
Epigenetic Regulation of Neutrophils in ARDS

 ,
,
Abstract
1. Introduction
2. Epigenetic Mechanisms
2.1. Overview of Epigenetic Mechanisms
2.2. DNA Methylation Mechanisms
2.3. Histone Modification Mechanisms
3. Neutrophil Function and Dysfunction
3.1. Normal Role of Neutrophils in the Immune System
3.2. Dysregulation of Neutrophil Function in ARDS
4. Neutrophil Epigenetic Biomarkers in ARDS
4.1. DNA Methylation
4.2. Histone Modifications
4.3. Non-Coding RNAs
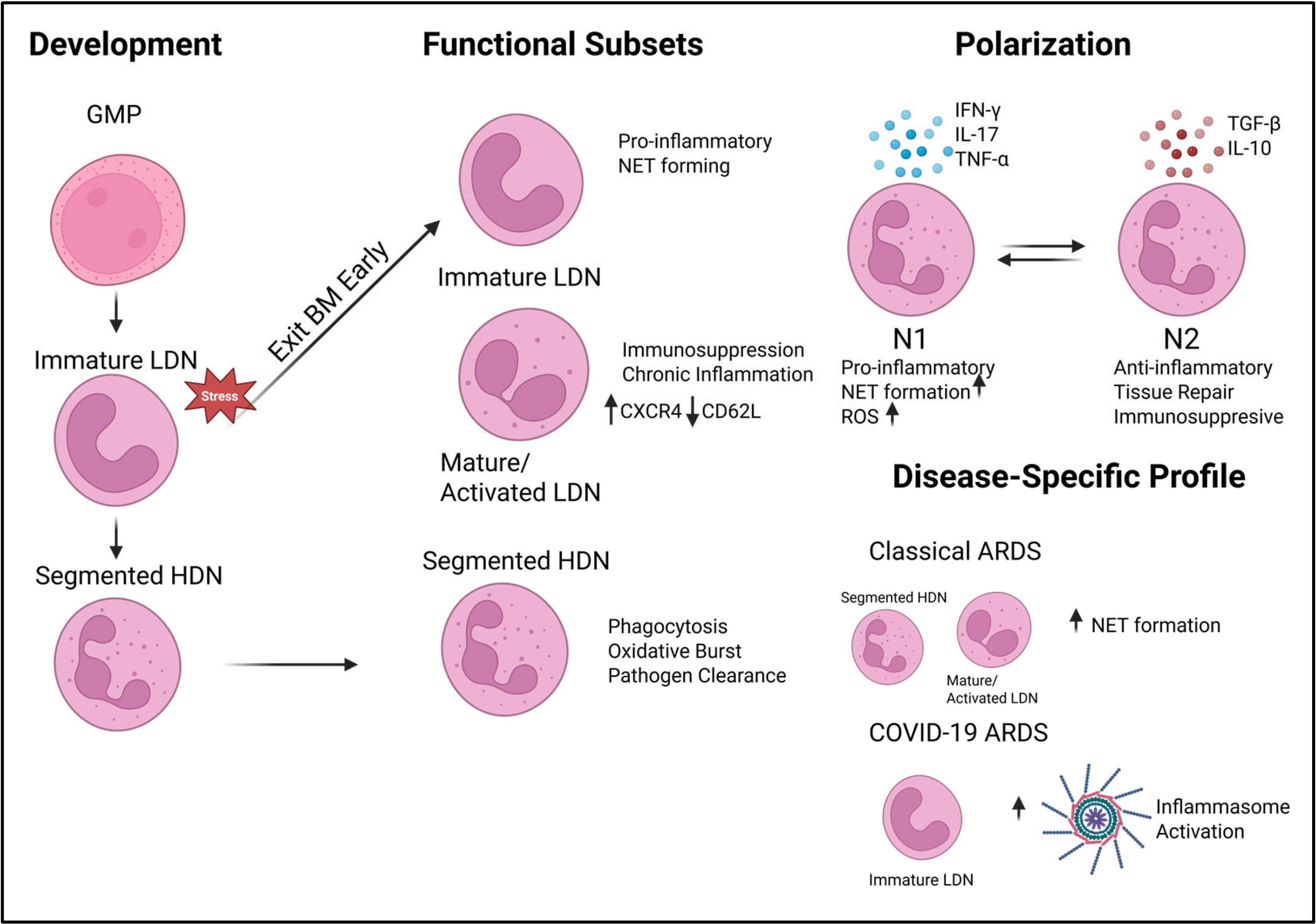
5. Neutrophil Diversity and ARDS Heterogeneity
5.1. Defining Neutrophil Heterogeneity: HDNs and NDNs
5.2. Single-Cell Insights into Neutrophil Subsets
Profiles in COVID-19 ARDS vs. Classical ARDS
6. Epigenetic Therapeutics in ARDS
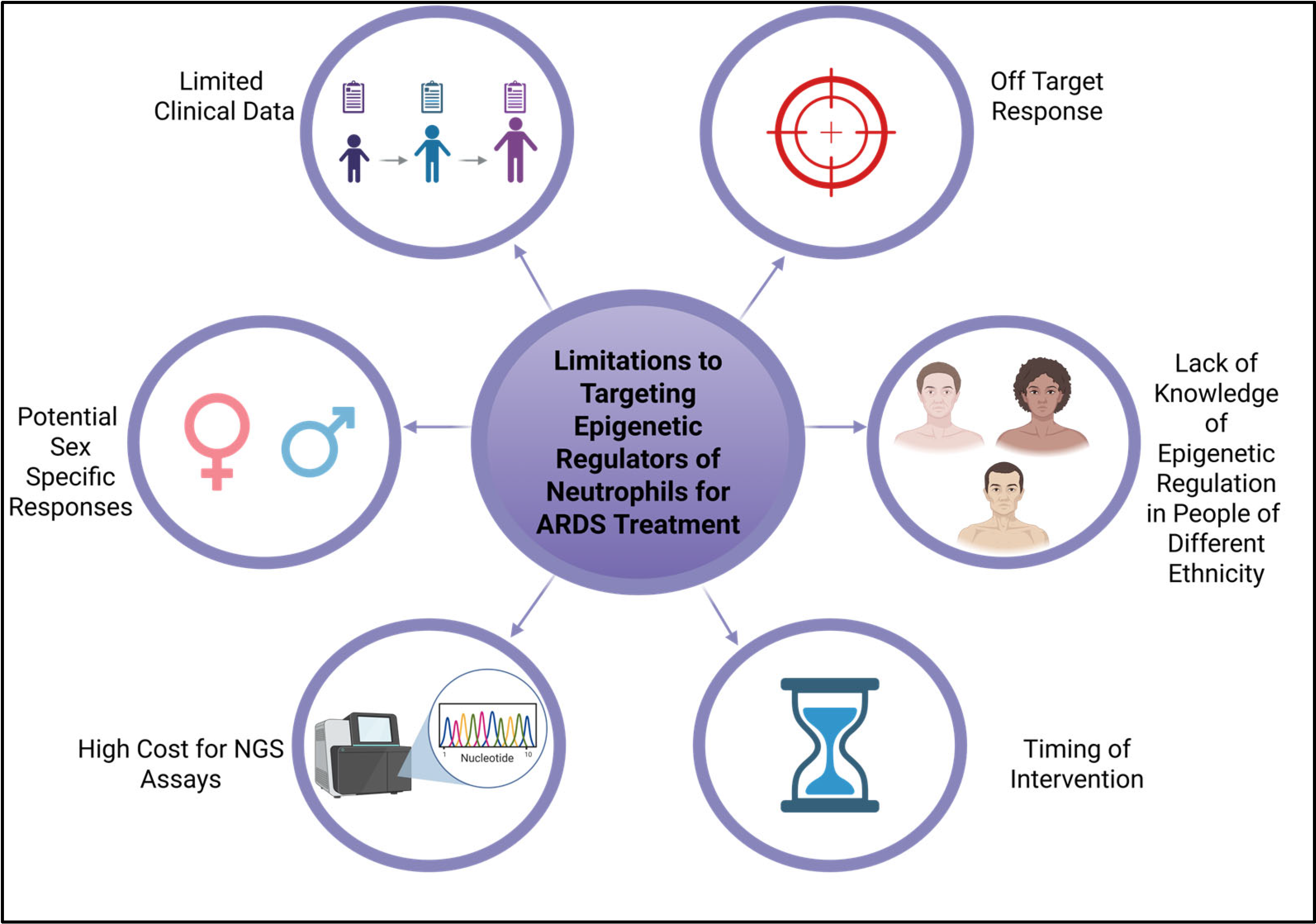
7. Limitations of Current Studies and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramji, H.F.; Hafiz, M.; Altaq, H.H.; Hussain, S.T.; Chaudry, F. Acute Respiratory Distress Syndrome; A Review of Recent Updates and a Glance into the Future. Diagnostics 2023, 13, 1528. [Google Scholar] [CrossRef]
- Yang, P.; Wu, T.; Yu, M.; Chen, F.; Wang, C.; Yuan, J.; Xu, J.; Zhang, G. A new method for identifying the acute respiratory distress syndrome disease based on noninvasive physiological parameters. PLoS ONE 2020, 15, e0226962. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Sjoding, M.W. Acute Respiratory Distress Syndrome: Definition, Diagnosis, and Routine Management. Crit. Care Clin. 2024, 40, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.C.; Zippoli, M.; Marsiglia, C.; Gavioli, E.M.; Cremonesi, G.; Khan, A.; Mantelli, F.; Allegretti, M.; Balk, R. Neutrophil activation and neutrophil extracellular traps (NETs) in COVID-19 ARDS and immunothrombosis. Eur. J. Immunol. 2023, 53, e2250010. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Williams, A.E.; Jose, R.J.; Mercer, P.F.; Brealey, D.; Parekh, D.; Thickett, D.R.; O’Kane, C.; McAuley, D.F.; Chambers, R.C. Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax 2017, 72, 66–73. [Google Scholar] [CrossRef]
- Ohms, M.; Möller, S.; Laskay, T. An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro. Front. Immunol. 2020, 11, 532. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Calle-Fabregat, C.; Morante-Palacios, O.; Ballestar, E. Understanding the Relevance of DNA Methylation Changes in Immune Differentiation and Disease. Genes 2020, 11, 110. [Google Scholar] [CrossRef]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef]
- Ostuni, R.; Piccolo, V.; Barozzi, I.; Polletti, S.; Termanini, A.; Bonifacio, S.; Curina, A.; Prosperini, E.; Ghisletti, S.; Natoli, G. Latent enhancers activated by stimulation in differentiated cells. Cell 2013, 152, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Prokhortchouk, E.; Defossez, P.-A. The cell biology of DNA methylation in mammals. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2008, 1783, 2167–2173. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell. Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Histone Ubiquitination: Triggering Gene Activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.F.; Gruber, M.A. Neutrophils-From Bone Marrow to First-Line Defense of the Innate Immune System. Front. Immunol. 2021, 12, 767175. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Borregaard, N.; Sørensen, O.E.; Theilgaard-Mönch, K. Neutrophil granules: A library of innate immunity proteins. Trends Immunol. 2007, 28, 340–345. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Dwivedi, A.; Ui Mhaonaigh, A.; Carroll, M.; Khosravi, B.; Batten, I.; Ballantine, R.S.; Hendricken Phelan, S.; O’Doherty, L.; George, A.M.; Sui, J.; et al. Emergence of dysfunctional neutrophils with a defect in arginase-1 release in severe COVID-19. JCI Insight 2024, 9, e171659. [Google Scholar] [CrossRef]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Ngo, V.L.; Geem, D.; Harusato, A.; Hirota, S.A.; Parkos, C.A.; Lukacs, N.W.; Nusrat, A.; Gaboriau-Routhiau, V.; Cerf-Bensussan, N.; et al. IL-17A-mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol. 2017, 10, 673–684. [Google Scholar] [CrossRef]
- Fu, Y.; Wen, Z.; Fan, J. Interaction of low-density neutrophils with other immune cells in the mechanism of inflammation. Mol. Med. 2025, 31, 133. [Google Scholar] [CrossRef]
- Sprenkeler, E.G.G.; Tool, A.T.J.; Henriet, S.S.V.; van Bruggen, R.; Kuijpers, T.W. Formation of neutrophil extracellular traps requires actin cytoskeleton rearrangements. Blood 2022, 139, 3166–3180. [Google Scholar] [CrossRef]
- Zhang, H.; Meng, L.; Liu, Y.; Jiang, J.; He, Z.; Qin, J.; Wang, C.; Yang, M.; He, K.; Yang, J.; et al. Sfxn5 Regulation of Actin Polymerization for Neutrophil Spreading Depends on a Citrate-Cholesterol-PI(4,5)P2 Pathway. J. Immunol. 2023, 211, 462–473. [Google Scholar] [CrossRef]
- Nong, Q.; Wu, Y.; Liu, S.; Tang, Y.; Wu, J.; Huang, H.; Hong, J.; Qin, Y.; Xu, R.; Zhao, W.; et al. Lead-induced actin polymerization aggravates neutrophil extracellular trap formation and contributes to vascular inflammation. Ecotoxicol. Environ. Saf. 2025, 290, 117598. [Google Scholar] [CrossRef] [PubMed]
- Grommes, J.; Soehnlein, O. Contribution of Neutrophils to Acute Lung Injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yao, Z.; Xu, S.; Yao, L.; Yu, Z. Glucocorticoid therapy for acute respiratory distress syndrome: Current concepts. J. Intensive Med. 2024, 4, 417–432. [Google Scholar] [CrossRef]
- Yang, L.; Gao, C.; He, Y.; Wang, X.; Yang, L.; Guo, S.; Chen, J.; He, S.; Sun, Y.; Gao, Y.; et al. The Neutrophil-to-Lymphocyte Ratio is Associated with the Requirement and the Duration of Invasive Mechanical Ventilation in Acute Respiratory Distress Syndrome Patients: A Retrospective Study. Can. Respir. J. 2022, 2022, 1581038. [Google Scholar] [CrossRef]
- Belchamber, K.B.R.; Thein, O.S.; Hazeldine, J.; Grudzinska, F.S.; Faniyi, A.A.; Hughes, M.J.; Jasper, A.E.; Yip, K.P.; Crowley, L.E.; Lugg, S.T.; et al. Dysregulated Neutrophil Phenotype and Function in Hospitalised Non-ICU COVID-19 Pneumonia. Cells 2022, 11, 2901. [Google Scholar] [CrossRef]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1. [Google Scholar] [CrossRef]
- Grégoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52, 1702590. [Google Scholar] [CrossRef]
- Bradic, M.; Taleb, S.; Thomas, B.; Chidiac, O.; Robay, A.; Hassan, N.; Malek, J.; Ait Hssain, A.; Abi Khalil, C. DNA methylation predicts the outcome of COVID-19 patients with acute respiratory distress syndrome. J. Transl. Med. 2022, 20, 526. [Google Scholar] [CrossRef]
- Feng, J.; Pang, J.; He, D.; Wu, Z.; Li, Q.; Ji, P.; He, C.; Zhong, Z.; Li, H.; Zhang, J. Identification of Genes with Altered Methylation and Its Role in Early Diagnosis of Sepsis-Induced Acute Respiratory Distress Syndrome. Int. J. Gen. Med. 2021, 14, 243–253. [Google Scholar] [CrossRef]
- Xu, X.; Sadiku, P.; Griffith, D.; Sánchez García, M.; Walmsley, S. S97 Neutrophil Epigenetic Signatures in the Context of Acute Respiratory Distress Syndrome. Thorax 2022, 77, A61. [Google Scholar] [CrossRef]
- Liu, X.; Li, T.; Chen, H.; Yuan, L.; Ao, H. Role and intervention of PAD4 in NETs in acute respiratory distress syndrome. Respir. Res. 2024, 25, 63. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Pui-Yan Ma, V.; Di Gioia, M.; Broggi, A.; Benamar, M.; Chen, Q.; Mazitschek, R.; Haggarty, S.J.; Chatila, T.A.; Karp, J.M.; et al. Zinc-dependent histone deacetylases drive neutrophil extracellular trap formation and potentiate local and systemic inflammation. iScience 2021, 24, 103256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, Y. MiR-223 within neutrophil axis promotes Th17 expansion by PI3K-AKT pathway in systemic lupus erythematosus. Arthritis Res. Ther. 2025, 27, 21. [Google Scholar] [CrossRef]
- Xu, W.; Wang, Y.; Ma, Y.; Yang, J. MiR-223 plays a protecting role in neutrophilic asthmatic mice through the inhibition of NLRP3 inflammasome. Respir. Res. 2020, 21, 116. [Google Scholar] [CrossRef]
- Almuntashiri, S.; Han, Y.; Youngblood, H.A.; Chase, A.; Zhu, Y.; Wang, X.; Linder, D.F.; Siddiqui, B.; Sikora, A.; Liu, Y.; et al. Identification of circulating microvesicle-encapsulated miR-223 as a potential novel biomarker for ARDS. Physiol. Rep. 2022, 10, e15494. [Google Scholar] [CrossRef]
- Arroyo, A.B.; Fernández-Pérez, M.P.; Del Monte, A.; Águila, S.; Méndez, R.; Hernández-Antolín, R.; García-Barber, N.; de Los Reyes-García, A.M.; González-Jiménez, P.; Arcas, M.I.; et al. miR-146a is a pivotal regulator of neutrophil extracellular trap formation promoting thrombosis. Haematologica 2021, 106, 1636–1646. [Google Scholar] [CrossRef]
- Gysemans, C.; Beya, M.; Pedace, E.; Mathieu, C. Exploring Neutrophil Heterogeneity and Plasticity in Health and Disease. Biomedicines 2025, 13, 597. [Google Scholar] [CrossRef]
- Hassani, M.; Hellebrekers, P.; Chen, N.; van Aalst, C.; Bongers, S.; Hietbrink, F.; Koenderman, L.; Vrisekoop, N. On the origin of low-density neutrophils. J. Leukoc. Biol. 2020, 107, 809–818. [Google Scholar] [CrossRef]
- He, W.; Yan, L.; Hu, D.; Hao, J.; Liou, Y.C.; Luo, G. Neutrophil heterogeneity and plasticity: Unveiling the multifaceted roles in health and disease. MedComm (2020) 2025, 6, e70063. [Google Scholar] [CrossRef]
- Panda, R.; Castanheira, F.V.; Schlechte, J.M.; Surewaard, B.G.; Shim, H.B.; Zucoloto, A.Z.; Slavikova, Z.; Yipp, B.G.; Kubes, P.; McDonald, B. A functionally distinct neutrophil landscape in severe COVID-19 reveals opportunities for adjunctive therapies. JCI Insight 2022, 7, e152291. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Anders, H.-J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the dark-NETs of neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Calfee, C.S.; Cherian, S.; Brealey, D.; Cutler, S.; King, C.; Killick, C.; Richards, O.; Cheema, Y.; Bailey, C.; et al. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: A prospective observational study. Lancet Respir. Med. 2020, 8, 1209–1218. [Google Scholar] [CrossRef]
- Cabrera, L.E.; Jokiranta, S.T.; Mäki, S.; Miettinen, S.; Kant, R.; Kareinen, L.; Sironen, T.; Pietilä, J.-P.; Kantele, A.; Kekäläinen, E.; et al. The assembly of neutrophil inflammasomes during COVID-19 is mediated by type I interferons. PLOS Pathog. 2024, 20, e1012368. [Google Scholar] [CrossRef]
- Surolia, R.; Li, F.J.; Wang, Z.; Kashyap, M.; Srivastava, R.K.; Traylor, A.M.; Singh, P.; Dsouza, K.G.; Kim, H.; Pittet, J.F.; et al. NETosis in the pathogenesis of acute lung injury following cutaneous chemical burns. JCI Insight 2021, 6, 147564. [Google Scholar] [CrossRef]
- Bonilha, C.S.; Veras, F.P.; Dos Santos Ramos, A.; Gomes, G.F.; Rodrigues Lemes, R.M.; Arruda, E.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q. PAD4 inhibition impacts immune responses in SARS-CoV-2 infection. Mucosal Immunol. 2025, in press. [CrossRef]
- Deng, H.; Lin, C.; Garcia-Gerique, L.; Fu, S.; Cruz, Z.; Bonner, E.E.; Rosenwasser, M.; Rajagopal, S.; Sadhu, M.N.; Gajendran, C.; et al. A Novel Selective Inhibitor JBI-589 Targets PAD4-Mediated Neutrophil Migration to Suppress Tumor Progression. Cancer Res. 2022, 82, 3561–3572. [Google Scholar] [CrossRef]
- Gajendran, C.; Fukui, S.; Sadhu, N.M.; Zainuddin, M.; Rajagopal, S.; Gosu, R.; Gutch, S.; Fukui, S.; Sheehy, C.E.; Chu, L.; et al. Alleviation of arthritis through prevention of neutrophil extracellular traps by an orally available inhibitor of protein arginine deiminase 4. Sci. Rep. 2023, 13, 3189. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.-F.; Wang, J.; Yan, X.-L.; Tian, F.; Zhao, J.-B.; Wang, Y.-J.; Jiang, T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir. Res. 2010, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Mercer, P.F.; Williams, A.E.; Scotton, C.J.; José, R.J.; Sulikowski, M.; Moffatt, J.D.; Murray, L.A.; Chambers, R.C. Proteinase-activated receptor-1, CCL2, and CCL7 regulate acute neutrophilic lung inflammation. Am. J. Respir. Cell Mol. Biol. 2014, 50, 144–157. [Google Scholar] [CrossRef]
- Cui, S.N.; Chen, L.; Yang, Y.Y.; Wang, Y.X.; Li, S.N.; Zhou, T.; Xiao, H.R.; Qin, L.; Yang, W.; Yuan, S.Y.; et al. Activation of death-associated protein kinase 1 promotes neutrophil apoptosis to accelerate inflammatory resolution in acute respiratory distress syndrome. Lab. Investig. 2019, 99, 1143–1156. [Google Scholar] [CrossRef]
- Eruslanov, E.B.; Singhal, S.; Albelda, S.M. Mouse versus Human Neutrophils in Cancer: A Major Knowledge Gap. Trends Cancer 2017, 3, 149–160. [Google Scholar] [CrossRef]
- Pilling, D.; Consalvo, K.M.; Kirolos, S.A.; Gomer, R.H. Differences between human male and female neutrophils with respect to which RNAs are present in polysomes. bioRxiv 2025. [Google Scholar] [CrossRef]
- Lat, T.I.; McGraw, M.K.; White, H.D. Gender Differences in Critical Illness and Critical Care Research. Clin. Chest Med. 2021, 42, 543–555. [Google Scholar] [CrossRef]
- Gujski, M.; Jankowski, M.; Rabczenko, D.; Gorynski, P.; Juszczyk, G. The Prevalence of Acute Respiratory Distress Syndrome (ARDS) and Outcomes in Hospitalized Patients with COVID-19-A Study Based on Data from the Polish National Hospital Register. Viruses 2022, 14, 76. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Faidah, H.; Al-Maiahy, T.J.; Cruz-Martins, N.; Batiha, G.E. The Looming Effects of Estrogen in COVID-19: A Rocky Rollout. Front. Nutr. 2021, 8, 649128. [Google Scholar] [CrossRef]
- Ishikawa, M.; Murakami, H.; Higashi, H.; Inoue, T.; Fujisaki, N.; Kohama, K. Sex Differences of Neutrophil Extracellular Traps on Lipopolysaccharide-Stimulated Human Neutrophils. Surg. Infect. 2024, 25, 505–512. [Google Scholar] [CrossRef]
- Papoutsi, E.; Kremmydas, P.; Tsolaki, V.; Kyriakoudi, A.; Routsi, C.; Kotanidou, A.; Siempos, I.I. Racial and ethnic minority participants in clinical trials of acute respiratory distress syndrome. Intensive Care Med. 2023, 49, 1479–1488. [Google Scholar] [CrossRef]
- Constantinescu, A.E.; Hughes, D.A.; Bull, C.J.; Fleming, K.; Mitchell, R.E.; Zheng, J.; Kar, S.; Timpson, N.J.; Amulic, B.; Vincent, E.E. A genome-wide association study of neutrophil count in individuals associated to an African continental ancestry group facilitates studies of malaria pathogenesis. Hum. Genom. 2024, 18, 26. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | a Preclinical Evidence Strength | b Clinical Development Status | c Role in Neutrophil Biology | d Example Inhibitor Compounds |
|---|---|---|---|---|
| PAD4 | Strong | Preclinical | Drives NETosis through histone citrullination | JBI-589, GSK199, GSK484, BMS-P5 |
| HDAC 1/2/3 | Strong | Clinical (Oncology) | Pro-inflammatory gene expression and NETosis | Entinostat, Vorinostat |
| HDAC 6 | Strong | Clinical (Oncology) | NET formation | Ricolinostat, TubastatinA |
| DNMT3A | Moderate | Approved (Oncology) | Loss of methylation leads to inflammation | Azacitidine |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, J.E.; Mauya, Z.; Walkup, V.; Adderley, S.; Evans, C.; Wilson, K. Epigenetic Regulation of Neutrophils in ARDS. Cells 2025, 14, 1151. https://doi.org/10.3390/cells14151151
Williams JE, Mauya Z, Walkup V, Adderley S, Evans C, Wilson K. Epigenetic Regulation of Neutrophils in ARDS. Cells. 2025; 14(15):1151. https://doi.org/10.3390/cells14151151
Chicago/Turabian StyleWilliams, Jordan E., Zannatul Mauya, Virginia Walkup, Shaquria Adderley, Colin Evans, and Kiesha Wilson. 2025. "Epigenetic Regulation of Neutrophils in ARDS" Cells 14, no. 15: 1151. https://doi.org/10.3390/cells14151151
APA StyleWilliams, J. E., Mauya, Z., Walkup, V., Adderley, S., Evans, C., & Wilson, K. (2025). Epigenetic Regulation of Neutrophils in ARDS. Cells, 14(15), 1151. https://doi.org/10.3390/cells14151151