulaiman, I.; Imam, M.U. Neuroprotective effects of brown rice consumption in an iron-induced parkinsonism in Drosophila. Nutr. Neurosci. 2024, 28, 591–601. [Google Scholar] [CrossRef]
- Vastegani, S.M.; Khoshnam, S.E.; Ghafouri, S.; Bakhtiari, N.; Farbood, Y.; Sarkaki, A.; Ribeiro, W.L.C. Anethole attenuates motor dysfunctions, striatal neuronal activity deficiency and blood brain barrier permeability by decreasing striatal α-synuclein and oxidative stress in rotenone-induced Parkinson’s disease of male rats. PLoS ONE 2023, 18, e0294612. [Google Scholar] [CrossRef]
- Nunes, Y.C.; Mendes, N.M.; de Lima, E.P.; Chehadi, A.C.; Lamas, C.B.; Haber, J.F.S.; Bueno, M.d.S.; Araújo, A.C.; Catharin, V.C.S.; Detregiachi, C.R.P.; et al. Curcumin: A Golden Approach to Healthy Aging: A Systematic Review of the Evidence. Nutrients 2024, 16, 2721. [Google Scholar] [CrossRef]
- Neto, L.J.V.; de Araujo, M.R.; Junior, R.C.M.; Machado, N.M.; Joshi, R.K.; Buglio, D.d.S.; Lamas, C.B.; Direito, R.; Laurindo, L.F.; Tanaka, M.; et al. Investigating the Neuroprotective and Cognitive-Enhancing Effects of Bacopa monnieri: A Systematic Review Focused on Inflammation, Oxidative Stress, Mitochondrial Dysfunction, and Apoptosis. Antioxidants 2024, 13, 393. [Google Scholar] [CrossRef]
- Tan, A.H.; Lim, S.Y.; Lang, A.E. The microbiome–gut–brain axis in Parkinson disease—From basic research to the clinic. Nat. Rev. Neurol. 2022, 18, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Gao, V.; Crawford, C.V.; Burré, J. The Gut–Brain Axis in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2024, 15, a041618. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, K.W.; Lee, E.J. Gut–brain axis and environmental factors in Parkinson’s disease: Bidirectional link between disease onset and progression. Neural Regen. Res. 2024, 20, 3416–3429. [Google Scholar] [CrossRef] [PubMed]
- Dogra, N.; Mani, R.J.; Katare, D.P. The Gut-Brain Axis: Two Ways Signaling in Parkinson’s Disease. Cell. Mol. Neurobiol. 2021, 42, 315–332. [Google Scholar] [CrossRef]
- Bonaz, B. The gut-brain axis in Parkinson’s disease. Rev. Neurol. 2023, 180, 65–78. [Google Scholar] [CrossRef]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- dos Santos, J.C.C.; Rebouças, C.d.S.M.; Oliveira, L.F.; Cardoso, F.d.S.; Nascimento, T.d.S.; Oliveira, A.V.; Lima, M.P.P.; de Andrade, G.M.; Brito, G.A.d.C.; Viana, G.S.d.B. The role of gut-brain axis in a rotenone-induced rat model of Parkinson’s disease. Neurobiol. Aging 2023, 132, 185–197. [Google Scholar] [CrossRef]
- Soni, D.; Upadhayay, S.; Dhureja, M.; Arthur, R.; Kumar, P. Crosstalk between gut–brain axis: Unveiling the mysteries of gut ROS in progression of Parkinson’s disease. Inflammopharmacology 2024, 32, 2921–2941. [Google Scholar] [CrossRef]
- Caputi, V.; Giron, M.C. Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 1689. [Google Scholar] [CrossRef]
- Lucchesi, M.; Biso, L.; Bonaso, M.; Longoni, B.; Buchignani, B.; Battini, R.; Santorelli, F.M.; Doccini, S.; Scarselli, M. Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease. Int. J. Mol. Sci. 2025, 26, 4451. [Google Scholar] [CrossRef]
- Hattori, N.; Sato, S. Mitochondrial dysfunction in Parkinson’s disease. J. Neural Transm. 2024, 131, 1415–1428. [Google Scholar] [CrossRef]
- Vastegani, S.M.; Nasrolahi, A.; Ghaderi, S.; Belali, R.; Rashno, M.; Farzaneh, M.; Khoshnam, S.E. Mitochondrial Dysfunction and Parkinson’s Disease: Pathogenesis and Therapeutic Strategies. Neurochem. Res. 2023, 48, 2285–2308. [Google Scholar] [CrossRef]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2021, 8, 615461. [Google Scholar] [CrossRef] [PubMed]
- Bantle, C.M.; Hirst, W.D.; Weihofen, A.; Shlevkov, E. Mitochondrial Dysfunction in Astrocytes: A Role in Parkinson’s Disease? Front. Cell Dev. Biol. 2021, 8, 608026. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Liu, H.; Gao, Y.; Cao, G.; Wang, Y.; Li, Z. Ameliorating Mitochondrial Dysfunction for the Therapy of Parkinson’s Disease. Small 2024, 20, e2311571. [Google Scholar] [CrossRef] [PubMed]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Henrich, M.T.; Oertel, W.H.; Surmeier, D.J.; Geibl, F.F. Mitochondrial dysfunction in Parkinson’s disease—A key disease hallmark with therapeutic potential. Mol. Neurodegener. 2023, 18, 83. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Coelho-Júnior, H.J.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. [Google Scholar] [CrossRef]
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Chaudhuri, K.R.; Weintraub, D. Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Prim. 2021, 7, 47. [Google Scholar] [CrossRef]
- Pagotto, G.L.d.O.; dos Santos, L.M.O.; Osman, N.; Lamas, C.B.; Laurindo, L.F.; Pomini, K.T.; Guissoni, L.M.; de Lima, E.P.; Goulart, R.d.A.; Catharin, V.M.C.S.; et al. Ginkgo biloba: A Leaf of Hope in the Fight against Alzheimer’s Dementia: Clinical Trial Systematic Review. Antioxidants 2024, 13, 651. [Google Scholar] [CrossRef]
- Váradi, C. Clinical Features of Parkinson’s Disease: The Evolution of Critical Symptoms. Biology 2020, 9, 103. [Google Scholar] [CrossRef]
- Silva, A.B.R.L.; de Oliveira, R.W.G.; Diógenes, G.P.; Aguiar, M.F.d.C.; Sallem, C.C.; Lima, M.P.P.; Filho, L.B.d.A.; de Medeiros, S.D.P.; de Mendonça, L.L.P.; Filho, P.C.d.S.; et al. Premotor, nonmotor and motor symptoms of Parkinson’s Disease: A new clinical state of the art. Ageing Res. Rev. 2022, 84, 101834. [Google Scholar] [CrossRef]
- Morris, H.R.; Spillantini, M.G.; Sue, C.M.; Williams-Gray, C.H. The pathogenesis of Parkinson’s disease. Lancet 2024, 403, 293–304. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 95–121. [Google Scholar] [CrossRef] [PubMed]
- Bidesi, N.S.R.; Andersen, I.V.; Windhorst, A.D.; Shalgunov, V.; Herth, M.M. The role of neuroimaging in Parkinson’s disease. J. Neurochem. 2021, 159, 660–689. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Reddy, R.P.; Roghani, A.K.; Garcia, R.I.; Khemka, S.; Pattoor, V.; Jacob, M.; Reddy, P.H.; Sehar, U. Artificial intelligence in Parkinson’s disease: Early detection and diagnostic advancements. Ageing Res. Rev. 2024, 99, 102410. [Google Scholar] [CrossRef]
- Costa, H.N.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Parkinson’s Disease: A Multisystem Disorder. Neurosci. Bull. 2022, 39, 113–124. [Google Scholar] [CrossRef]
- Zhu, B.; Yin, D.; Zhao, H.; Zhang, L. The immunology of Parkinson’s disease. Semin. Immunopathol. 2022, 44, 659–672. [Google Scholar] [CrossRef]
- Delic, V.; Beck, K.D.; Pang, K.C.H.; Citron, B.A. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 2020, 8, 45. [Google Scholar] [CrossRef]
- Li, T.; Le, W.; Jankovic, J. Linking the cerebellum to Parkinson disease: An update. Nat. Rev. Neurol. 2023, 19, 645–654. [Google Scholar] [CrossRef]
- Liu, Z.; Cheung, H.-H. Stem Cell-Based Therapies for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 8060. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Mining imaging and clinical data with machine learning approaches for the diagnosis and early detection of Parkinson’s disease. npj Park. Dis. 2022, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Thakur, J.; Godad, A. Deciphering the role of neuropeptides as biomarkers for early diagnosis of Parkinson’s disease. Life Sci. 2025, 363, 123376. [Google Scholar] [CrossRef] [PubMed]
- Priyadharshini, S.; Ramkumar, K.; Vairavasundaram, S.; Narasimhan, K.; Venkatesh, S.; Amirtharajan, R.; Kotecha, K. A Comprehensive framework for Parkinson’s disease diagnosis using explainable artificial intelligence empowered machine learning techniques. Alex. Eng. J. 2024, 107, 568–582. [Google Scholar] [CrossRef]
- Gouda, N.A.; Elkamhawy, A.; Cho, J. Emerging Therapeutic Strategies for Parkinson’s Disease and Future Prospects: A 2021 Update. Biomedicines 2022, 10, 371. [Google Scholar] [CrossRef]
- Stoker, T.B.; Barker, R.A. Recent developments in the treatment of Parkinson’s Disease. F1000Research 2020, 9, 862. [Google Scholar] [CrossRef]
- Ntetsika, T.; Papathoma, P.-E.; Markaki, I. Novel targeted therapies for Parkinson’s disease. Mol. Med. 2021, 27, 17. [Google Scholar] [CrossRef]
- Akki, A.J.; Patil, S.A.; Hungund, S.; Sahana, R.; Patil, M.M.; Kulkarni, R.V.; Reddy, K.R.; Zameer, F.; Raghu, A.V. Advances in Parkinson’s disease research—A computational network pharmacological approach. Int. Immunopharmacol. 2024, 139, 112758. [Google Scholar] [CrossRef]
- Tanaka, M.; Szatmári, I.; Vécsei, L. Quinoline Quest: Kynurenic Acid Strategies for Next-Generation Therapeutics via Rational Drug Design. Pharmaceuticals 2025, 18, 607. [Google Scholar] [CrossRef]
- Alvarez, M.M.; Cano-Herrera, G.; Martínez, M.F.O.; Gonzales-Portillo, J.V.; Monroy, G.R.; Pérez, R.M.; Torres-Ríos, J.A.; van Tienhoven, X.A.; Bernot, E.M.G.; Salazar, F.E.; et al. A Comprehensive Approach to Parkinson’s Disease: Addressing Its Molecular, Clinical, and Therapeutic Aspects. Int. J. Mol. Sci. 2024, 25, 7183. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Sharma, K.; Agyeah, G.; Krüger, R.; Grünewald, A.; Fitzgerald, J.C. Neurodegeneration and Neuroinflammation in Parkinson’s Disease: A Self-Sustained Loop. Curr. Neurol. Neurosci. Rep. 2022, 22, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, P.; Amaral, J.; Rodrigues, C. Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef]
- Surguchov, A. Invertebrate Models Untangle the Mechanism of Neurodegeneration in Parkinson’s Disease. Cells 2021, 10, 407. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef]
- Coukos, R.; Krainc, D. Key genes and convergent pathogenic mechanisms in Parkinson disease. Nat. Rev. Neurosci. 2024, 25, 393–413. [Google Scholar] [CrossRef]
- Coleman, C.; Martin, I. Unraveling Parkinson’s Disease Neurodegeneration: Does Aging Hold the Clues? J. Park. Dis. 2022, 12, 2321–2338. [Google Scholar] [CrossRef]
- Koziorowski, D.; Figura, M.; Milanowski, Ł.M.; Szlufik, S.; Alster, P.; Madetko, N.; Friedman, A. Mechanisms of Neurodegeneration in Various Forms of Parkinsonism—Similarities and Differences. Cells 2021, 10, 656. [Google Scholar] [CrossRef]
- Kim, J.J.; Vitale, D.; Otani, D.V.; Lian, M.M.; Heilbron, K.; Iwaki, H.; Lake, J.; Solsberg, C.W.; Leonard, H.; Makarious, M.B.; et al. Multi-ancestry genome-wide association meta-analysis of Parkinson’s disease. Nat. Genet. 2023, 56, 27–36. [Google Scholar] [CrossRef]
- Schilder, B.M.; Raj, T. Fine-mapping of Parkinson’s disease susceptibility loci identifies putative causal variants. Hum. Mol. Genet. 2021, 31, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ortiz, M.E.; Seo, Y.; Posavi, M.; Cordon, M.C.; Clark, E.; Jain, N.; Charan, R.; Gallagher, M.D.; Unger, T.L.; Amari, N.; et al. GPNMB confers risk for Parkinson’s disease through interaction with α-synuclein. Science 2022, 377, eabk0637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kargilis, D.; Tropea, T.; Robinson, J.; Shen, J.; Brody, E.M.; Brinkmalm, A.; Sjödin, S.; Berndt, A.J.; Carceles-Cordon, M.; et al. Calcium modulating ligand confers risk for Parkinson’s disease and impacts lysosomes. Ann. Clin. Transl. Neurol. 2025, 12, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Bustos, B.I.; Billingsley, K.; Blauwendraat, C.; Gibbs, J.R.; Gan-Or, Z.; Krainc, D.; Singleton, A.B.; Lubbe, S.J.; International Parkinson’s Disease Genomics Consortium (IPDGC). Genome-wide contribution of common short-tandem repeats to Parkinson’s disease genetic risk. Brain 2022, 146, 65–74. [Google Scholar] [CrossRef]
- Ohlei, O.; Paul, K.; Nielsen, S.S.; Gmelin, D.; Dobricic, V.; Altmann, V.; Schilling, M.; Bronstein, J.M.; Franke, A.; Wittig, M.; et al. Genome-wide meta-analysis of short-tandem repeats for Parkinson’s disease risk using genotype imputation. Brain Commun. 2024, 6, fcae146. [Google Scholar] [CrossRef]
- Makarious, M.B.; Lake, J.; Pitz, V.; Fu, A.Y.; Guidubaldi, J.L.; Solsberg, C.W.; Bandres-Ciga, S.; Leonard, H.L.; Kim, J.J.; Billingsley, K.J.; et al. Large-scale rare variant burden testing in Parkinson’s disease. Brain 2023, 146, 4622–4632. [Google Scholar] [CrossRef]
- Chen, R.; Liu, J.; Li, S.; Li, X.; Huo, Y.; Yao, Y.-G.; Xiao, X.; Li, M.; Luo, X.-J. Functional genomics elucidates regulatory mechanisms of Parkinson’s disease-associated variants. BMC Med. 2022, 20, 68. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Lee, A.J.; Kim, C.; Park, S.; Joo, J.; Choi, B.; Yang, D.; Jun, K.; Eom, J.; Lee, S.-J.; Chung, S.J.; et al. Characterization of altered molecular mechanisms in Parkinson’s disease through cell type–resolved multiomics analyses. Sci. Adv. 2023, 9, eabo2467. [Google Scholar] [CrossRef]
- Gomes, L.C.; Galhoz, A.; Jain, G.; Roser, A.; Maass, F.; Carboni, E.; Barski, E.; Lenz, C.; Lohmann, K.; Klein, C.; et al. Multi-omic landscaping of human midbrains identifies disease-relevant molecular targets and pathways in advanced-stage Parkinson’s disease. Clin. Transl. Med. 2022, 12, e692. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Ansari, R.; Pestana, F.; Hebestreit, K.; Gasparyan, H.; Aleksanyan, R.; Hnatova, S.; Poovathingal, S.; Marneffe, C.; Thal, D.R.; et al. Unravelling cell type-specific responses to Parkinson’s Disease at single cell resolution. Mol. Neurodegener. 2024, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Kõks, S. The Exon-Based Transcriptomic Analysis of Parkinson’s Disease. Biomolecules 2025, 15, 440. [Google Scholar] [CrossRef] [PubMed]
- Irmady, K.; Hale, C.R.; Qadri, R.; Fak, J.; Simelane, S.; Carroll, T.; Przedborski, S.; Darnell, R.B. Blood transcriptomic signatures associated with molecular changes in the brain and clinical outcomes in Parkinson’s disease. Nat. Commun. 2023, 14, 3956. [Google Scholar] [CrossRef]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2022, 68, 125–130. [Google Scholar] [CrossRef]
- Haider, A.; Elghazawy, N.H.; Dawoud, A.; Gebhard, C.; Wichmann, T.; Sippl, W.; Hoener, M.; Arenas, E.; Liang, S.H. Translational molecular imaging and drug development in Parkinson’s disease. Mol. Neurodegener. 2023, 18, 3956. [Google Scholar] [CrossRef]
- Zaman, V.; Shields, D.C.; Shams, R.; Drasites, K.P.; Matzelle, D.; Haque, A.; Banik, N.L. Cellular and molecular pathophysiology in the progression of Parkinson’s disease. Metab. Brain Dis. 2021, 36, 815–827. [Google Scholar] [CrossRef]
- Tomkins, J.E.; Manzoni, C. Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiol. Dis. 2021, 155, 105395. [Google Scholar] [CrossRef]
- Mestre, T.A.; Fereshtehnejad, S.-M.; Berg, D.; Bohnen, N.I.; Dujardin, K.; Erro, R.; Espay, A.J.; Halliday, G.; van Hilten, J.J.; Hu, M.T.; et al. Parkinson’s Disease Subtypes: Critical Appraisal and Recommendations. J. Park. Dis. 2021, 11, 395–404. [Google Scholar] [CrossRef]
- Beaulieu-Jones, B.K.; Frau, F.; Bozzi, S.; Chandross, K.J.; Peterschmitt, M.J.; Cohen, C.; Coulovrat, C.; Kumar, D.; Kruger, M.J.; Lipnick, S.L.; et al. Disease progression strikingly differs in research and real-world Parkinson’s populations. npj Park. Dis. 2024, 10, 58. [Google Scholar] [CrossRef]
- Paul, E.; George, J.; Ward, S.; Fitzgerald, K.; Jones, G.; Magana, K.; Modi, J.; Magee, T.; Hughes, G.; Ford, A.I.; et al. Assessing uptake of the core outcome set in randomized controlled trials for Parkinson’s disease: A systematic review. Ageing Res. Rev. 2023, 91, 102081. [Google Scholar] [CrossRef]
- Hirschwald, J.; Hofacker, J.; Duncan, S.; Walshe, M. Swallowing outcomes in dysphagia interventions in Parkinson’s disease: A scoping review. BMJ Evid.-Based Med. 2022, 28, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Tabashum, T.; Snyder, R.C.; O’BRien, M.K.; Albert, M.V. Machine Learning Models for Parkinson Disease: Systematic Review. JMIR Public Health Surveill. 2024, 12, e50117. [Google Scholar] [CrossRef] [PubMed]
- Dzialas, V.; Doering, E.; Eich, H.; Strafella, A.P.; Vaillancourt, D.E.; Simonyan, K.; van Eimeren, T.; International Parkinson Movement Disorders Society-Neuroimaging Study Group. Houston, We Have AI Problem! Quality Issues with Neuroimaging-Based Artificial Intelligence in Parkinson’s Disease: A Systematic Review. Mov. Disord. 2024, 39, 2130–2143. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-C.; Lin, Y.-C.; Ng, S.-H.; Chen, Y.-L.; Cheng, J.-S.; Lu, C.-S.; Weng, Y.-H.; Lin, S.-H.; Chen, P.-Y.; Wu, Y.-M.; et al. A Method for the Prediction of Clinical Outcome Using Diffusion Magnetic Resonance Imaging: Application on Parkinson’s Disease. J. Clin. Med. 2020, 9, 647. [Google Scholar] [CrossRef]
- Lenka, A.; Jankovic, J. How should future clinical trials be designed in the search for disease-modifying therapies for Parkinson’s disease? Expert Rev. Neurother. 2023, 23, 107–122. [Google Scholar] [CrossRef]
- Mirelman, A.; Siderowf, A.; Chahine, L. Outcome Assessment in Parkinson Disease Prevention Trials. Neurology 2022, 99, 52–60. [Google Scholar] [CrossRef]
- Pigott, J.S.; Kane, E.J.; Ambler, G.; Walters, K.; Schrag, A. Systematic review and meta-analysis of clinical effectiveness of self-management interventions in Parkinson’s disease. BMC Geriatr. 2022, 22, 45. [Google Scholar] [CrossRef]
- Mari, Z.; Mestre, T.A. The Disease Modification Conundrum in Parkinson’s Disease: Failures and Hopes. Front. Aging Neurosci. 2022, 14, 810860. [Google Scholar] [CrossRef]
- Chocarro, J.; Lanciego, J.L. Adeno-associated viral vectors for modeling Parkinson’s disease in non-human primates. Neural Regen. Res. 2025, 21, 224–232. [Google Scholar] [CrossRef]
- Espay, A.J.; Kalia, L.V.; Gan-Or, Z.; Williams-Gray, C.H.; Bedard, P.L.; Rowe, S.M.; Morgante, F.; Fasano, A.; Stecher, B.; Kauffman, M.A.; et al. Disease modification and biomarker development in Parkinson disease. Neurology 2020, 94, 481–494. [Google Scholar] [CrossRef]
- Menozzi, E.; Schapira, A.H. Prospects for Disease Slowing in Parkinson Disease. Annu. Rev. Pharmacol. Toxicol. 2025, 65, 237–258. [Google Scholar] [CrossRef]
- Mahlknecht, P.; Poewe, W. Pharmacotherapy for Disease Modification in Early Parkinson’s Disease: How Early Should We Be? J. Park. Dis. 2024, 14, S407–S421. [Google Scholar] [CrossRef]
- Cong, C.; Milne-Ives, M.; Ananthakrishnan, A.; Maetzler, W.; Meinert, E. From past to future: Digital approaches to success of clinical drug trials for Parkinson’s disease. J. Park. Dis. 2025, 1877718X251330839. [Google Scholar] [CrossRef]
- Bouhadoun, S.; Delva, A.; Schwarzschild, M.A.; Postuma, R.B. Preparing for Parkinson’s disease prevention trials: Current progress and future directions. J. Park. Dis. 2025, 1877718X251334050. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, P.; Marini, K.; Werkmann, M.; Poewe, W.; Seppi, K. Prodromal Parkinson’s disease: Hype or hope for disease-modification trials? Transl. Neurodegener. 2022, 11, 11. [Google Scholar] [CrossRef]
- Vaccari, C.; Grotto, D.; Pereira, T.d.V.; de Camargo, J.L.V.; Lopes, L.C.; Wei, L.K. GLP-1 and GIP receptor agonists in the treatment of Parkinson’s disease: Translational systematic review and meta-analysis protocol of clinical and preclinical studies. PLoS ONE 2021, 16, e0255726. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Bhate, L.; Agrawal, Y.; Aspatwar, A. Advanced nutraceutical approaches to Parkinson’s disease: Bridging nutrition and neuroprotection. Nutr. Neurosci. 2025, 1–17. [Google Scholar] [CrossRef] [PubMed]
- El Nebrisi, E. Neuroprotective Activities of Curcumin in Parkinson’s Disease: A Review of the Literature. Int. J. Mol. Sci. 2021, 22, 11248. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, S.; Pan, I.; Issac, P.K.; Kumar, M.S.K.; Giri, J.; Guru, A.; Arockiaraj, J. Selenium Nanoparticles as Neuroprotective Agents: Insights into Molecular Mechanisms for Parkinson’s Disease Treatment. Mol. Neurobiol. 2024, 62, 6655–6682. [Google Scholar] [CrossRef]
- Godoy, A.C.F.; Frota, F.F.; Araújo, L.P.; Valenti, V.E.; Pereira, E.d.S.B.M.; Detregiachi, C.R.P.; Galhardi, C.M.; Caracio, F.C.; Haber, R.S.A.; Laurindo, L.F.; et al. Neuroinflammation and Natural Antidepressants: Balancing Fire with Flora. Biomedicines 2025, 13, 1129. [Google Scholar] [CrossRef] [PubMed]
- Juhász, L.; Spisák, K.; Szolnoki, B.Z.; Nászai, A.; Szabó, Á.; Rutai, A.; Tallósy, S.P.; Szabó, A.; Toldi, J.; Tanaka, M.; et al. The Power Struggle: Kynurenine Pathway Enzyme Knockouts and Brain Mitochondrial Respiration. J. Neurochem. 2025, 169, e70075. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Du, Z.R.; Wang, X.; Sun, X.R.; Zhao, Q.; Zhao, F.; Wong, W.T.; Wong, K.H.; Dong, X.-L. Polymannuronic acid prebiotic plus Lacticaseibacillus rhamnosus GG probiotic as a novel synbiotic promoted their separate neuroprotection against Parkinson’s disease. Food Res. Int. 2022, 155, 111067. [Google Scholar] [CrossRef] [PubMed]
- de Lima, E.P.; Laurindo, L.F.; Catharin, V.C.S.; Direito, R.; Tanaka, M.; German, I.J.S.; Lamas, C.B.; Guiguer, E.L.; Araújo, A.C.; Fiorini, A.M.R.; et al. Polyphenols, Alkaloids, and Terpenoids Against Neurodegeneration: Evaluating the Neuroprotective Effects of Phytocompounds Through a Comprehensive Review of the Current Evidence. Metabolites 2025, 15, 124. [Google Scholar] [CrossRef]
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Chigurupati, S.; Alrashdi, I.; Bungau, S.G. Elucidating the Neuroprotective Role of PPARs in Parkinson’s Disease: A Neoteric and Prospective Target. Int. J. Mol. Sci. 2021, 22, 10161. [Google Scholar] [CrossRef]
- Shtilbans, A.; Esneault, E.; Simon, F.; Mazzulli, J.R.; Quiriconi, D.J.; Rom, D.; Reintsch, W.E.; Krahn, A.I.; Durcan, T.M. Evaluation of Additive Neuroprotective Effect of Combination Therapy for Parkinson’s Disease Using In Vitro Models. Antioxidants 2025, 14, 396. [Google Scholar] [CrossRef]
- Bougea, A. Some Novel Therapies in Parkinson’s Disease: A Promising Path Forward or Not Yet? A Systematic Review of the Literature. Biomedicines 2024, 12, 549. [Google Scholar] [CrossRef]
- Church, F.C. Treatment Options for Motor and Non-Motor Symptoms of Parkinson’s Disease. Biomolecules 2021, 11, 612. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.-K.; Chao, Y.-X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Lopez-Gonzalez del Rey, N. Animal Models of Parkinson’s Disease; IntechOpen: London, UK, 2016. [Google Scholar]
- Dovonou, A.; Bolduc, C.; Linan, V.S.; Gora, C.; Iii, M.R.P.; Lévesque, M. Animal models of Parkinson’s disease: Bridging the gap between disease hallmarks and research questions. Transl. Neurodegener. 2023, 12, 36. [Google Scholar] [CrossRef]
- Ke, M.; Chong, C.-M.; Zhu, Q.; Zhang, K.; Cai, C.-Z.; Lu, J.-H.; Qin, D.; Su, H. Comprehensive Perspectives on Experimental Models for Parkinson’s Disease. Aging Dis. 2021, 12, 223–246. [Google Scholar] [CrossRef]
- Yamakado, H.; Takahashi, R. Experimental Animal Models of Prodromal Parkinson’s Disease. J. Park. Dis. 2024, 14, S369–S379. [Google Scholar] [CrossRef]
- Saponjic, J.; Mejías, R.; Nikolovski, N.; Dragic, M.; Canak, A.; Papoutsopoulou, S.; Gürsoy-Özdemir, Y.; Fladmark, K.E.; Ntavaroukas, P.; Muluk, N.B.; et al. Experimental Models to Study Immune Dysfunction in the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 4330. [Google Scholar] [CrossRef]
- Altunlu, Ö.; Topatan, E.; Al-yaqoobi, Z.; Burul, F.; Bayram, C.; Sezen, S.; Okkay, I.F.; Okkay, U.; Hacımüftüoğlu, A. Ex-perimental Models in Parkinson’s Disease: Advantages and Disadvantages. Ağrı Tıp Fakültesi Dergisi 2024, 2, 80–87. [Google Scholar] [CrossRef]
- Magalhães, P.; Lashuel, H.A. Opportunities and challenges of alpha-synuclein as a potential biomarker for Parkinson’s disease and other synucleinopathies. npj Park. Dis. 2022, 8, 93. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, T.; Zhao, W.; Zhao, X.; Xue, Y.; Deng, Q. Current trends in blood biomarkers detection and neuroimaging for Parkinson’s disease. Ageing Res. Rev. 2025, 104, 102658. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, X.; Yang, H.; Liu, Y. Review of Metabolomics-Based Biomarker Research for Parkinson’s Disease. Mol. Neurobiol. 2021, 59, 1041–1057. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.H.; Tennagels, S.; Gold, R.; Gerwert, K.; Beyer, L.; Tönges, L. Update on CSF Biomarkers in Parkinson’s Disease. Biomolecules 2022, 12, 329. [Google Scholar] [CrossRef] [PubMed]
- Karayel, O.; Winter, S.V.; Padmanabhan, S.; Kuras, Y.I.; Vu, D.T.; Tuncali, I.; Merchant, K.; Wills, A.-M.; Scherzer, C.R.; Mann, M. Proteome profiling of cerebrospinal fluid reveals biomarker candidates for Parkinson’s disease. Cell Rep. Med. 2022, 3, 100661. [Google Scholar] [CrossRef]
- Zarkali, A.; Thomas, G.E.C.; Zetterberg, H.; Weil, R.S. Neuroimaging and fluid biomarkers in Parkinson’s disease in an era of targeted interventions. Nat. Commun. 2024, 15, 5661. [Google Scholar] [CrossRef]
- Mitchell, T.; Lehéricy, S.; Chiu, S.Y.; Strafella, A.P.; Stoessl, A.J.; Vaillancourt, D.E. Emerging Neuroimaging Biomarkers Across Disease Stage in Parkinson Disease. JAMA Neurol. 2021, 78, 1262–1272. [Google Scholar] [CrossRef]
- Ameli, A.; Peña-Castillo, L.; Usefi, H. Assessing the reproducibility of machine-learning-based biomarker discovery in Par-kinson’s disease. Comput. Biol. Med. 2024, 174, 108407. [Google Scholar] [CrossRef]
- Sigcha, L.; Borzì, L.; Amato, F.; Rechichi, I.; Ramos-Romero, C.; Cárdenas, A.; Gascó, L.; Olmo, G. Deep learning and wearable sensors for the diagnosis and monitoring of Parkinson’s disease: A systematic review. Expert Syst. Appl. 2023, 229, 120541. [Google Scholar] [CrossRef]
- Giannakopoulou, K.-M.; Roussaki, I.; Demestichas, K. Internet of Things Technologies and Machine Learning Methods for Parkinson’s Disease Diagnosis, Monitoring and Management: A Systematic Review. Sensors 2022, 22, 1799. [Google Scholar] [CrossRef]
- Lei, H.; Lei, Y.; Chen, Z.; Li, S.; Huang, Z.; Zhou, F.; Tan, E.-L.; Xiao, X.; Lei, Y.; Hu, H.; et al. Early diagnosis and clinical score prediction of Parkinson’s disease based on longitudinal neuroimaging data. Neural Comput. Appl. 2023, 35, 16429–16455. [Google Scholar] [CrossRef]
- Adeniyi, O.J.; Ayankoya, F.Y.; Kuyoro, S.O.; Akwaronwu, B.G.; Abiodun, A.G. Machine Learning Models for Predicting Parkinson’s Disease Progression Using Longitudinal Data: A Systematic Review. Asian J. Res. Comput. Sci. 2025, 18, 274–294. [Google Scholar] [CrossRef]
- Puig-Davi, A.; Martinez-Horta, S.; Pérez-Carasol, L.; Horta-Barba, A.; Ruiz-Barrio, I.; Aracil-Bolaños, I.; Pérez-González, R.; Rivas-Asensio, E.; Sampedro, F.; Campolongo, A.; et al. Prediction of Cognitive Heterogeneity in Parkinson’s Disease: A 4-Year Longitudinal Study Using Clinical, Neuroimaging, Biological and Electrophysiological Biomarkers. Ann. Neurol. 2024, 96, 981–993. [Google Scholar] [CrossRef]
- Dadu, A.; Satone, V.; Kaur, R.; Hashemi, S.H.; Leonard, H.; Iwaki, H.; Makarious, M.B.; Billingsley, K.J.; Bandres-Ciga, S.; Sargent, L.J.; et al. Identification and prediction of Parkinson’s disease subtypes and progression using machine learning in two cohorts. npj Park. Dis. 2022, 8, 172. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Xin, R.; Shao, J.-Y.; Wang, S.-H.; Yang, H.-Q.; Zhang, H.-J.; Zhang, J.-W.; Chen, S. A predictive model for longitudinal cognitive subtypes in Parkinson’s disease. Neurol. Sci. 2025, 46, 2627–2635. [Google Scholar] [CrossRef]
- Macklin, E.A.; Coffey, C.S.; Brumm, M.C.; Seibyl, J.P. Statistical Considerations in the Design of Clinical Trials Targeting Prodromal Parkinson Disease. Neurology 2022, 99, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Joza, S.; Hu, M.T.; Jung, K.-Y.; Kunz, D.; Stefani, A.; Dušek, P.; Terzaghi, M.; Arnaldi, D.; Videnovic, A.; Schiess, M.C.; et al. Progression of clinical markers in prodromal Parkinson’s disease and dementia with Lewy bodies: A multicentre study. Brain 2023, 146, 3258–3272. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, C.E.; Farmer, C.M.; Ryan, J.; Ernecoff, N. Clinical Outcome Assessments and Digital Health Technologies Supporting Clinical Trial Endpoints in Early Parkinson’s Disease: Roundtable Proceedings and Roadmap for Research; RAND: Santa Monica, CA, USA, 2024; Volume 11. [Google Scholar]
- Roeben, B.; Liepelt-Scarfone, I.; Lerche, S.; Zimmermann, M.; Wurster, I.; Sünkel, U.; Schulte, C.; Deuschle, C.; Eschweiler, G.W.; Maetzler, W.; et al. Longitudinal cognitive decline characterizes the profile of non-PD-manifest GBA1 mutation carriers. npj Park. Dis. 2024, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.; Kim, C.; Guzy, S.; Yoneyama, T.; Gharahi, H.; Siripuram, V.K.; Iadevaia, S.; Barua, D.; Hang, Y.; Iakovleva, T.; et al. Establishment of a Biomarker-Directed Clinical Endpoint Model for Early-Stage Parkinson’s Disease Patients. Clin. Pharmacol. Ther. 2025, 117, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Benz, H.L.; Caldwell, B.; Ruiz, J.P.; Saha, A.; Ho, M.; Christopher, S.; Bardot, D.; Sheehan, M.; Donnelly, A.; McLaughlin, L.; et al. Patient-Centered Identification of Meaningful Regulatory Endpoints for Medical Devices to Treat Parkinson’s Disease. MDM Policy Pract. 2021, 6, 23814683211021380. [Google Scholar] [CrossRef]
- Luz, M.; Whone, A.; Bassani, N.; Wyse, R.K.; Stebbins, G.T.; Mohr, E. The Parkinson’s Disease Comprehensive Response (PDCORE): A composite approach integrating three standard outcome measures. Brain Commun. 2020, 2, fcaa046. [Google Scholar] [CrossRef]
- de Aquino, C.H. Methodological Issues in Randomized Clinical Trials for Prodromal Alzheimer’s and Parkinson’s Disease. Front. Neurol. 2021, 12, 694329. [Google Scholar] [CrossRef]
- Morel, T.; Cleanthous, S.; Andrejack, J.; Barker, R.A.; Blavat, G.; Brooks, W.; Burns, P.; Cano, S.; Gallagher, C.; Gosden, L.; et al. Patient Experience in Early-Stage Parkinson’s Disease: Using a Mixed Methods Analysis to Identify Which Concepts Are Cardinal for Clinical Trial Outcome Assessment. Neurol. Ther. 2022, 11, 1319–1340. [Google Scholar] [CrossRef]
- Mestre, T.A.; Stebbins, G.T.; Stephenson, D.; Dexter, D.; Lee, K.K.; Xiao, Y.; Dam, T.; Kopil, C.M.; Simuni, T. Patient-centered development of clinical outcome assessments in early Parkinson disease: Key priorities and advances. npj Park. Dis. 2024, 10, 101. [Google Scholar] [CrossRef]
- George, J.; Paul, E.; Ward, S.; Fitzgerald, K.; Jones, G.; Magana, K.; Ford, A.; Vassar, M. ASSESSING THE UPTAKE OF CORE OUTCOME SETS IN RANDOMIZED CONTROLLED TRIALS FOR PARKINSON’S DISEASE. Innov. Aging 2023, 7, 990. [Google Scholar] [CrossRef]
- Mohaghegh, M.; Peng, N. Predicting Parkinson’s Disease Severity Using Patient-Reported Outcomes and Genetic Information. In Neural Information Processing, 29th International Conference, ICONIP 2022, Virtual Event, 22–26 November 2022; Springer: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Xu, Z.; Jin, L.; Chen, W.; Hu, T.; Li, S.; Liang, X.; Han, X.; Chen, Y.; Tang, Y.; Wang, J.; et al. Using a smartphone-based self-management platform to study sex differences in Parkinson’s disease: Multicenter, cross-sectional pilot study. BMC Med. Informatics Decis. Mak. 2024, 24, 176. [Google Scholar] [CrossRef]
- Shoulson, I.; Arbatti, L.; Hosamath, A.; Eberly, S.W.; Oakes, D. Longitudinal Cohort Study of Verbatim-Reported Postural Instability Symptoms as Outcomes for Online Parkinson’s Disease Trials. J. Park. Dis. 2022, 12, 1969–1978. [Google Scholar] [CrossRef]
- Kim, H.; Shulman, L.M.; Shakya, S.; Gruber-Baldini, A. The effects of medical comorbidity, cognition, and age on patient-reported outcomes in Parkinson’s disease. Park. Relat. Disord. 2023, 116, 105892. [Google Scholar] [CrossRef] [PubMed]
- Kurpershoek, E.; Visser, L.N.; Malekzadeh, A.; de Bie, R.M.; Dijk, J.M.; Hillen, M.A. How Information Affects Patients with Parkinson’s Disease: A Scoping Review of the Literature. J. Park. Dis. 2024, 14, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Pérez, S.A.; Koppelmans, V.; Duff, K.M.; Ruitenberg, M.F. One-year practice effects predict long-term cognitive outcomes in Parkinson’s disease. J. Park. Dis. 2025, 15, 858–867. [Google Scholar] [CrossRef]
- Rábano-Suárez, P.; del Campo, N.; Benatru, I.; Moreau, C.; Desjardins, C.; Sánchez-Ferro, Á.; Fabbri, M. Digital Outcomes as Biomarkers of Disease Progression in Early Parkinson’s Disease: A Systematic Review. Mov. Disord. 2024, 40, 184–203. [Google Scholar] [CrossRef]
- Zuo, C.; Suo, X.; Lan, H.; Pan, N.; Wang, S.; Kemp, G.J.; Gong, Q. Global Alterations of Whole Brain Structural Connectome in Parkinson’s Disease: A Meta-analysis. Neuropsychol. Rev. 2022, 33, 783–802. [Google Scholar] [CrossRef]
- Palakayala, A.R.; Kuppusamy, P.; Kothandaraman, D.; Gunakala, A.; Gera, J. HAMF: A novel Hierarchical Attention based Multi-modal Fusion model for Parkinson’s Disease Classification and Severity Prediction. IEEE Access 2025. [Google Scholar] [CrossRef]
- Huang, Z.; Li, J.; Yang, J.; Wan, J.; Chen, J.; Yang, Z.; Shi, M.; Zhou, R.; Gan, H. Parkinson’s disease classification and prediction via adaptive sparse learning from multiple modalities. Biomed. Signal Process. Control. 2024, 100, 107061. [Google Scholar] [CrossRef]
- Ryan, B.; Marioni, R.; Simpson, T.I.; Roy, S. An integrative network approach for longitudinal stratification in Parkinson’s disease. PLoS Comput. Biol. 2025, 21, e1012857. [Google Scholar] [CrossRef]
- Yan, H.; Coughlin, C.; Smolin, L.; Wang, J. Unraveling the Complexity of Parkinson’s Disease: Insights into Pathogenesis and Precision Interventions. Adv. Sci. 2024, 11, e2405309. [Google Scholar] [CrossRef]
- Rajan, R.; Brennan, L.; Bloem, B.R.; Dahodwala, N.; Gardner, J.; Goldman, J.G.; Grimes, D.A.; Iansek, R.; Kovács, N.; McGinley, J.; et al. Integrated Care in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2020, 35, 1509–1531. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Henderson, E.J.; Dorsey, E.R.; Okun, M.S.; Okubadejo, N.; Chan, P.; Andrejack, J.; Darweesh, S.K.L.; Munneke, M. Integrated and patient-centred management of Parkinson’s disease: A network model for reshaping chronic neurological care. Lancet Neurol. 2020, 19, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Aquino, S. Exploring the Viability of a Speech-Based Diagnostic Tool in Parkinson’s Disease in An Integrated Service. Int. J. Integr. Care 2025, 25, 651. [Google Scholar] [CrossRef]
- Hunt, J.; Coulson, E.J.; Rajnarayanan, R.; Oster, H.; Videnovic, A.; Rawashdeh, O. Sleep and circadian rhythms in Parkinson’s disease and preclinical models. Mol. Neurodegener. 2022, 17, 2. [Google Scholar] [CrossRef]
- Lal, R.; Singh, A.; Watts, S.; Chopra, K. Experimental models of Parkinson’s disease: Challenges and Opportunities. Eur. J. Pharmacol. 2024, 980, 176819. [Google Scholar] [CrossRef]
- Junaid, M.; Ali, S.; Eid, F.; El-Sappagh, S.; Abuhmed, T. Explainable machine learning models based on multimodal time-series data for the early detection of Parkinson’s disease. Comput. Methods Programs Biomed. 2023, 234, 107495. [Google Scholar] [CrossRef]
- Makarious, M.B.; Leonard, H.L.; Vitale, D.; Iwaki, H.; Sargent, L.; Dadu, A.; Violich, I.; Hutchins, E.; Saffo, D.; Bandres-Ciga, S.; et al. Multi-modality machine learning predicting Parkinson’s disease. npj Park. Dis. 2022, 8, 35. [Google Scholar] [CrossRef]
- Gunduz, H. An efficient dimensionality reduction method using filter-based feature selection and variational autoencoders on Parkinson’s disease classification. Biomed. Signal Process. Control. 2021, 66, 102452. [Google Scholar] [CrossRef]
- Lummer, C.; Eggers, C.; Becker, A.; Demandt, F.; Warnecke, T.; Parkinson Netzwerke Deutschland eV. Interdisciplinary network care collaboration in Parkinson’s disease: A baseline evaluation in Germany. Neurol. Res. Pract. 2024, 6, 5. [Google Scholar] [CrossRef]
- Kerkemeyer, L.; PNM + steering committee; Claus, I.; Kutscher, M.; von Stülpnagel, V.; Nieden, P.Z.; Huchtemann, T.; Warnecke, T. Strengthening Communication and Collaboration in the Fragmented German Healthcare System: A Mixed-Method Evaluation of an Interdisciplinary Network for Parkinson’s Disease. J. Park. Dis. 2022, 12, 1307–1317. [Google Scholar] [CrossRef]
- van Munster, M.; Tönges, L.; Loewenbrück, K.F.; Warnecke, T.; Eggers, C. Building a Parkinson-Network–Experiences from Germany. J. Clin. Med. 2020, 9, 2743. [Google Scholar] [CrossRef] [PubMed]
- Rose, O.; Happe, S.; Huchtemann, T.; Mönig, C.; Ohms, M.; Schwalbe, O.; Warnecke, T.; Erzkamp, S. Enhancing medication therapy in Parkinson’s disease by establishing an interprofessional network including pharmacists. Pharm. Weekbl. 2021, 43, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Onyiuke, F.G.; Makins, S.K.; Nelson, J.; Martin, A. Feasibility of interprofessional partnerships between neurologists and dentists for oral health management of Parkinson’s disease. J. Interprof. Care 2024, 38, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Navarta-Sánchez, M.V.; Palmar-Santos, A.; Pedraz-Marcos, A.; Reidy, C.; Soilemezi, D.; Haahr, A.; Sørensen, D.; Smidt, H.R.; Bragstad, L.K.; Hjelle, E.G.; et al. Perspectives of people with Parkinson’s disease and family carers about disease management in community settings: A cross-country qualitative study. J. Clin. Nurs. 2023, 32, 5201–5218. [Google Scholar] [CrossRef]
- Bayen, S.; Bayen, M.; Moreau, C.; Defebvre, L.; Devos, D.; Messaadi, N. Parkinson’s disease: Improving person-centered care coordination by interdisciplinary communication and teamwork with patients: A qualitative mixed method study. Int. J. Health Manag. 2021, 15, 238–245. [Google Scholar] [CrossRef]
- Pirtošek, Z. Breaking barriers in Parkinson’s care: The multidisciplinary team approach. J. Neural Transm. 2024, 131, 1349–1361. [Google Scholar] [CrossRef]
- Vollstedt, E.-J.; Madoev, H.; Aasly, A.; Ahmad-Annuar, A.; Al-Mubarak, B.; Alcalay, R.N.; Alvarez, V.; Amorin, I.; Annesi, G.; Arkadir, D.; et al. Establishing an online resource to facilitate global collaboration and inclusion of underrepresented populations: Experience from the MJFF Global Genetic Parkinson’s Disease Project. PLoS ONE 2023, 18, e0292180. [Google Scholar] [CrossRef]
- The Global Parkinson’s Genetics Program. GP2: The Global Parkinson’s Genetics Program. Mov. Disord. 2021, 36, 842–851. [Google Scholar] [CrossRef]
- Schumacher-Schuh, A.F.; Bieger, A.; Okunoye, O.; Mok, K.; Lim, S.-Y.; Bardien, S.; Annuar, A.A.; Lopes-Santos, B.; Strelow, M.Z.; Salama, M. Underrepresented Populations in Parkinson’s Genetics Research: Current Landscape and Future Directions. Mov. Disord. 2022, 37, 1593–1604. [Google Scholar] [CrossRef]
- Rizig, M.; Bandres-Ciga, S.; Makarious, M.B.; Ojo, O.O.; Crea, P.W.; Abiodun, O.V.; Levine, K.S.; Abubakar, S.A.; Achoru, C.O.; Vitale, D.; et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: A genome-wide association study. Lancet Neurol. 2023, 22, 1015–1025. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, L.; Pan, H.; Liu, Z.; Jiang, L.; He, Y.; Zeng, Q.; Zhou, X.; Zhou, X.; Zhou, Y.; et al. The role of genetics in Parkinson’s disease: A large cohort study in Chinese mainland population. Brain 2020, 143, 2220–2234. [Google Scholar] [CrossRef] [PubMed]
- Ba, D.P.L.; Horimoto, A.R.V.R.; Heilbron, K.; Sarihan, E.I.; Bs, M.I.-M.; Bs, E.M.; Cornejo-Olivas, M.; Torres, L.; Mazzetti, P.; Cosentino, C.; et al. Characterizing the Genetic Architecture of Parkinson’s Disease in Latinos. Ann. Neurol. 2021, 90, 353–365. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Alcalay, R.N. Genetics of Parkinson’s Disease in Underrepresented Populations: New Studies Pave the Way. Mov. Disord. 2024, 39, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Khani, M.; Cerquera-Cleves, C.; Kekenadze, M.; Crea, P.W.; Singleton, A.B.; Bandres-Ciga, S. Towards a Global View of Parkinson’s Disease Genetics. Ann. Neurol. 2024, 95, 831–842. [Google Scholar] [CrossRef]
- Liang, M.; Chu, L.; Yue, Z. New Multiomic Studies Shed Light on Cellular Diversity and Neuronal Susceptibility in Parkinson’s Disease. Mov. Disord. 2025, 40, 431–437. [Google Scholar] [CrossRef]
- Lau, Y.H.; Podlewska, A.; Ocloo, J.; Gupta, A.; Gonde, C.; Bloem, B.R.; Chaudhuri, K.R. Does Ethnicity Influence Recruitment into Clinical Trials of Parkinson’s Disease? J. Park. Dis. 2022, 12, 975–981. [Google Scholar] [CrossRef]
- Tsai, C.; Tao, B.; Lin, V.; Lo, J.; Brahmbhatt, S.; Kalo, C.; Marras, C.; Khosa, F. Representational Disparities in the Enrollment of Parkinson’s Disease Clinical Trials. Mov. Disord. Clin. Pract. 2025, 12, 878–881. [Google Scholar] [CrossRef]
- Di Luca, D.G.; Sambursky, J.A.; Margolesky, J.; Cordeiro, J.G.; Diaz, A.; Shpiner, D.S.; Moore, H.P.; Singer, C.; Luca, C. Minority Enrollment in Parkinson’s Disease Clinical Trials: Meta-Analysis and Systematic Review of Studies Evaluating Treatment of Neuropsychiatric Symptoms. J. Park. Dis. 2020, 10, 1709–1716. [Google Scholar] [CrossRef]
- Adrissi, J.; Fleisher, J. Moving the Dial Toward Equity in Parkinson’s Disease Clinical Research: A Review of Current Literature and Future Directions in Diversifying PD Clinical Trial Participation. Curr. Neurol. Neurosci. Rep. 2022, 22, 475–483. [Google Scholar] [CrossRef]
- Vaswani, P.A.; Tropea, T.F.; Dahodwala, N. Overcoming Barriers to Parkinson Disease Trial Participation: Increasing Diversity and Novel Designs for Recruitment and Retention. Neurotherapeutics 2020, 17, 1724–1735. [Google Scholar] [CrossRef]
- Kłosowska, D.; Fiszer, U.; Dulski, J.; Górski, A.; Borysowski, J. Exclusion of older patients from randomized clinical trials in Parkinson’s disease. GeroScience 2024, 46, 3819–3830. [Google Scholar] [CrossRef]
- Smilowska, K.; Carvalho, V.; Szejko, N.; Costa, J.; Moro, E.; Antonini, A. How well is the female population represented in clinical trials with infusion therapies for Parkinson’s disease? A systematic review and metanalysis. Eur. J. Neurol. 2025, 32, e70024. [Google Scholar] [CrossRef]
- Sanchez, A.V.; Ison, J.M.; Hemley, H.; Willis, A.; Siddiqi, B.; Macklin, E.A.; Ulysse, C.; Reynolds, M.; Schwarzschild, M.A.; Jackson, J.D. Designing the Fostering Inclusivity in Research Engagement for Underrepresented Populations in Parkinson’s Disease study. Contemp. Clin. Trials 2022, 115, 106713. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Rafaloff, G.; Baptista, M.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2022 Update. J. Park. Dis. 2022, 12, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Zanardi, A.P.J.; da Silva, E.S.; Costa, R.R.; Passos-Monteiro, E.; dos Santos, I.O.; Kruel, L.F.M.; Peyré-Tartaruga, L.A. Gait parameters of Parkinson’s disease compared with healthy controls: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 752. [Google Scholar] [CrossRef] [PubMed]
- de Lima, E.P.; Tanaka, M.; Lamas, C.B.; Quesada, K.; Detregiachi, C.R.P.; Araújo, A.C.; Guiguer, E.L.; Catharin, V.M.C.S.; de Castro, M.V.M.; Junior, E.B.; et al. Vascular Impairment, Muscle Atrophy, and Cognitive Decline: Critical Age-Related Conditions. Biomedicines 2024, 12, 2096. [Google Scholar] [CrossRef]
- Auffret, M.; Weiss, D.; Stocchi, F.; Vérin, M.; Jost, W.H. Access to device-aided therapies in advanced Parkinson’s disease: Navigating clinician biases, patient preference, and prognostic uncertainty. J. Neural Transm. 2023, 130, 1411–1432. [Google Scholar] [CrossRef]
- Vuttipittayamongkol, P.; Elyan, E. Neighbourhood-based undersampling approach for handling imbalanced and overlapped data. Inf. Sci. 2020, 509, 47–70. [Google Scholar]
- Gelissen, L.M.Y.; Bergh, R.v.D.; Talebi, A.H.; Geerlings, A.D.; Maas, B.R.; Burgler, M.M.; Kroeze, Y.; Smink, A.; Bloem, B.R.; Munneke, M.; et al. Assessing the validity of a Parkinson’s care evaluation: The PRIME-NL study. Eur. J. Epidemiol. 2024, 39, 811–825. [Google Scholar] [CrossRef]
- Paul, S.; Maindarkar, M.; Saxena, S.; Saba, L.; Turk, M.; Kalra, M.; Krishnan, P.R.; Suri, J.S. Bias Investigation in Artificial Intelligence Systems for Early Detection of Parkinson’s Disease: A Narrative Review. Diagnostics 2022, 12, 166. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, L.; Liu, H.; Su, H.; Jiang, J.; Qiang, C.; Wang, Q.; Qu, X.; Sun, W.; Bi, H. Efficacy of non-pharmacological interventions on depressive symptoms in patients with Parkinson’s disease: A study protocol for a systematic review and network meta-analysis. BMJ Open 2023, 13, e068019. [Google Scholar] [CrossRef]
- Karimi, A.; Elmi, M.; Shiri, Z.; Baharvand, H. Therapeutic potential of pluripotent stem cell-derived dopaminergic progenitors in Parkinson’s disease: A systematic review protocol. Syst. Rev. 2021, 10, 188. [Google Scholar] [CrossRef]
- Jun, P.; Zhao, H.; Jung, I.C.; Kwon, O.; Han, C.-H.; Won, J.; Jang, J.-H. Efficacy of herbal medicine treatment based on syndrome differentiation for Parkinson’s disease: A systematic review and meta-analysis of randomized placebo-controlled clinical trials. Front. Pharmacol. 2023, 14, 1108407. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, K.; Rudakou, U.; Gan-Or, Z. New therapeutic approaches to Parkinson’s disease targeting GBA, LRRK2 and Parkin. Neuropharmacology 2022, 202, 108822. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.; Godad, A. An update on novel and emerging therapeutic targets in Parkinson’s disease. Metab. Brain Dis. 2024, 39, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Singh, S. Neuromodulation in Parkinson’s disease targeting opioid and cannabinoid receptors, understanding the role of NLRP3 pathway: A novel therapeutic approach. Inflammopharmacology 2023, 31, 1605–1627. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Liu, X.; Ye, Y.; Yan, X.; Cheng, Y.; Zhao, L.; Chen, F.; Ling, Z. Gut Microbiota: A Novel Therapeutic Target for Parkinson’s Disease. Front. Immunol. 2022, 13, 937555. [Google Scholar] [CrossRef]
- Douma, E.H.; Smidt, M.P.; van der Heide, L.P. Boosting endogenous dopamine production: A novel therapeutic approach for Parkinson’s disease. Trends Mol. Med. 2024, 30, 800–803. [Google Scholar] [CrossRef]
- Soni, R.; Delvadia, P.; Joharapurkar, A.; Shah, J. Uncovering Novel Therapeutic Targets for Parkinson’s Disease. ACS Chem. Neurosci. 2023, 14, 1935–1949. [Google Scholar] [CrossRef]
- Pingale, T.; Gupta, G.L. Current and emerging therapeutic targets for Parkinson’s disease. Metab. Brain Dis. 2020, 36, 13–27. [Google Scholar] [CrossRef]
- Raza, A.; Raina, J.; Sahu, S.K.; Wadhwa, P. Genetic mutations in kinases: A comprehensive review on marketed inhibitors and unexplored targets in Parkinson’s disease. Neurol. Sci. 2025, 46, 1509–1524. [Google Scholar] [CrossRef]
- Tanaka, M. From Serendipity to Precision: Integrating AI, Multi-Omics, and Human-Specific Models for Personalized Neuropsychiatric Care. Biomedicines 2025, 13, 167. [Google Scholar] [CrossRef]
- Tanaka, M. Beyond the boundaries: Transitioning from categorical to dimensional paradigms in mental health diagnostics. Adv. Clin. Exp. Med. 2024, 33, 1295–1301. [Google Scholar] [CrossRef]
- Fox, S.; Brotchie, J. Special Issue on new therapeutic approaches to Parkinson disease. Neuropharmacology 2022, 208, 108998. [Google Scholar] [CrossRef] [PubMed]
- Akgüller, Ö.; Balcı, M.A.; Cioca, G. A Multi-Modal Graph Neural Network Framework for Parkinson’s Disease Therapeutic Discovery. Int. J. Mol. Sci. 2025, 26, 4453. [Google Scholar] [CrossRef]
- Unnithan, D.; Sartaj, A.; Iqubal, M.K.; Ali, J.; Baboota, S. A neoteric annotation on the advances in combination therapy for Parkinson’s disease: Nanocarrier-based combination approach and future anticipation. Part II: Nanocarrier design and development in focus. Expert Opin. Drug Deliv. 2024, 21, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Malhotra, M.; Sharma, A.; Singh, A.P.; Singh, A.P. A Holistic Approach to Parkinson’s Disease: Integrating Ad-vances in Pathophysiology, Diagnosis, and Therapy. J. Drug Deliv. Ther. 2024, 14, 141–147. [Google Scholar] [CrossRef]
- Kola, S.; Subramanian, I. Updates in Parkinson’s Disease Integrative Therapies: An Evidence-Based Review. Curr. Neurol. Neurosci. Rep. 2023, 23, 717–726. [Google Scholar] [CrossRef]
- Saluja, A.; Goyal, V.; Dhamija, R.K. Multi-Modal Rehabilitation Therapy in Parkinson’s Disease and Related Disorders. Ann. Indian Acad. Neurol. 2023, 26, S15–S25. [Google Scholar] [CrossRef]
- Jászberényi, M.; Thurzó, B.; Bagosi, Z.; Vécsei, L.; Tanaka, M. The Orexin/Hypocretin System, the Peptidergic Regulator of Vigilance, Orchestrates Adaptation to Stress. Biomedicines 2024, 12, 448. [Google Scholar] [CrossRef]
- Alrawis, M.; Al-Ahmadi, S.; Mohammad, F. Bridging Modalities: A Multimodal Machine Learning Approach for Parkinson’s Disease Diagnosis Using EEG and MRI Data. Appl. Sci. 2024, 14, 3883. [Google Scholar] [CrossRef]
- Al-Tam, R.M.; Hashim, F.A.; Maqsood, S.; Abualigah, L.; Alwhaibi, R.M. Enhancing Parkinson’s Disease Diagnosis Through Stacking Ensemble-Based Machine Learning Approach. IEEE Access 2024, 12, 79549–79567. [Google Scholar] [CrossRef]
- Mennecke, A.; Khakzar, K.M.; German, A.; Herz, K.; Fabian, M.S.; Liebert, A.; Blümcke, I.; Kasper, B.S.; Nagel, A.M.; Laun, F.B.; et al. 7 tricks for 7 T CEST: Improving the reproducibility of multipool evaluation provides insights into the effects of age and the early stages of Parkinson’s disease. NMR Biomed. 2022, 36, e4717. [Google Scholar] [CrossRef] [PubMed]
- Panahi, M.; Habibi, M.; Hosseini, M.S. Enhancing MRI radiomics feature reproducibility and classification performance in Parkinson’s disease: A harmonization approach to gray-level discretization variability. Magn. Reson. Mater. Phys. Biol. Med. 2024, 38, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Panahi, M.; Hosseini, M.S. Impact of Harmonization on MRI Radiomics Feature Variability Across Preprocessing Methods for Parkinson’s Disease Motor Subtype Classification. J. Imaging Inform. Med. 2024, 1–14. [Google Scholar] [CrossRef]
- Salmanpour, M.R.; Shamsaei, M.; Saberi, A.; Hajianfar, G.; Soltanian-Zadeh, H.; Rahmim, A. Robust identification of Parkinson’s disease subtypes using radiomics and hybrid machine learning. Comput. Biol. Med. 2021, 129, 104142. [Google Scholar] [CrossRef]
- Germani, E.; Bhagwat, N.; Dugré, M.; Gau, R.; Montillo, A.A.; Nguyen, K.P.; Sokolowski, A.; Sharp, M.; Poline, J.-B.; Glatard, T.; et al. Predicting Parkinson’s disease trajectory using clinical and functional MRI features: A reproduction and replication study. PLoS ONE 2025, 20, e0317566. [Google Scholar] [CrossRef]
- Liloia, D.; Zamfira, D.A.; Tanaka, M.; Manuello, J.; Crocetta, A.; Keller, R.; Cozzolino, M.; Duca, S.; Cauda, F.; Costa, T. Disentangling the role of gray matter volume and concentration in autism spectrum disorder: A meta-analytic investigation of 25 years of voxel-based morphometry research. Neurosci. Biobehav. Rev. 2024, 164, 105791. [Google Scholar] [CrossRef]
- Hopfner, F.; Höglinger, G.; Trenkwalder, C. Definition and diagnosis of Parkinson’s disease: Guideline “Parkinson’s disease” of the German Society of Neurology. J. Neurol. 2024, 271, 7102–7119. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Lawton, M.; Heslegrave, A.J.; Guo, T.; Tan, M.; Jabbari, E.; Real, R.; Woodside, J.; Grosset, K.; Chelban, V.; et al. Combining biomarkers for prognostic modelling of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2022, 93, 707–715. [Google Scholar] [CrossRef]
- Vieira, S.R.; Toffoli, M.; Campbell, P.; Schapira, A.H. Biofluid Biomarkers in Parkinson’s Disease: Clarity Amid Controversy. Mov. Disord. 2020, 35, 1128–1133. [Google Scholar] [CrossRef]
- Gaetani, L.; Paoletti, F.P.; Mechelli, A.; Bellomo, G.; Parnetti, L. Research advancement in fluid biomarkers for Parkinson’s disease. Expert Rev. Mol. Diagn. 2024, 24, 885–898. [Google Scholar] [CrossRef]
- Xylaki, M.; Chopra, A.; Weber, S.; Bartl, M.; Outeiro, T.F.; Mollenhauer, B. Extracellular Vesicles for the Diagnosis of Parkinson’s Disease: Systematic Review and Meta-Analysis. Mov. Disord. 2023, 38, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Afshari, M.; Almeida, Q.; Amundsen-Huffmaster, S.; Balfany, K.; Camicioli, R.; Christiansen, C.; Dale, M.L.; Dibble, L.E.; Earhart, G.M.; et al. Digital gait biomarkers in Parkinson’s disease: Susceptibility/risk, progression, response to exercise, and prognosis. npj Park. Dis. 2025, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Yousif, M.A.M.; Elhassan, M.; Elhussein, Y.; Badawi, D.; Gabralla, S.; Abdelrahman, N. Genetic and serological biomarkers as predictors of Parkinson’s disease cognitive impairment: A systematic review. Innov. Aging 2024, 8, 1247. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. A Decade of Dedication: Pioneering Perspectives on Neurological Diseases and Mental Illnesses. Biomedicines 2024, 12, 1083. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Battaglia, S.; Giménez-Llort, L.; Chen, C.; Hepsomali, P.; Avenanti, A.; Vécsei, L. Innovation at the Intersection: Emerging Translational Research in Neurology and Psychiatry. Cells 2024, 13, 790. [Google Scholar] [CrossRef]
- Towns, C.; Richer, M.; Jasaityte, S.; Stafford, E.J.; Joubert, J.; Antar, T.; Martinez-Carrasco, A.; Makarious, M.B.; Casey, B.; Vitale, D.; et al. Defining the causes of sporadic Parkinson’s disease in the global Parkinson’s genetics program (GP2). npj Park. Dis. 2023, 9, 131. [Google Scholar] [CrossRef]
- Lim, S.-Y.; Tan, A.H.; Ahmad-Annuar, A.; Okubadejo, N.U.; Lohmann, K.; Morris, H.R.; Toh, T.S.; Tay, Y.W.; Lange, L.M.; Bandres-Ciga, S.; et al. Uncovering the genetic basis of Parkinson’s disease globally: From discoveries to the clinic. Lancet Neurol. 2024, 23, 1267–1280. [Google Scholar] [CrossRef]
- Schiess, N.; Cataldi, R.; Okun, M.S.; Fothergill-Misbah, N.; Dorsey, E.R.; Bloem, B.R.; Barretto, M.; Bhidayasiri, R.; Brown, R.; Chishimba, L.; et al. Six Action Steps to Address Global Disparities in Parkinson Disease. JAMA Neurol. 2022, 79, 929–936. [Google Scholar] [CrossRef]
- Lowell, E.R.; Macpherson, C.; Villarreal-Cavazos, K.; Chandrana, A.; Sevitz, J.S.; Veit, K.; Dakin, A.; Quinn, L.; Troche, M.S. An interdisciplinary approach to rehabilitation in Parkinson’s disease: Case series. Neurodegener. Dis. Manag. 2024, 14, 217–226. [Google Scholar] [CrossRef]
- McComish, S.F.; Copas, A.N.M.; Caldwell, M.A. Human Brain-Based Models Provide a Powerful Tool for the Advancement of Parkinson’s Disease Research and Therapeutic Development. Front. Neurosci. 2022, 16, 851058. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lv, X.; Ye, T.; Zhao, M.; Chen, Z.; Zhang, Y.; Yang, W.; Xie, H.; Zhan, L.; Chen, L.; et al. Microbiota-microglia crosstalk between Blautia producta and neuroinflammation of Parkinson’s disease: A bench-to-bedside translational approach. Brain Behav. Immun. 2024, 117, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Bellini, G.; D’ANtongiovanni, V.; Palermo, G.; Antonioli, L.; Fornai, M.; Ceravolo, R.; Bernardini, N.; Derkinderen, P.; Pellegrini, C. α-Synuclein in Parkinson’s Disease: From Bench to Bedside. Med. Res. Rev. 2024, 45, 909–946. [Google Scholar] [CrossRef] [PubMed]
- Augustine, A.; Winstanley, C.A.; Krishnan, V. Impulse Control Disorders in Parkinson’s Disease: From Bench to Bedside. Front. Neurosci. 2021, 15, 654238. [Google Scholar] [CrossRef]
- Hilmer, S. Drug development and evaluation: Age, sex, and frailty considerations to facilitate the fortitude factor. Innov. Aging 2024, 8 (Suppl. 1), 503. [Google Scholar] [CrossRef]
- Russillo, M.C.; Andreozzi, V.; Erro, R.; Picillo, M.; Amboni, M.; Cuoco, S.; Barone, P.; Pellecchia, M.T. Sex Differences in Parkinson’s Disease: From Bench to Bedside. Brain Sci. 2022, 12, 917. [Google Scholar] [CrossRef]
- Klæstrup, I.H.; Just, M.K.; Holm, K.L.; Alstrup, A.K.O.; Romero-Ramos, M.; Borghammer, P.; Berge, N.V.D. Impact of aging on animal models of Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 909273. [Google Scholar] [CrossRef]
- Vaidya, B.; Gupta, P.; Biswas, S.; Laha, J.K.; Roy, I.; Sharma, S.S. Effect of Clemizole on Alpha-Synuclein-Preformed Fibrils-Induced Parkinson’s Disease Pathology: A Pharmacological Investigation. NeuroMolecular Med. 2024, 26, 19. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. From Lab to Life: Exploring Cutting-Edge Models for Neurological and Psychiatric Disorders. Biomedicines 2024, 12, 613. [Google Scholar] [CrossRef]
- Pan, C.; Tian, Y.; Ma, L.; Zhou, T.; Ouyang, S.; Li, J. CISL-PD: A deep learning framework of clinical intervention strategies for Parkinson’s disease based on directional counterfactual Dual GANs. Expert Syst. Appl. 2024, 261, 125506. [Google Scholar] [CrossRef]
- Latourelle, J.C.; Beste, M.T.; Hadzi, T.C.; Miller, R.E.; Oppenheim, J.N.; Valko, M.P.; Wuest, D.M.; Church, B.W.; Khalil, I.G.; Hayete, B.; et al. Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: A longitudinal cohort study and validation. Lancet Neurol. 2017, 16, 908–916. [Google Scholar] [CrossRef]
- Jung, K.; Florin, E.; Patil, K.R.; Caspers, J.; Rubbert, C.; Eickhoff, S.B.; Popovych, O.V. Whole-brain dynamical modelling for classification of Parkinson’s disease. Brain Commun. 2022, 5, fcac331. [Google Scholar] [CrossRef]
- West, T.O.; Berthouze, L.; Farmer, S.F.; Cagnan, H.; Litvak, V. Inference of brain networks with approximate Bayesian computation—Assessing face validity with an example application in Parkinsonism. NeuroImage 2021, 236, 118020. [Google Scholar] [CrossRef]
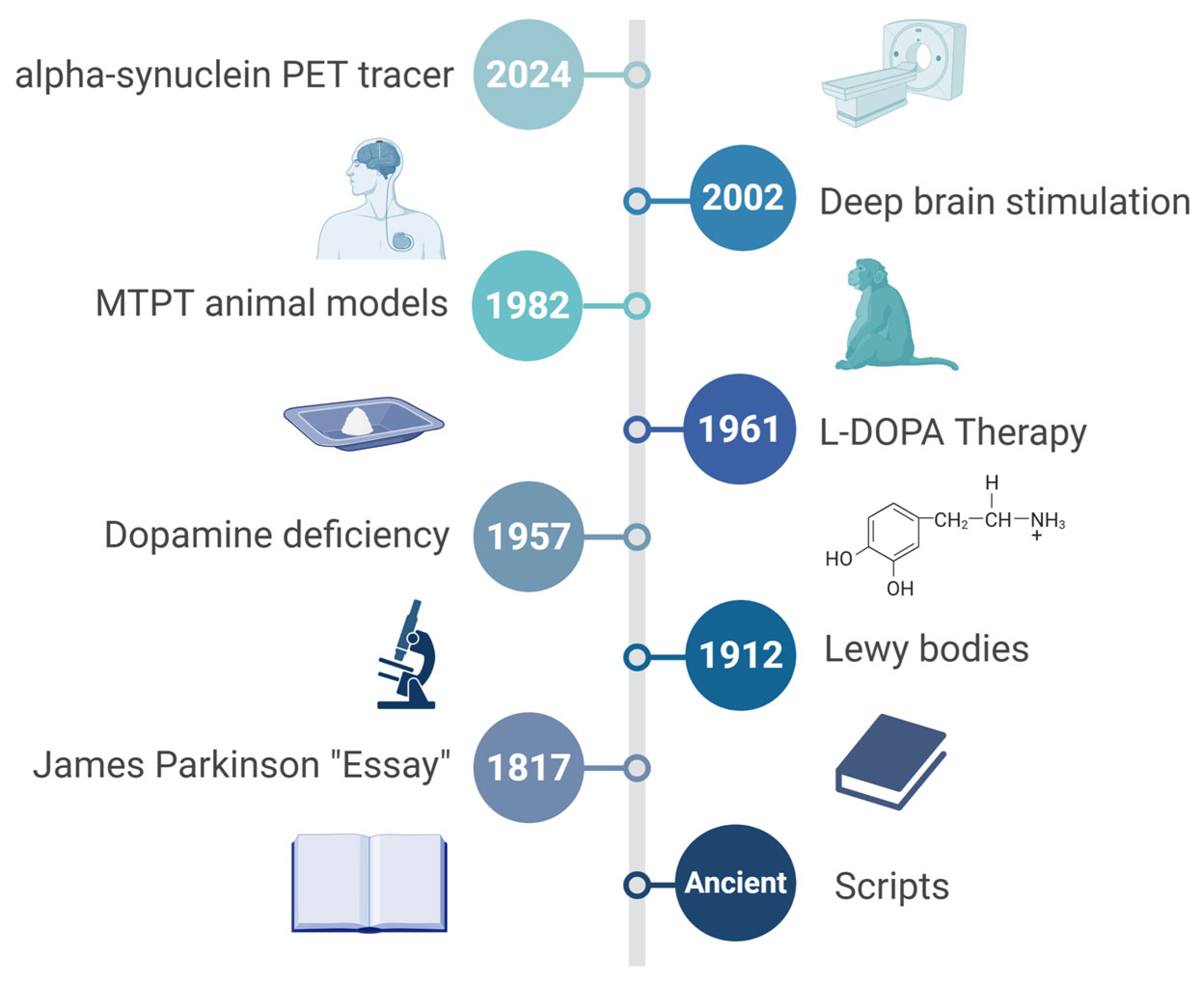
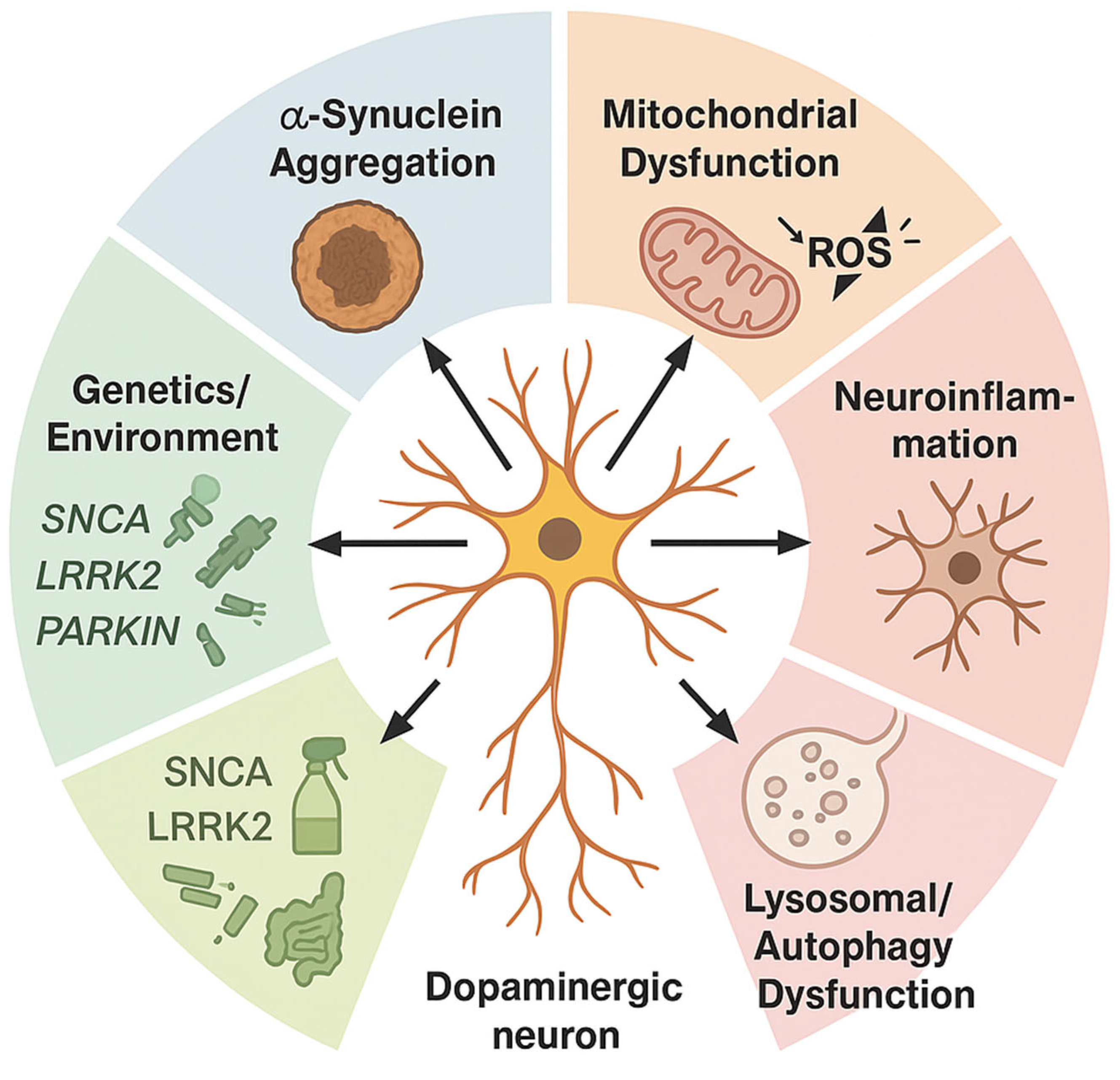
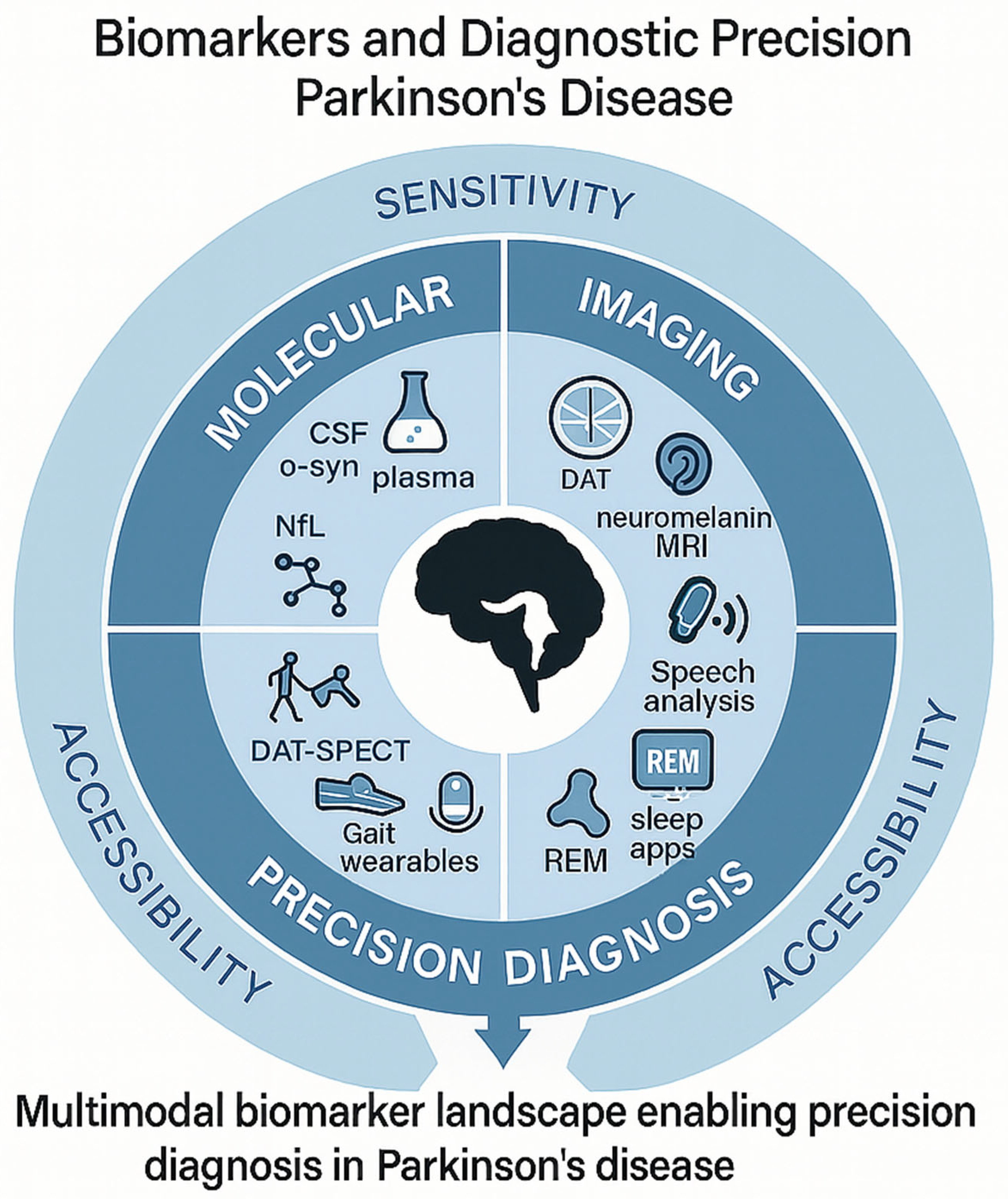


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene/Locus | Penetrance/Effect Size | Notable Phenotype or Pathway |
|---|---|---|---|
| Mendelian Genes | SNCA | High (autosomal dominant) | Early-onset, rapid progression; α-syn aggregation |
| LRRK2 | Moderate to high (age-dependent) | Variable onset; kinase signaling, autophagy dysregulation | |
| PRKN | High (recessive) | Juvenile onset, slow progression; mitochondrial quality control | |
| PINK1 | High (recessive) | Early-onset with dystonia; mitophagy dysfunction | |
| GBA1 | Moderate (heterozygous), high (biallelic) | Cognitive decline risk; lysosomal storage pathway | |
| Top GWAS Loci | MAPT (17q21) | OR ~1.3 | Tau processing, microtubule stabilization |
| BST1 (4p15) | OR ~1.2 | Immune regulation and calcium signaling | |
| GCH1 (14q22) | OR ~1.1–1.3 | Dopamine biosynthesis (tetrahydrobiopterin synthesis) | |
| TMEM175 | OR ~1.1 | Lysosomal function, linked to GBA1 network | |
| HLA-DRB5 | OR ~1.2 | Immune/inflammatory modulation, MHC class II region | |
| Polygenic Risk Tools | – | Aggregated PRS (AUC ~0.65–0.70) | Integrate >80 loci; used in research stratification, biomarker enrichment |
| Exposure Type | Example/Source | Relative Risk (RR) | Primary Mechanistic Link |
|---|---|---|---|
| Pesticides | Paraquat, rotenone | 1.5–2.5 | Mitochondrial complex-I inhibition, oxidative stress |
| Solvents | TCE, perchloroethylene | 1.3–2.0 | Dopaminergic neuron degeneration, α-syn aggregation |
| Metals | Manganese, lead | 1.2–1.8 | Oxidative damage, metal-induced neuroinflammation |
| Air Pollution | PM2.5, NO2 | 1.1–1.6 | Microglial activation, systemic inflammation |
| Head Trauma | Repeated concussions, TBI | 1.5–3.0 | BBB disruption, tauopathy |
| Diet | High dairy, low antioxidants | 0.8–1.3 | Gut–brain axis, mitochondrial stress |
| Exercise | Moderate-to-vigorous activity | 0.6–0.8 (protective) | Neurotrophic support, mitochondrial biogenesis |
| Pathway/Target | Mechanistic Role | Therapeutic Lead (≥Phase I) | Mechanism of Modulation |
|---|---|---|---|
| TLR4 (Toll-like receptor 4) | Innate immune activation, microglial priming | ApTOLL (TLR4 antagonist) | Blocks pro-inflammatory signaling cascade |
| NLRP3 inflammasome | IL-1β/IL-18 maturation, pyroptosis | Inzomelid (Inflazome/Roche) | Selective NLRP3 inhibition |
| NRF2 | Antioxidant transcriptional response | Dimethyl fumarate, PB125 | Activates NRF2-ARE pathway |
| Mitochondrial Complex I | Site of rotenone toxicity, ROS overproduction | UBIAD1 analogs, IACS-010759 | Stabilize complex I/enhance respiratory flux |
| GSK-3β | Crosstalk between inflammation and oxidative stress | Tideglusib, LY2090314 | Inhibits GSK-3β to restore redox and immune balance |
| NOX2 (NADPH oxidase 2) | ROS generation in activated microglia | GSK2795039 (NOX2 inhibitor) | Attenuates microglia-derived oxidative burst |
| Core Gap | How It Skews Evidence | Concrete Fix Suggested in Review |
|---|---|---|
| Diagnostic variability | Inflates cohort heterogeneity; undercuts power in early-phase trials | Apply multidimensional stratification (clinical + molecular) |
| Animal–human mismatch | Overpredicts efficacy; fails to capture complex non-motor pathology | Employ human iPSC-derived neurons/organoids and aged animal models to replicate human, age-dependent Parkinson’s pathology |
| Biomarker drift | Leads to irreproducible panels; fails external validation | Use longitudinal anchoring and cross-platform harmonization |
| Phase II–III collapse | Promising leads fail at scale; endpoint misalignment | Integrate target engagement biomarkers + adaptive designs |
| Underrepresentation | Skews generalizability; neglects frailty and late-life phenotypes | Mandate inclusive recruitment across age, ethnicity, frailty |
| Compartmentalized datasets | Blocks integration across imaging, omics, clinical tools | Build multimodal, federated data architectures |
| Biomarker/Tool | Modality | Primary Target/Read-out | Current Validation Phase |
|---|---|---|---|
| CSF α-syn RT-QuIC | Biofluid (CSF) | Seed-competent α-syn aggregates | Phase II (multicenter observational cohorts) |
| Plasma p-α-syn | Biofluid (blood) | Phosphorylated α-syn species | Phase I (assay standardization) |
| Plasma (NfL) | Biofluid (blood/CSF) | Axonal integrity marker | Clinically qualified for prognostics in ALS; Phase II for PD |
| DAT-SPECT | Molecular Imaging | Striatal dopamine-transporter binding | Clinically qualified (diagnostic aid) |
| Neuromelanin-sensitive MRI | Advanced MRI | Nigral neuromelanin loss | Phase I/II (single-site reproducibility) |
| α-syn PET Tracers | Molecular Imaging | Fibrillar/oligomeric α-syn in vivo | Preclinical → early Phase I (first-in-human safety) |
| Wearable-derived Gait Metrics | Digital/Sensor | Step amplitude, stride dynamics | Phase I/II (analytical validity, small cohorts) |
| Speech-analytics (smartphone) | Digital/AI | Articulatory rate, pitch variability | Phase I (algorithm development) |
| Research Domain | Current Non-European Participation | Active Inclusion Initiatives |
|---|---|---|
| Genetic Studies | <15% globally; <5% in GWAS meta-analyses | GP2—expanding genomic data from Africa, Asia, Latin America |
| Biomarker Cohorts | <10% in most CSF/imaging studies | MJFF Global PD Initiative—building diverse biosample banks and imaging pipelines |
| Clinical Trials | Typically <8% non-European enrolment | FIRE-UP PD—focused on equitable recruitment, community engagement, and outcome relevance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, M. Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation. Cells 2025, 14, 1161. https://doi.org/10.3390/cells14151161
Tanaka M. Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation. Cells. 2025; 14(15):1161. https://doi.org/10.3390/cells14151161
Chicago/Turabian StyleTanaka, Masaru. 2025. "Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation" Cells 14, no. 15: 1161. https://doi.org/10.3390/cells14151161
APA StyleTanaka, M. (2025). Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation. Cells, 14(15), 1161. https://doi.org/10.3390/cells14151161