
Estradiol Downregulates MicroRNA-193a to Mediate Its Angiogenic Actions

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Transfection of HUVECs with MicroRNA-193a Mimic and Antimir
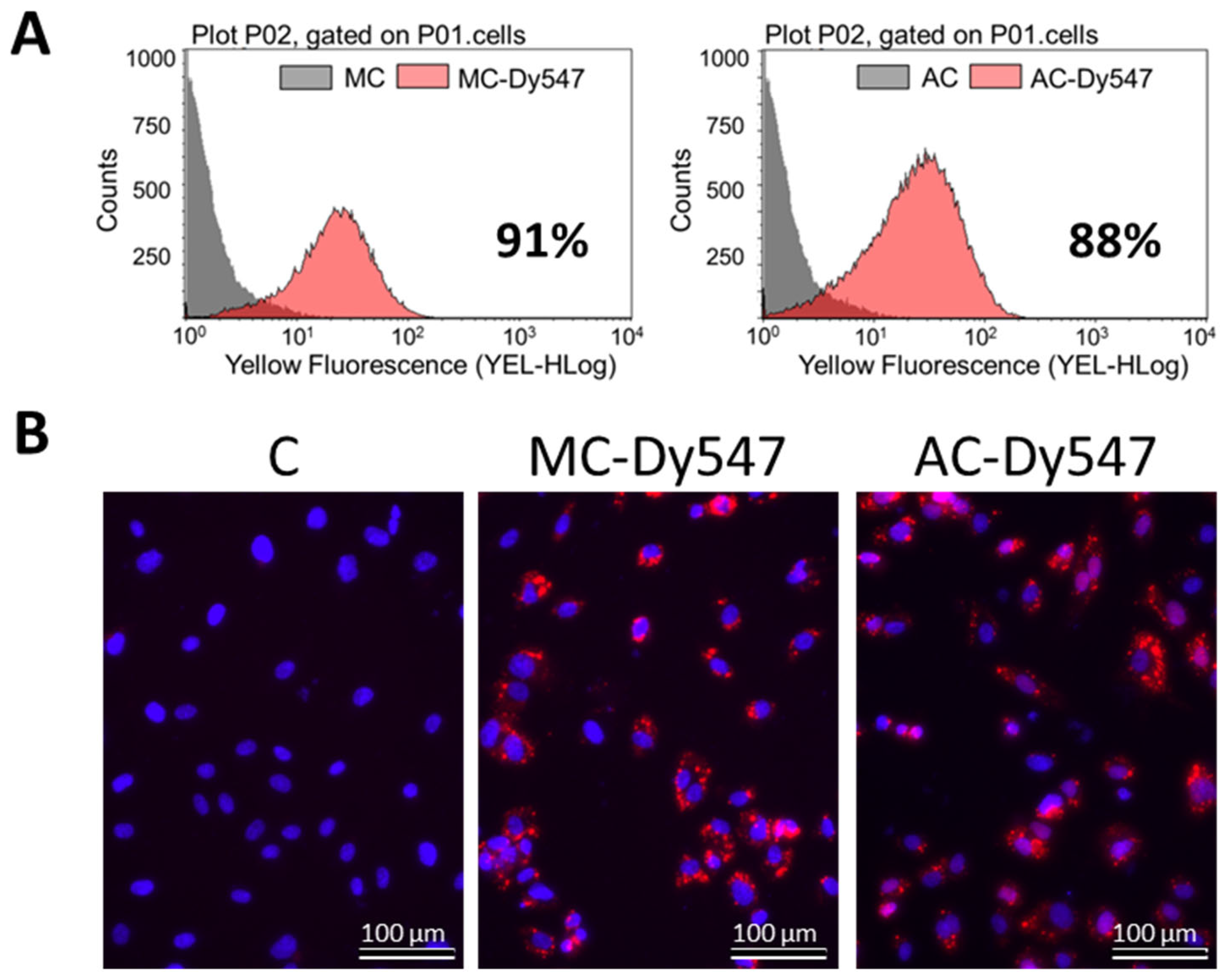
2.3. Transfection Efficiency
2.4. Expression of Pri- and Mature MiRNA by qRT-PCR
2.5. HUVEC Growth Studies: Microvessel Formation, Migration, and Proliferation
2.6. Protein Expression Studies
2.7. Ovariectomized Model C57Bl6 and Matrigel Plug Assay
2.8. Analysis of Hemoglobin from Matrigel Plugs by the Drabkin Method
2.9. Human Samples
2.10. Statistical Analysis
3. Results
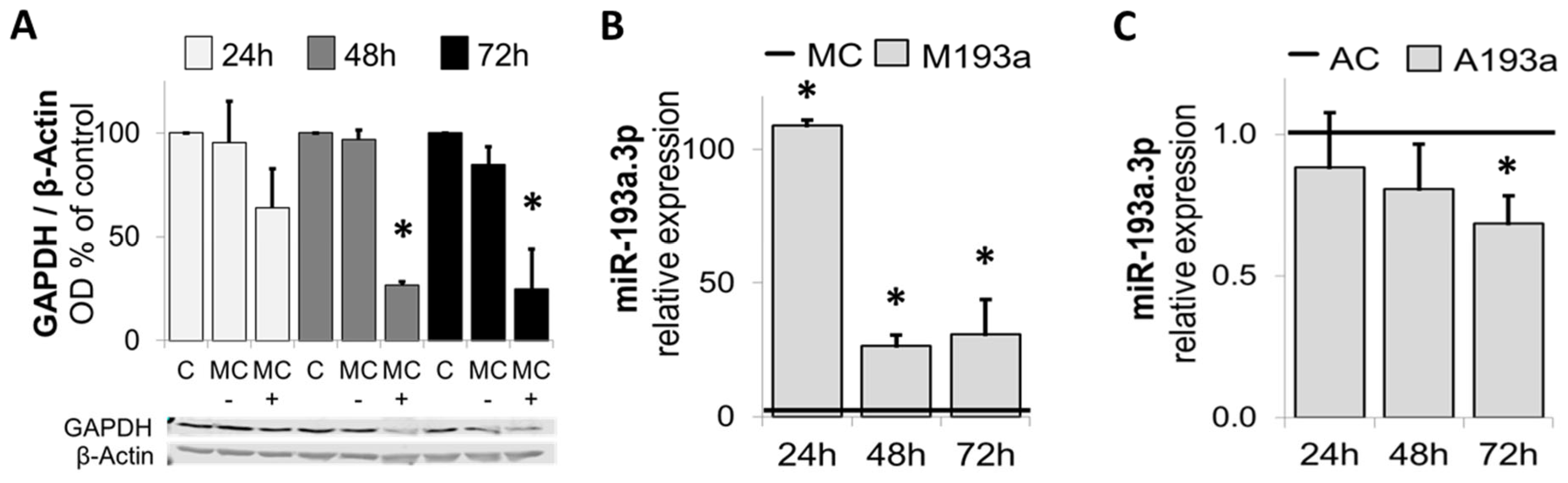
3.1. E2 Downregulates miR-193a but Not miR-146, miR-409-5p, and miR-494 in HUVECs
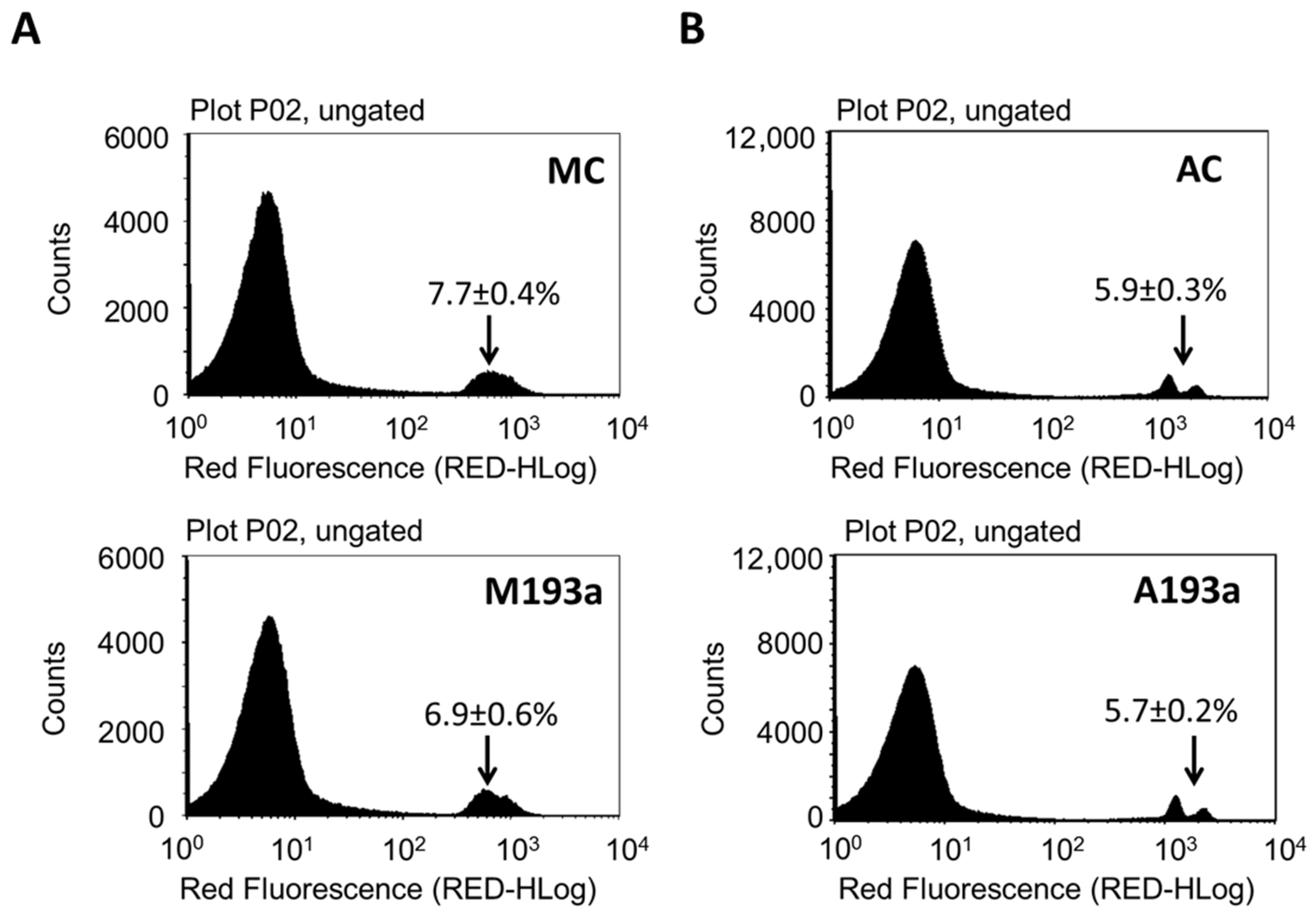
3.2. MiR-193a-3p Transfection Efficiency in HUVECS
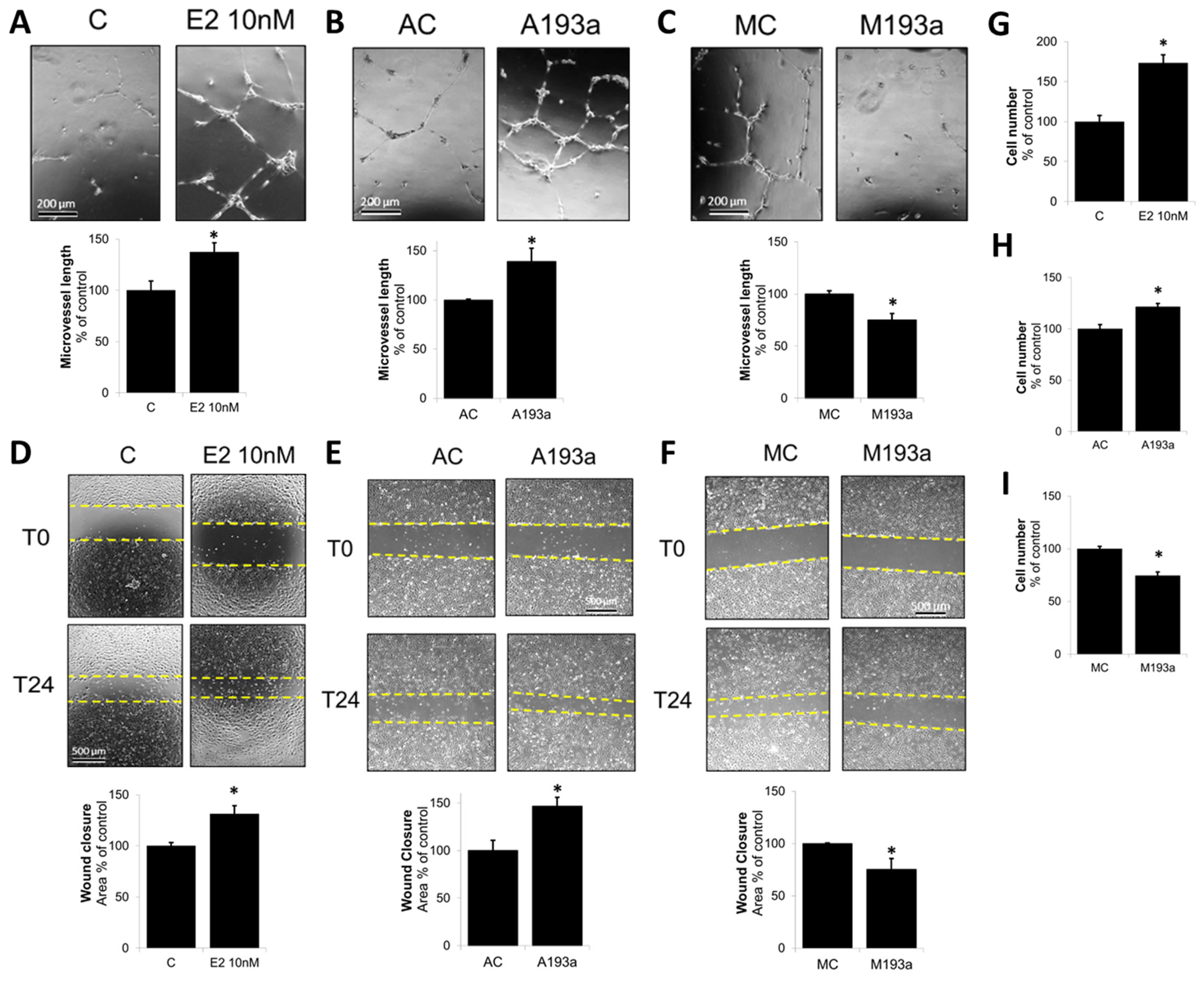
3.3. MiR-193a-3p Mimic Inhibits EC Microvessel Formation, Migration, and Proliferation Whereas Its Antimir Has Pro-Growth/Capillary-Stimulating Effects Similar to E2
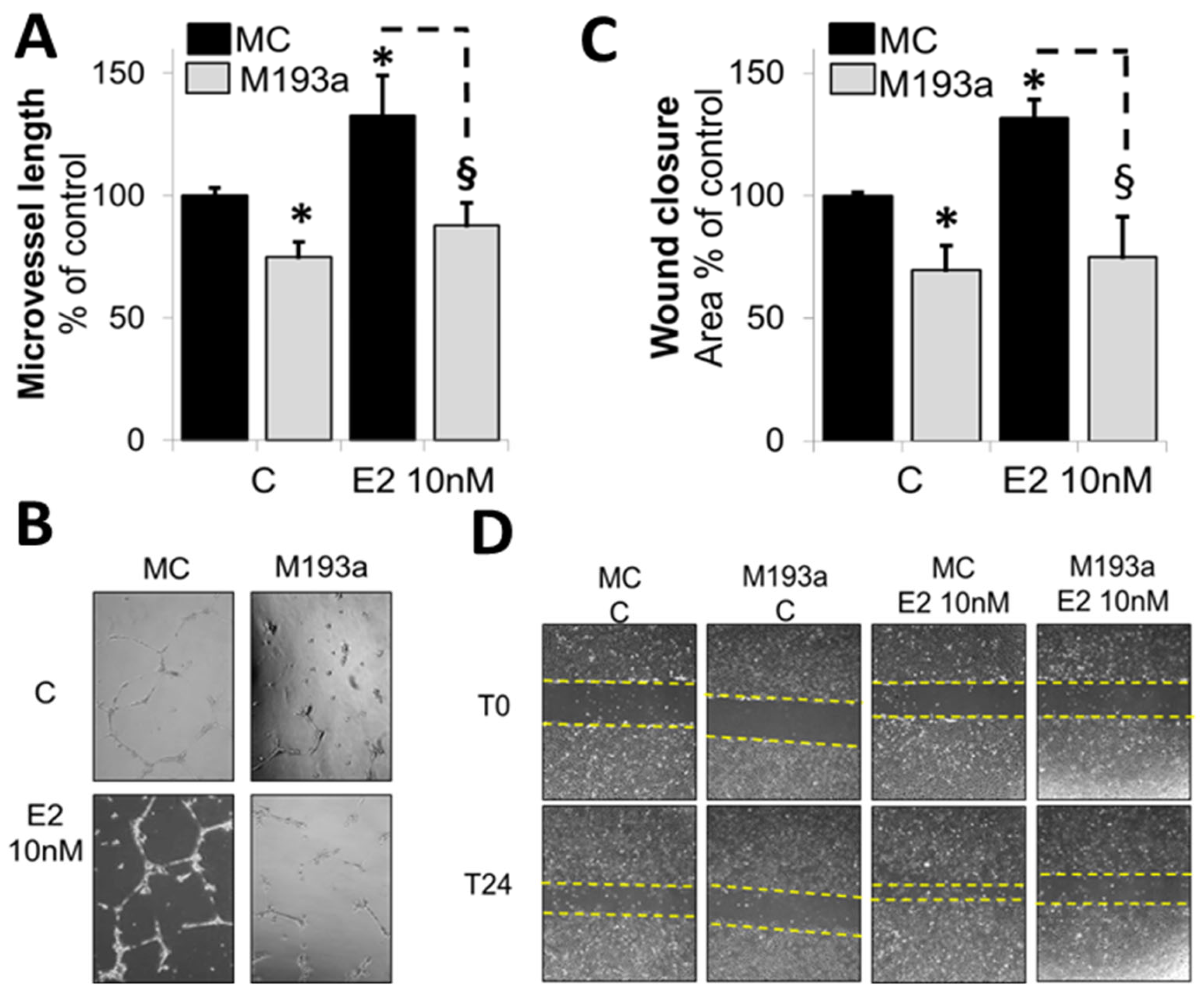
3.4. E2 Promotes EC Migration and Capillary Formation by Downregulating the Expression of miR-193a-3p
3.5. MiR-193a-3p Does Not Affect EC Viability
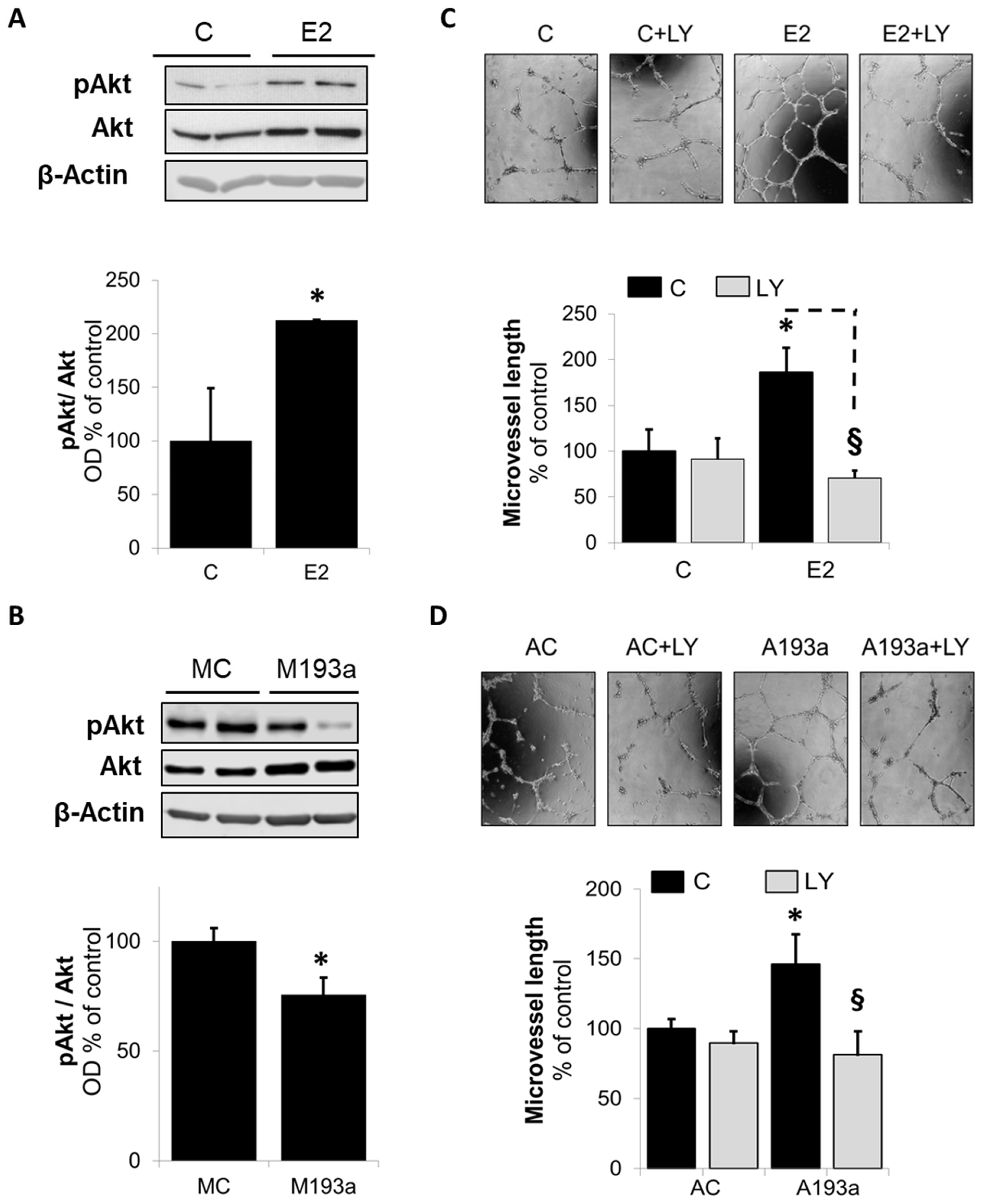
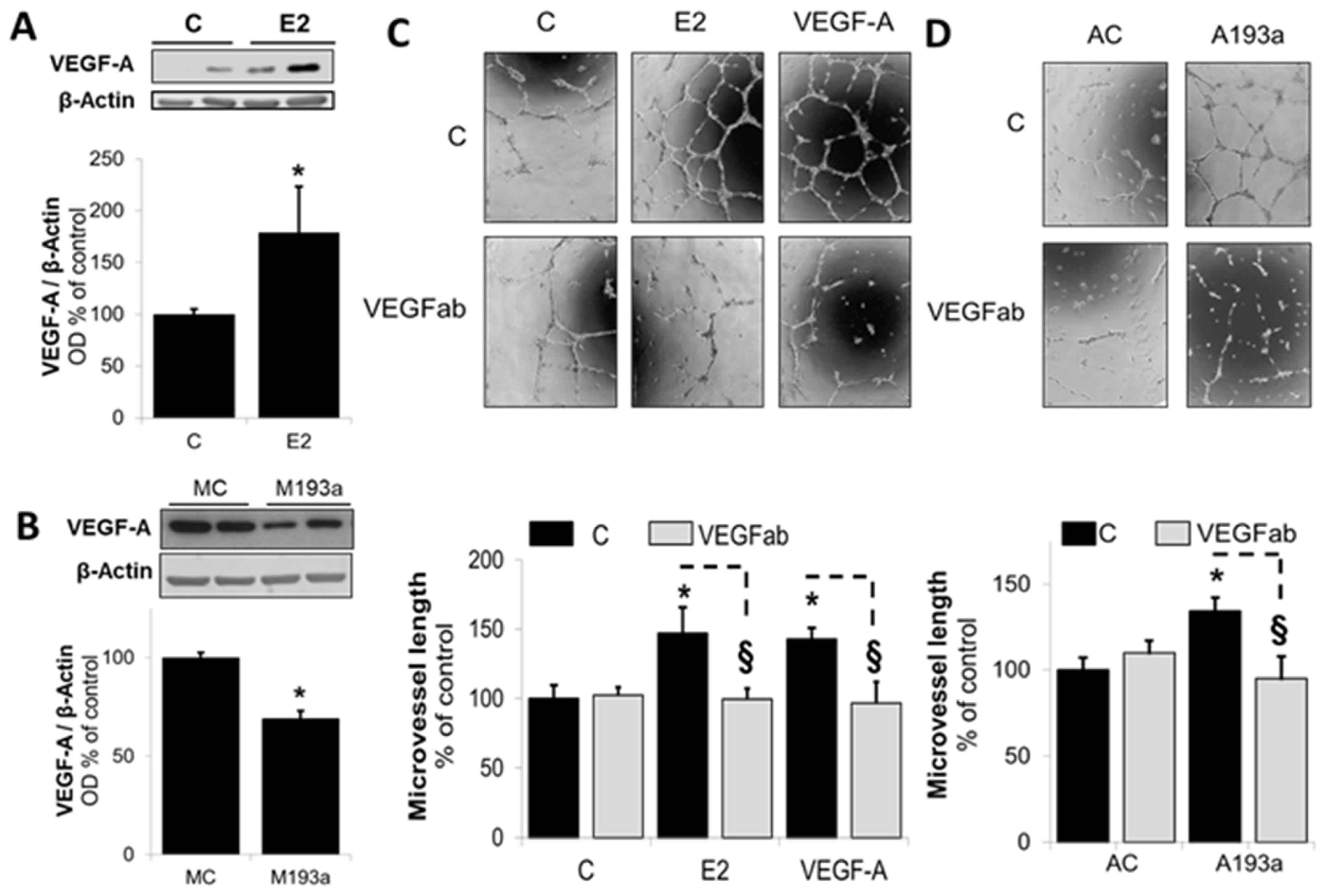
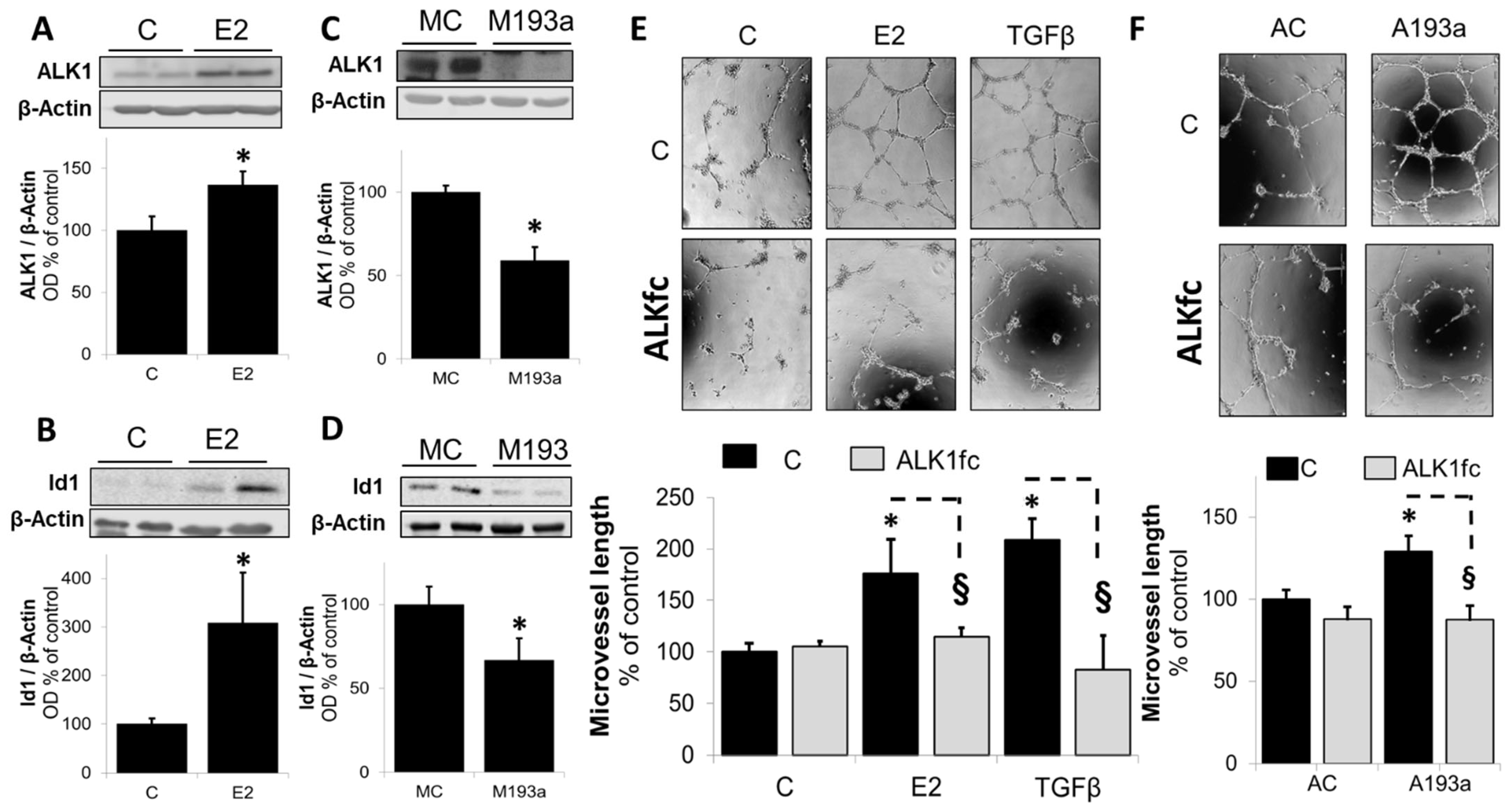
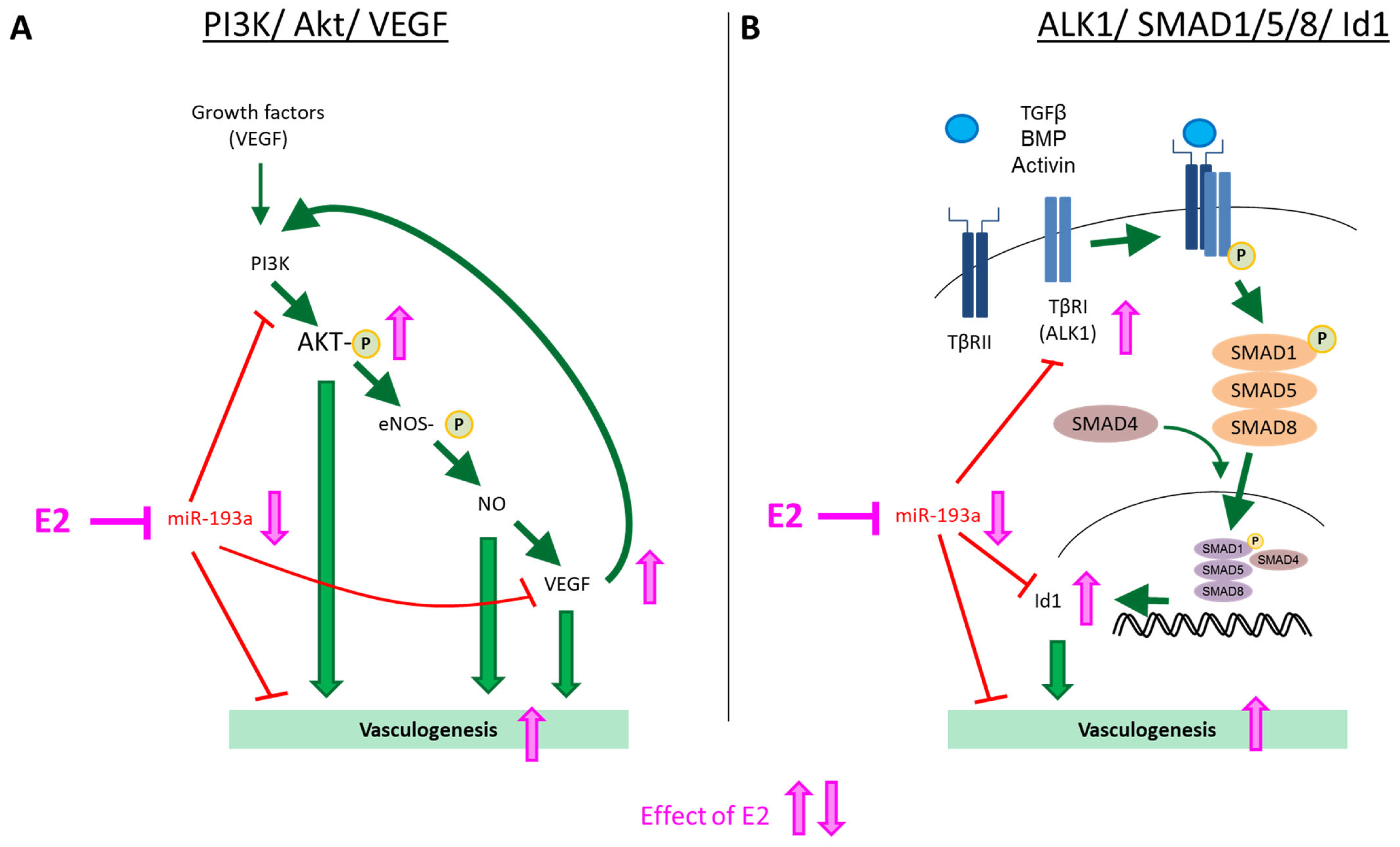
3.6. Intracellular Mechanisms of MiR-193a-3p-Mediated Vasculogenesis
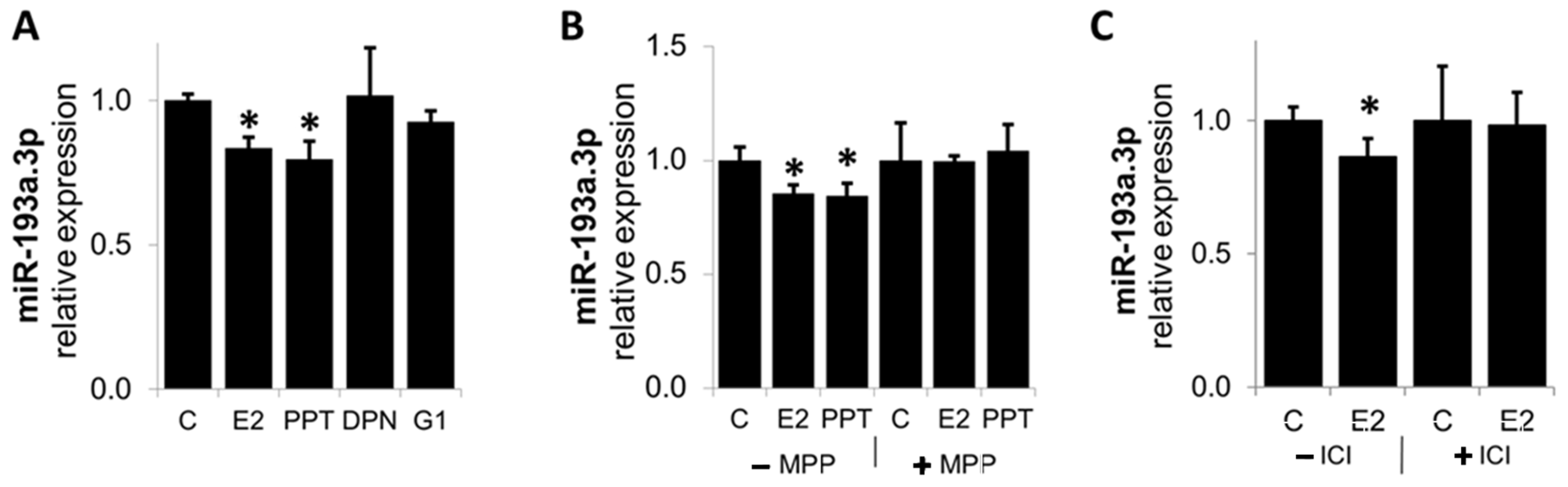
3.7. E2 Downregulates MiR-193a-3p Expression via ER-α
3.8. E2 Does Not Influence Pri-MiR-193a-3p Expression
3.9. Methoxyestradiol, Endogenous E2 Metabolite Upregulates MiR-193a-3p Expression in HUVECs
3.10. MiR-193a-3p Mimic Inhibits Basal and E2-Induced Angiogenesis In Vivo
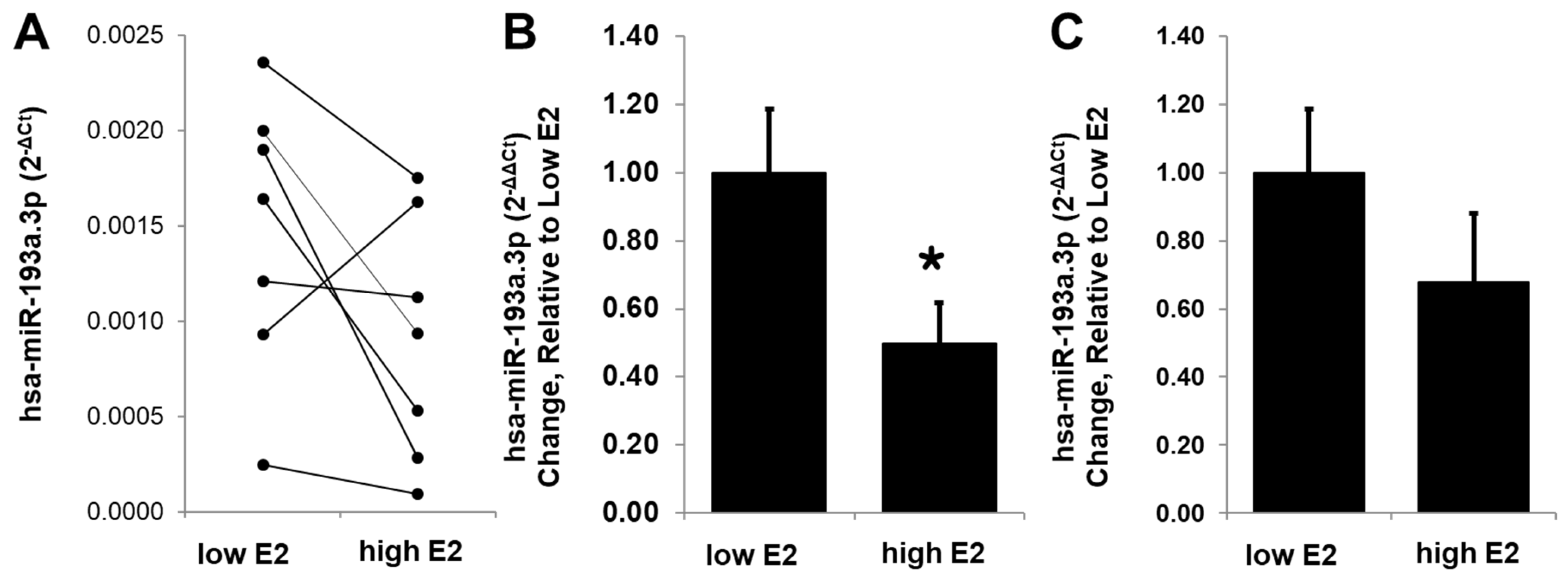
3.11. E2 Modulates Circulating miR-193a Levels in Humans
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tunstall-Pedoe, H.; Kuulasmaa, K.; Amouyel, P.; Arveiler, D.; Rajakangas, A.M.; Pajak, A. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents. Circulation 1994, 90, 583–612. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.M.; Fowlkes, L.P. The clinical aspects of estrogen and the cardiovascular system. Obstet. Gynecol. 1996, 87, 36S–43S. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.M.C.; Vitale, C.; Marazzi, G.; Volterrani, M. Menopause and cardiovascular disease: The evidence. Climacteric 2007, 10 (Suppl. S1), 19–24. [Google Scholar] [CrossRef] [PubMed]
- Gouva, L.; Tsatsoulis, A. The role of estrogens in cardiovascular disease in the aftermath of clinical trials. Hormones 2004, 3, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Mikkola, T.S.; Tuomikoski, P.; Lyytinen, H.; Korhonen, P.; Hoti, F.; Vattulainen, P.; Gissler, M.; Ylikorkala, O. Estradiol-based postmenopausal hormone therapy and risk of cardiovascular and all-cause mortality. Menopause 2015, 22, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Oparil, S. Effects of Sex Steroids in Vascular Injury. In Endocrinology of Cardiovascular Function; Levin, E.R., Nadler, J.L., Eds.; Springer: Boston, MA, USA, 1998; pp. 45–59. [Google Scholar] [CrossRef]
- Nie, G.; Yang, X.; Wang, Y.; Liang, W.; Li, X.; Luo, Q.; Yang, H.; Liu, J.; Wang, J.; Guo, Q.; et al. The Effects of Menopause Hormone Therapy on Lipid Profile in Postmenopausal Women: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022, 13, 850815. [Google Scholar] [CrossRef] [PubMed]
- Rubanyi, G.M.; Johns, A.; Kauser, K. Effect of estrogen on endothelial function and angiogenesis. Vasc. Pharmacol. 2002, 38, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Cossette, É.; Cloutier, I.; Tardif, K.; DonPierre, G.; Tanguay, J.-F. Estradiol inhibits vascular endothelial cells pro-inflammatory activation induced by C-reactive protein. Mol. Cell. Biochem. 2013, 373, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Krasinski, K.; Spyridopoulos, I.; Asahara, T.; van der Zee, R.; Isner, J.M.; Losordo, D.W. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation 1997, 95, 1768–1772. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, M.E.; Karas, R.H. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.E.; McGowan, K.A.; Grant, D.S.; Maheshwari, S.; Bhartiya, D.; Cid, M.C.; Kleinman, H.K.; Schnaper, H.W. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 1995, 91, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Nozell, S.; Chen, Y.-F.; Hage, F.; Oparil, S. Estrogen and Mechanisms of Vascular Protection. Arter. Thromb. Vasc. Biol. 2009, 29, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Al-Shami, K.; Awadi, S.; Khamees, A.; Alsheikh, A.M.; Al-Sharif, S.; Bereshy, R.A.; Al-Eitan, S.F.; Banikhaled, S.H.; Al-Qudimat, A.R.; Al-Zoubi, R.M.; et al. Estrogens and the risk of breast cancer: A narrative review of literature. Heliyon 2023, 9, e20224. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E.; Hunter, S.; Bracht, J. MicroRNAs: A developing story. Curr. Opin. Genet. Dev. 2005, 15, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hata, A. Functions of microRNAs in cardiovascular biology and disease. Annu. Rev. Physiol. 2013, 75, 69–93. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.; Curcio, A.; Indolfi, C. Emerging role of microRNAs in cardiovascular diseases. Circ. J. 2014, 78, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Jahantigh, M.; Wei, Y.; Schober, A. The role of microRNAs in arterial remodelling. Thromb. Haemost. 2012, 107, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogen Regulation of MicroRNA Expression. Curr. Genom. 2009, 10, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Catalucci, D.; Gallo, P.; Condorelli, G. MicroRNAs in Cardiovascular Biology and Heart Disease. Circ. Cardiovasc. Genet. 2009, 2, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cremades, D.; Mompeón, A.; Vidal-Gómez, X.; Hermenegildo, C.; Novella, S. miRNA as a New Regulatory Mechanism of Estrogen Vascular Action. Int. J. Mol. Sci. 2018, 19, 473. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Wang, F.; Nie, X.; Du, H.; Zhao, Y.; Yin, Z.; Li, H.; Fan, J.; Wen, Z.; Wang, D.W.; et al. The Cell Type–Specific Functions of miR-21 in Cardiovascular Diseases. Front. Genet. 2020, 11, 563166. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wei, J.; Li, X.; Cheng, Y.; Chen, W.; Cui, Y.; Simoncini, T.; Gu, Z.; Yang, J.; Fu, X. 17β-Estradiol Enhances Vascular Endothelial Ets-1/miR-126-3p Expression: The Possible Mechanism for Attenuation of Atherosclerosis. J. Clin. Endocrinol. Metab. 2017, 102, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Azzarito, G.; Kurmann, L.; Leeners, B.; Dubey, R.K. Micro-RNA193a-3p Inhibits Breast Cancer Cell Driven Growth of Vascular Endothelial Cells by Altering Secretome and Inhibiting Mitogenesis: Transcriptomic and Functional Evidence. Cells 2022, 11, 2967. [Google Scholar] [CrossRef] [PubMed]
- Kroh, E.M.; Parkin, R.K.; Mitchell, P.S.; Tewari, M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods 2010, 50, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, I.; Barchiesi, F.; Jackson, E.K.; Imthurn, B.; Stiller, R.; Kim, J.-H.; Schaufelberger, S.; Rosselli, M.; Hughes, C.C.W.; Dubey, R.K. Estradiol stimulates capillary formation by human endothelial progenitor cells: Role of estrogen receptor-{alpha}/{beta}, heme oxygenase 1, and tyrosine kinase. Hypertension 2010, 56, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Haas, E.; Prossnitz, E.R.; Barton, M. Non-genomic regulation of vascular cell function and growth by estrogen. Mol. Cell. Endocrinol. 2009, 308, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Imbrie, G.A.; Baur, W.E.; Iyer, L.K.; Aronovitz, M.J.; Kershaw, T.B.; Haselmann, G.M.; Lu, Q.; Karas, R.H. Estrogen receptor-mediated regulation of microRNA inhibits proliferation of vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2013, 33, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Gasparini, P.; Piovan, C.; Ngankeu, A.; Garofalo, M.; Taccioli, C.; Iorio, M.V.; Li, M.; Volinia, S.; Alder, H. MicroRNA cluster 221-222 and estrogen receptor alpha interactions in breast cancer. J. Natl. Cancer Inst. 2010, 102, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Jackson, E.K. Potential Vascular Actions of 2-Methoxyestradiol. Trends Endocrinol. Metab. 2009, 20, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.-G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Furneaux, H.; White, B.A. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol. 2007, 21, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Khordadmehr, M.; Shahbazi, R.; Sadreddini, S.; Baradaran, B. miR-193: A new weapon against cancer. J. Cell. Physiol. 2019, 234, 16861–16872. [Google Scholar] [CrossRef] [PubMed]
- Khoo, C.P.; Roubelakis, M.G.; Schrader, J.B.; Tsaknakis, G.; Konietzny, R.; Kessler, B.; Harris, A.L.; Watt, S.M. miR-193a-3p interaction with HMGB1 downregulates human endothelial cell proliferation and migration. Sci. Rep. 2017, 7, 44137. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.; Salvi, A.; Abeni, E.; Marchina, E.; De Petro, G. Biological Function of MicroRNA193a-3p in Health and Disease. Int. J. Genom. 2017, 2017, 5913195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Feng, J.; Xu, B.; Niu, Y.; Zheng, Y. From Bone Remodeling to Wound Healing: An miR-146a-5p-Loaded Nanocarrier Targets Endothelial Cells to Promote Angiogenesis. ACS Appl. Mater. Interfaces 2024, 16, 32992–33004. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, Y.; Li, W. Compression loading of osteoclasts attenuated microRNA-146a-5p expression, which promotes angiogenesis by targeting adiponectin. Sci. China Life Sci. 2022, 65, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Bestepe, F.; Fritsche, C.; Lakhotiya, K.; Niosi, C.E.; Ghanem, G.F.; Martin, G.L.; Pal-Ghosh, R.; Becker-Greene, D.; Weston, J.; Hollan, I. Deficiency of miR-409-3p improves myocardial neovascularization and function through modulation of DNAJB9/p38 MAPK signaling. Mol. Ther. Nucleic Acids 2023, 32, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Knoepp, K.; Dutzmann, J.; Kalies, K.; Rieckmann, M.; Daniel, J.M.; Bauersachs, J.; Sedding, D.G. 261Decisive role of microRNA-494 in smooth muscle cell proliferation and vascular remodeling. Eur. Heart J. 2019, 40, ehz747.0077. [Google Scholar] [CrossRef]
- Cui, R.; Ye, S.; Zhong, J.; Liu, L.; Li, S.; Lin, X.; Yuan, L.; Yi, L. MicroRNA-494 inhibits apoptosis of murine vascular smooth muscle cells in vitro. Mol. Med. Rep. 2019, 19, 4457–4467. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, G.; Miao, H.; Tang, R.; Song, Y.; Hu, Y.; Wang, Z.; Hou, Y. MicroRNA-494 inhibits the growth and angiogenesis-regulating potential of mesenchymal stem cells. FEBS Lett. 2015, 589, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Welten, S.M.; Bastiaansen, A.J.; De Jong, R.C.M.; De Vries, M.R.; Peters, E.A.; Boonstra, M.C.; Sheikh, S.P.; La Monica, N.; Kandimalla, E.R.; Quax, P.H.A.; et al. Inhibition of 14q32 MicroRNAs miR-329, miR-487b, miR-494, and miR-495 Increases Neovascularization and Blood Flow Recovery After Ischemia. Circ. Res. 2014, 115, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Kurmann, L.; Azzarito, G.; Leeners, B.; Rosselli, M.; Dubey, R.K. 17β-Estradiol Abrogates TNF-α-Induced Human Brain Vascular Pericyte Migration by Downregulating miR-638 via ER-β. Int. J. Mol. Sci. 2024, 25, 11416. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-P.; Zhao, W.; Du, J.-K.; Ni, X.; Zhu, X.-Y.; Lu, J.-Q. miR-494 Contributes to Estrogen Protection of Cardiomyocytes Against Oxidative Stress via Targeting (NF-κB) Repressing Factor. Front. Endocrinol. 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Rotem, A.; Struhl, K. Inhibition of miR-193a Expression by Max and RXRα Activates K-Ras and PLAU to Mediate Distinct Aspects of Cellular Transformation. Cancer Res. 2011, 71, 5144–5153. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Yamada, Y.; Miyazawa, T.; Yoshida, T. Gain-of-function microRNA screens identify miR-193a regulating proliferation and apoptosis in epithelial ovarian cancer cells. Int. J. Oncol. 2013, 42, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-D.; Chen, L.; Zhang, M.; Qi, H.-J.; Chen, L.; Chen, H.-F.; Zhong, M.-K.; Shi, X.-J.; Li, Q.-Y. Downregulation of ERRα inhibits angiogenesis in human umbilical vein endothelial cells through regulating VEGF production and PI3K/Akt/STAT3 signaling pathway. Eur. J. Pharmacol. 2015, 769, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, L.; Luo, X.; Wang, L.; Gao, X.; Wang, W.; Sun, J.; Dou, L.; Li, J.; Xu, C.; et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 2013, 121, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Brüne, B. miR-193a-3p increases glycolysis under hypoxia by facilitating Akt phosphorylation and PFKFB3 activation in human macrophages. Cell. Mol. Life Sci. 2022, 79, 89. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, J.; Yan, M.; Liu, L.; Lin, H.; Zhao, F.; Sun, L.; Zhang, Y.; Cui, Y.; Zhang, F.; et al. MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene 2015, 34, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Azzarito, G.; Henry, M.; Rotshteyn, T.; Leeners, B.; Dubey, R.K. Transcriptomic and Functional Evidence That miRNA193a-3p Inhibits Lymphatic Endothelial Cell (LEC) and LEC + MCF-7 Spheroid Growth Directly and by Altering MCF-7 Secretome. Cells 2023, 12, 389. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-N.; Lin, J.; Li, Y.-H.; Gao, L.; Wang, X.-R.; Wang, W.; Kang, H.-Y.; Yan, G.-T.; Wang, L.-L.; Yu, L. MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene 2011, 30, 3416–3428. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.; Lu, Y.; Angle, M.; Magder, S. Estrogen induced changes in Akt-dependent activation of endothelial nitric oxide synthase and vasodilation. Steroids 2004, 69, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Haynes, M.P.; Sinha, D.; Russell, K.S.; Collinge, M.; Fulton, D.; Morales-Ruiz, M.; Sessa, W.C.; Bender, J.R. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ. Res. 2000, 87, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Esumi, H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim. Pol. 2003, 50, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.A.; Koos, R.D. Estrogen-induced activation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor expression, and edema in the uterus are mediated by the phosphatidylinositol 3-kinase/Akt pathway. Endocrinology 2007, 148, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Losordo, D.W.; Isner, J.M. Estrogen and angiogenesis: A review. Arter. Thromb. Vasc. Biol. 2001, 21, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, C.-D.; Yan, B.; Zhao, J.-L.; Wang, Z.-H. miRNA-directed regulation of VEGF in tilapia under hypoxia condition. Biochem. Biophys. Res. Commun. 2014, 454, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yi, Y.; Li, L.; Zhang, W.; Wang, J. Osteopontin induces vascular endothelial growth factor expression in articular cartilage through PI3K/AKT and ERK1/2 signaling. Mol. Med. Rep. 2015, 12, 4708–4712. [Google Scholar] [CrossRef] [PubMed]
- Malek, D.; Gust, R.; Kleuser, B. 17-Beta-estradiol inhibits transforming-growth-factor-beta-induced MCF-7 cell migration by Smad3-repression. Eur. J. Pharmacol. 2006, 534, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Tramontana, A. Potential Role of TFG-Beta1 in Estradiol Mediated Angiogenesis and Endothleial Cell Growth. Medical Thesis, Medizinische Universität Wien, Vienna, Austria, 2012. [Google Scholar]
- Unterleutner, E. Role of G-Protein Coupled Estrogen Receptor in Mediating the Vasoprotective Actions of Estradiol. Ph.D. Thesis, ETHZ Zürich, Zürich, Switzerland, 2016. [Google Scholar]
- Ju, B.; Nie, Y.; Yang, X.; Wang, X.; Li, F.; Wang, M.; Wang, C.; Zhang, H. miR-193a/b-3p relieves hepatic fibrosis and restrains proliferation and activation of hepatic stellate cells. J. Cell. Mol. Med. 2019, 23, 3824–3832. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Dai, C.-Y.; Mei, Z.; Jiang, M.-J.; Gu, D.-N.; Huang, Q.; Tian, L. microRNA-193a stimulates pancreatic cancer cell repopulation and metastasis through modulating TGF-β2/TGF-βRIII signalings. J. Exp. Clin. Cancer Res. 2018, 37, 25. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Hinske, L.C.; França, G.S.; Torres, H.A.M.; Ohara, D.T.; Lopes-Ramos, C.M.; Heyn, J.; Reis, L.F.L.; Ohno-Machado, L.; Kreth, S.; Galante, P.A.F. miRIAD—Integrating microRNA inter- and intragenic data. Database 2014, 2014, bau099. [Google Scholar] [CrossRef] [PubMed]
- Karas, R.H.; Hodgin, J.B.; Kwoun, M.; Krege, J.H.; Aronovitz, M.; Mackey, W.; Gustafsson, J.Å.; Korach, K.S.; Smithies, O.; Mendelsohn, M.E. Estrogen inhibits the vascular injury response in estrogen receptor β-deficient female mice. Proc. Natl. Acad. Sci. USA 1999, 96, 15133–15136. [Google Scholar] [CrossRef] [PubMed]
- Pare, G.; Krust, A.; Karas, R.H.; Dupont, S.; Aronovitz, M.; Chambon, P.; Mendelsohn, M.E. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ. Res. 2002, 90, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Toyama, T.; Sugiura, H.; Fujii, Y.; Yamashita, H. miR-206 Expression is down-regulated in estrogen receptor alpha-positive human breast cancer. Cancer Res. 2008, 68, 5004–5008. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Lu, Q.; Baur, W.; Aronovitz, M.J.; Karas, R.H. Rapid estrogen receptor signaling mediates estrogen-induced inhibition of vascular smooth muscle cell proliferation. Arter. Thromb. Vasc. Biol. 2013, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-H.; Fu, X.-D.; Yang, D.; Tan, Z.; Pan, J.-Y. Membrane estrogen receptor mediates the rapid nongenomic activation of endothelial nitric oxide synthase by estrogen. Sheng Li Xue Bao 2003, 55, 213–218. [Google Scholar] [PubMed]
- Leivonen, S.-K.; Mäkelä, R.; Östling, P.; Kohonen, P.; Haapa-Paananen, S.; Kleivi, K.; Enerly, E.; Aakula, A.; Hellström, K.; Sahlberg, N.; et al. Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene 2009, 28, 3926–3936. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.H.C.; Aguda, B.D.; Tsai, J.-C.; Kochańczyk, M.; Lin, J.M.J.; Chen, G.C.W.; Lai, H.-C.; Nephew, K.P.; Hwang, T.-W.; Chan, M.W.Y.; et al. A Mathematical Model of Bimodal Epigenetic Control of miR-193a in Ovarian Cancer Stem Cells. PLoS ONE 2014, 9, e116050. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Fu, X.; Alves, P.; Gerstein, M. mRNA expression profiles show differential regulatory effects of microRNAs between estrogen receptor-positive and estrogen receptor-negative breast cancer. Genome Biol. 2009, 10, R90. [Google Scholar] [CrossRef] [PubMed]
- Sobrino, A.; Mata, M.; Laguna-Fernandez, A.; Novella, S.; Oviedo, P.J.; García-Pérez, M.A.; Tarín, J.J.; Cano, A.; Hermenegildo, C.; Callaerts, P. Estradiol Stimulates Vasodilatory and Metabolic Pathways in Cultured Human Endothelial Cells. PLoS ONE 2009, 4, e8242. [Google Scholar] [CrossRef] [PubMed]
- Wortham, N.C.; Ahamed, E.; Nicol, S.M.; Thomas, R.S.; Periyasamy, M.; Jiang, J.; Ochocka, A.M.; Shousha, S.; Huson, L.; Bray, S.E.; et al. The DEAD-box protein p72 regulates ERα-/oestrogen-dependent transcription and cell growth, and is associated with improved survival in ERα-positive breast cancer. Oncogene 2009, 28, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Paris, O.; Ferraro, L.; Grober, O.M.V.; Ravo, M.; De Filippo, M.R.; Giurato, G.; Nassa, G.; Tarallo, R.; Cantarella, C.; Rizzo, F.; et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor beta in hormone-responsive breast cancer. Oncogene 2012, 31, 4196–4206. [Google Scholar] [CrossRef] [PubMed]
- Nothnick, W.B.; Healy, C.; Hong, X. Steroidal regulation of uterine miRNAs is associated with modulation of the miRNA biogenesis components Exportin-5 and Dicer1. Endocrine 2010, 37, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Redfern, A.D.; Colley, S.M.; Beveridge, D.J.; Ikeda, N.; Epis, M.R.; Li, X.; Foulds, C.E.; Stuart, L.M.; Barker, A.; Russell, V.J.; et al. RNA-induced silencing complex (RISC) Proteins PACT, TRBP, and Dicer are SRA binding nuclear receptor coregulators. Proc. Natl Acad. Sci. USA 2013, 110, 6536–6541. [Google Scholar] [CrossRef] [PubMed]
- Grelier, G.; Voirin, N.; Ay, A.-S.; Cox, D.G.; Chabaud, S.; Treilleux, I.; Léon-Goddard, S.; Rimokh, R.; Mikaelian, I.; Venoux, C.; et al. Prognostic value of Dicer expression in human breast cancers and association with the mesenchymal phenotype. Br. J. Cancer 2009, 101, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Claffey, K.P.; White, B.A. Argonaute-2 expression is regulated by epidermal growth factor receptor and mitogen-activated protein kinase signaling and correlates with a transformed phenotype in breast cancer cells. Endocrinology 2009, 150, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.K.; Linscott, M.L.; Flury, S.; Zhang, M.; Newby, M.L.; Pak, T.R. 17β-Estradiol Regulates miR-9-5p and miR-9-3p Stability and Function in the Aged Female Rat Brain. Noncoding RNA 2021, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, M.; Imthurn, B.; Keller, P.J.; Jackson, E.K.; Dubey, R.K. Circulating nitric oxide (nitrite/nitrate) levels in postmenopausal women substituted with 17 beta-estradiol and norethisterone acetate. A two-year follow-up study. Hypertension 1995, 25, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Kotecki, N.; Opdam, F.; Robbrecht, D.; Strijbos, M.; Kroon, K.; Janicot, M.; Yahyanejad, S.; Telford, B.; Bosch, M.v.D.; Alemdehy, F.; et al. Phase I/Ib study with INT-1B3, a novel LNP-formulated micro-RNA (miR-193a-3p mimic) therapeutic for patients with advanced solid cancer. J. Clin. Oncol. 2021, 39, TPS2666. [Google Scholar] [CrossRef]
- Christopher, A.F.; Bansal, P.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68–74. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug Discov. 2012, 11, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Jin, J.-P.; Wang, J.-Q.; Zhang, Z.-G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Balasubramanian, S.; Rajasingh, S.; Patel, U.; Dhanasekaran, A.; Dawn, B.; Rajasingh, J. MicroRNA: A new therapeutic strategy for cardiovascular diseases. Trends Cardiovasc. Med. 2016, 26, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Croce, C.M. MicroRNA: Trends in clinical trials of cancer diagnosis and therapy strategies. Exp. Mol. Med. 2023, 55, 1314–1321. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigassi, L.; Popa, M.A.; Stiller, R.; Leeners, B.; Rosselli, M.; Dubey, R.K. Estradiol Downregulates MicroRNA-193a to Mediate Its Angiogenic Actions. Cells 2025, 14, 1134. https://doi.org/10.3390/cells14151134
Rigassi L, Popa MA, Stiller R, Leeners B, Rosselli M, Dubey RK. Estradiol Downregulates MicroRNA-193a to Mediate Its Angiogenic Actions. Cells. 2025; 14(15):1134. https://doi.org/10.3390/cells14151134
Chicago/Turabian StyleRigassi, Lisa, Mirel Adrian Popa, Ruth Stiller, Brigitte Leeners, Marinella Rosselli, and Raghvendra Krishna Dubey. 2025. "Estradiol Downregulates MicroRNA-193a to Mediate Its Angiogenic Actions" Cells 14, no. 15: 1134. https://doi.org/10.3390/cells14151134
APA StyleRigassi, L., Popa, M. A., Stiller, R., Leeners, B., Rosselli, M., & Dubey, R. K. (2025). Estradiol Downregulates MicroRNA-193a to Mediate Its Angiogenic Actions. Cells, 14(15), 1134. https://doi.org/10.3390/cells14151134