
Prosaposin: A Multifaceted Protein Orchestrating Biological Processes and Diseases


Abstract
1. Introduction
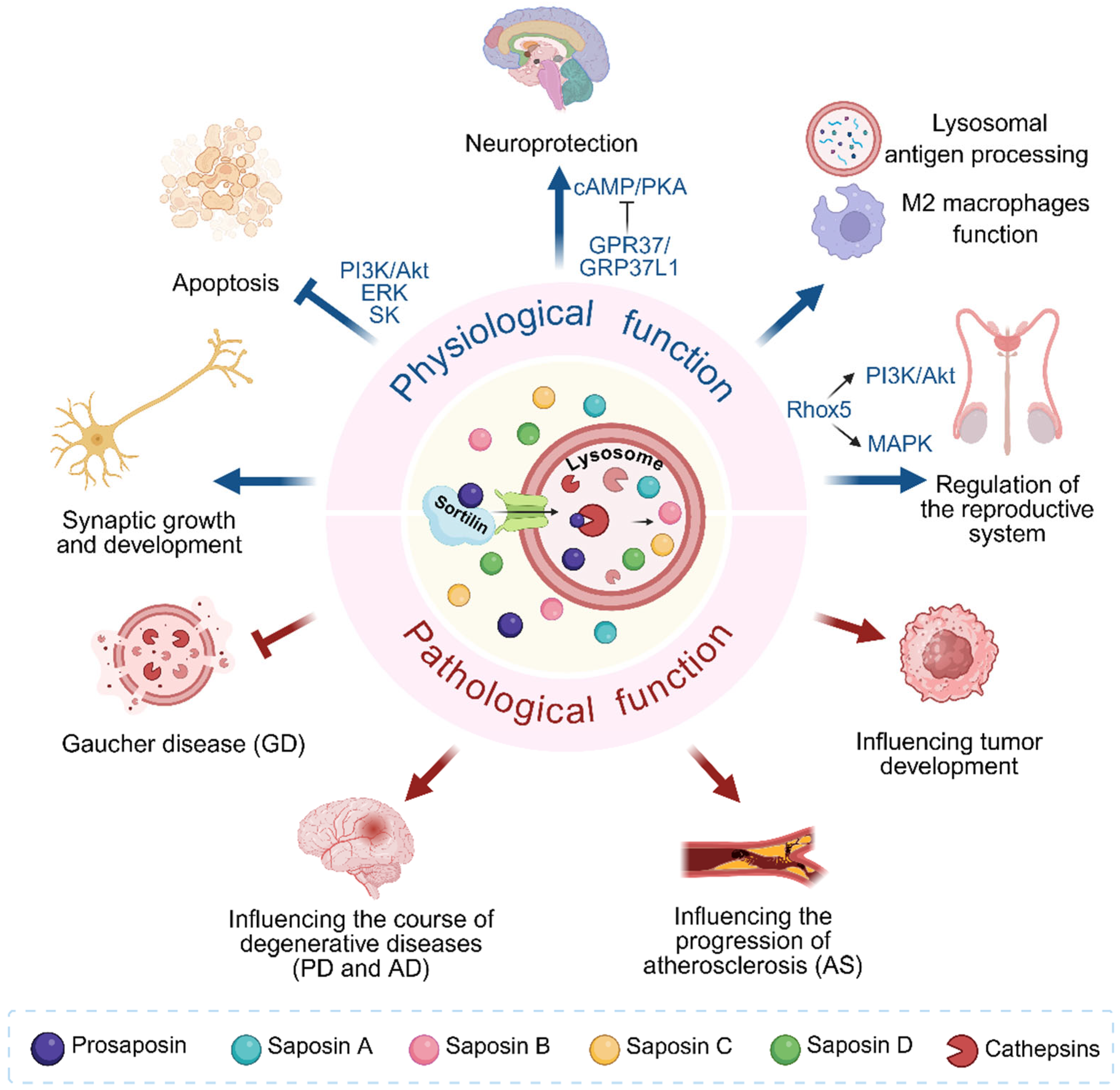
2. The Role of PSAP in Biological Processes
2.1. Prosaposin
2.2. Structure and Function of Saposins
2.2.1. Saposin A
2.2.2. Saposin B
2.2.3. Saposin C
2.2.4. Saposin D
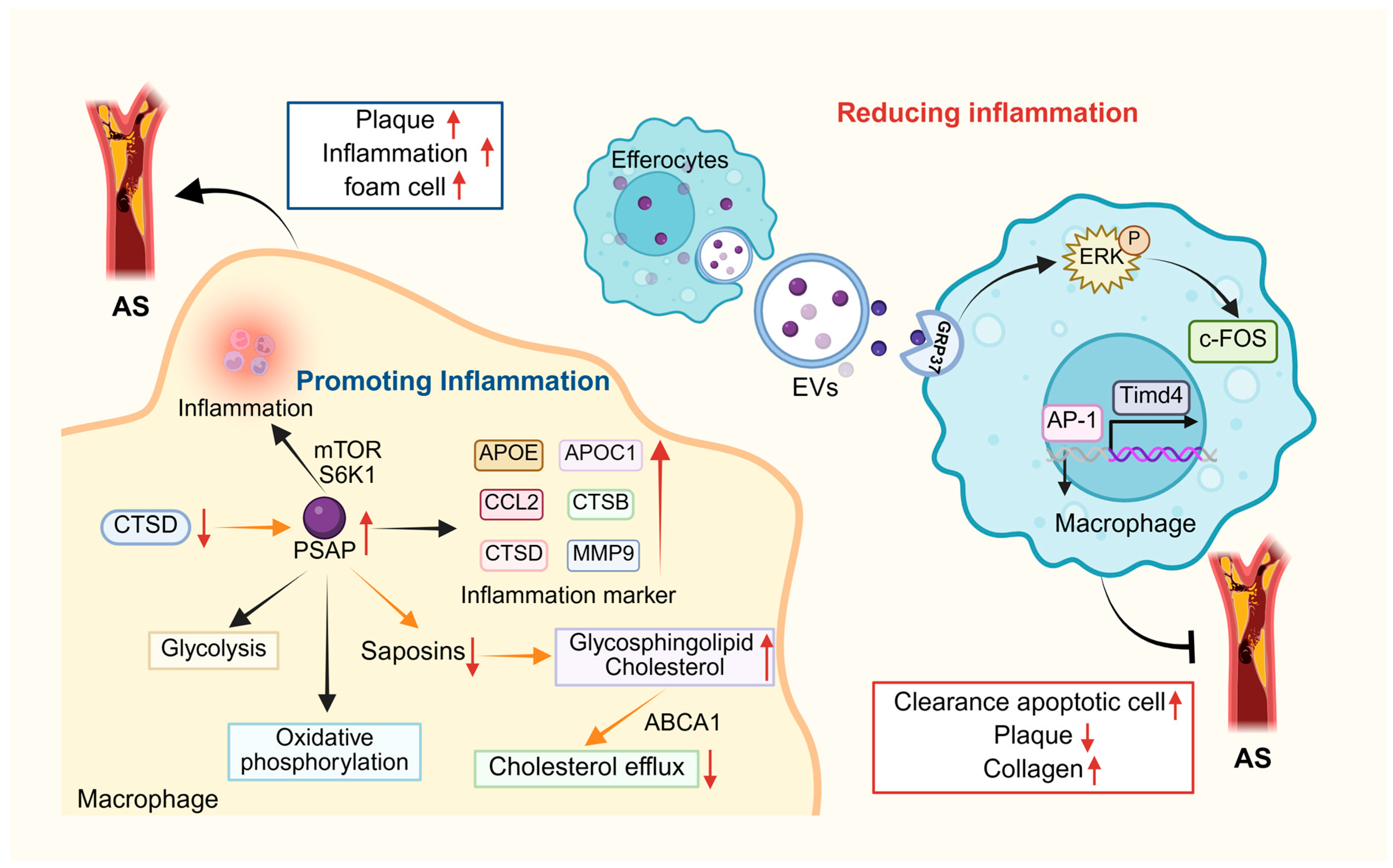
3. Pathophysiological Roles of Prosaposin
3.1. Gaucher Disease
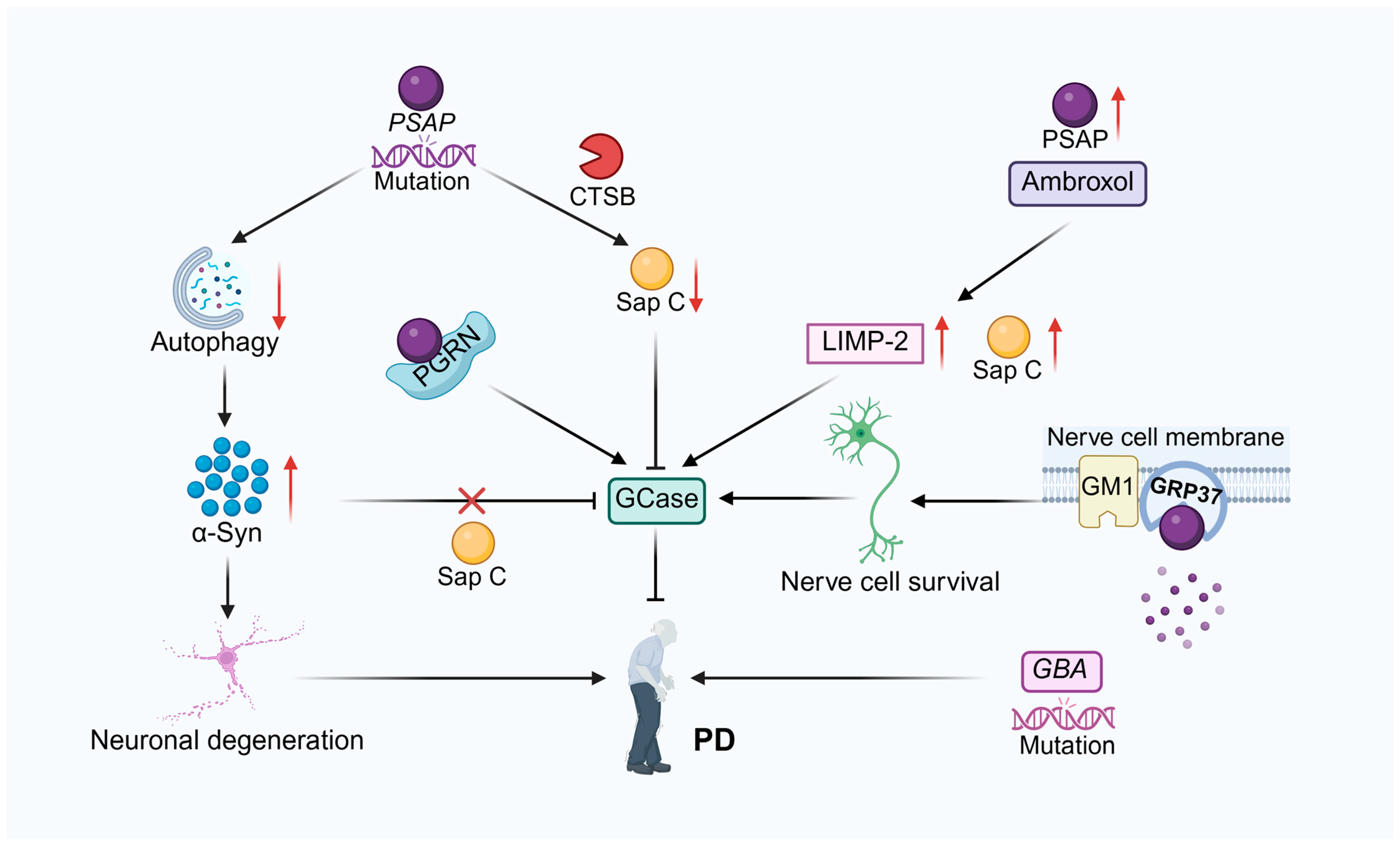
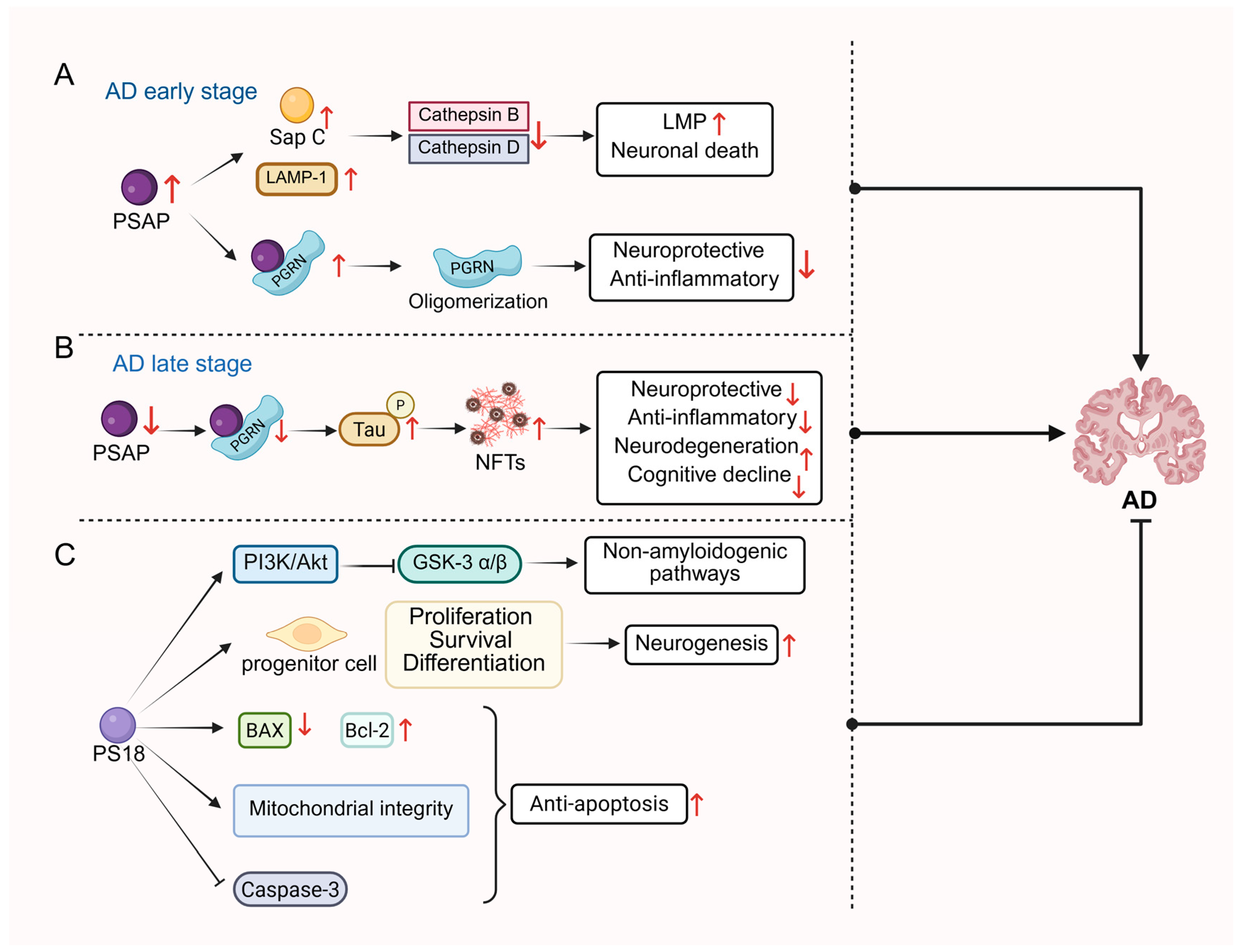
3.2. Neurodegenerative Disease
3.2.1. Parkinson’s Disease
3.2.2. Alzheimer’s Disease
3.3. Atherosclerosis
3.4. Cancer
3.4.1. Breast Cancer
3.4.2. Ovarian Cancer
3.4.3. Prostate Cancer
3.4.4. Other Cancers
Gliomas and Glioblastoma
Gastric Cancer
Colorectal Cancer
Gallbladder Cancer
Hepatocellular Carcinoma
Pancreatic Ductal Adenocarcinoma
Malignant Pleural Mesothelioma
Fibrosarcoma
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| AUC | Analytical ultracentrifugation |
| AD | Alzheimer’s disease |
| ADPD | Autosomal dominant Parkinson’s disease |
| AI | Androgen-independent |
| AR | Androgen receptor |
| ARs | Activity of hormone receptors |
| ASMase | Acid sphingomyelinase |
| α-Syn | α-synuclein |
| BMP | bis (monoacylglycero) phosphate |
| CAFs | Cancer-associated fibroblasts |
| CTSB | Cathepsin B |
| CTSD | Cathepsin D |
| CMS4 | Consensus molecular subtype 4 |
| CoQ | Coenzyme Q (ubiquinone) |
| CRC | Colorectal cancer |
| CRPC | Castration-resistant prostate cancer |
| CRIS-B | Colorectal cancer intrinsic subtype B |
| CSF | Cerebrospinal fluid |
| CSPC | Castration-sensitive prostate cancer |
| DA | Dopamine |
| DNs | Dystrophic neurites |
| EC | Endothelial cell |
| ECB | Encapsulated cell biodelivery |
| EMT | Epithelial–mesenchymal transition |
| EOPD | Early-onset Parkinson’s disease |
| ER | Estrogen receptor |
| ER α | Estrogen receptor α |
| ERKs | Extracellular signal-regulated kinases |
| ERK | Extracellular signal-regulated kinase |
| EVs | Extracellular vesicles |
| FPD | Familial Parkinson’s disease |
| FTD | Frontotemporal dementia |
| GalEAG | Galactosylalkylacylglycerol |
| Gb3 | Globotriaosylceramide |
| GD | Gaucher disease |
| GBC | Gallbladder cancer |
| GCase | Glucocerebrosidase |
| GC | Gastric cancer |
| Gln | Glucosamine |
| GSL | Glycosphingolipids |
| GM1 | Ganglioside GM1 |
| GPR37 | G protein-coupled receptor 37 |
| GPR37L1 | G protein-coupled receptor 37L1 |
| HCC | Hepatocellular carcinoma |
| HPLC | High-performance liquid chromatography |
| HRE | Hormone-responsive element |
| KO | Knockout |
| KD2 | Second Kunitz-type domain |
| LacCer | Lactoceramide |
| LAMP1 | Lysosome-associated membrane protein 1 |
| LIMP-2 | Lysosomal integral membrane protein 2 |
| LPC | Localized prostate cancer |
| LRP | Lipoprotein receptor-associated protein |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| LSDs | Lysosomal storage disease |
| M6P | Mannose-6-phosphate |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| MCI | Mild cognitive impairment |
| MEK | Mitogen-activated extracellular signal-regulated kinase |
| mCRPCa | Metastatic castrate-resistant prostate cancer |
| MPC | Metastatic prostate cancer |
| MPM | Malignant pleural mesothelioma |
| MMP-2 | Matrix metalloproteinase-2 |
| NFTs | Neurofibrillary tangles |
| OXPHOS | Oxidative phosphorylation |
| ox-LDL | Oxidized low-density lipoprotein |
| PCa | Prostate cancer |
| PD | Parkinson’s disease |
| PDAC | Pancreatic ductal adenocarcinoma |
| PGRN | Progranulin |
| PI3K | Phosphatidylinositol 3-kinase |
| POPC | Palmitoyloleoylphosphatidylcholine |
| PrSt | Prostate stromal cells |
| proCath-D | Procathepsin D |
| PSAP | Prosaposin |
| QSOX1 | Quiescin Q6 sulfhydryl oxidase 1 |
| RAP | Receptor-associated protein |
| ROS | Reactive oxygen species |
| S-1-P | Sphingosine-1-phosphate |
| Sap A | Saposin A |
| Sap B | Saposin B |
| Sap C | Saposin C |
| Sap D | Saposin D |
| SAPK/JNK | Stress-activated protein kinase/c-jun n-terminal kinase |
| SG | Spiral ganglion |
| SK | Sphingosine kinase |
| SGP-1 | Sulfated glycoprotein-1 |
| SPD | Sporadic Parkinson’s disease |
| STAD | Stomach adenocarcinoma |
| TCGA | The Cancer Genome Atlas |
| TFPI-2 | Tissue factor pathway inhibitor-2 |
| TGF-β | Transforming growth factor-β |
| TGF-β1/Smad | Transforming growth factor-β1/Smad |
| TISIDB | Tumor Immune System Interaction Database |
| TMT | Tandem mass spectrometry tagging |
| TNFα | Tumor necrosis factor alpha |
| TSP-1 | Anti-tumor protein thrombospondin-1 |
| uPA | Urokinase-type fibrinogen activator |
| uPAR | Urokinase-type fibrinogen activator receptor |
| UPR | Unfolded protein response |
| 6-OHDA | 6-hydroxydopamine |
References
- Kishimoto, Y.; Hiraiwa, M.; O’Brien, J.S. Saposins: Structure, function, distribution, and molecular genetics. J. Lipid Res. 1992, 33, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Tadano-Aritomi, K.; Matsuda, J.; Fujimoto, H.; Suzuki, K.; Ishizuka, I. Seminolipid and its precursor/degradative product, galactosylalkylacylglycerol, in the testis of saposin A- and prosaposin-deficient mice. J. Lipid Res. 2003, 44, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.C.; Giddens, M.M.; Coleman, B.M.; Hall, R.A. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014, 1585, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Darmoise, A.; Maschmeyer, P.; Winau, F. The immunological functions of saposins. Adv. Immunol. 2010, 105, 25–62. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Halvorsen, O.J.; Gravdal, K.; Bhattacharya, N.; Lee, J.M.; Liu, N.W.; Johnston, B.T.; Johnston, A.B.; Haukaas, S.A.; Aamodt, K.; et al. Prosaposin inhibits tumor metastasis via paracrine and endocrine stimulation of stromal p53 and Tsp-1. Proc. Natl. Acad. Sci. USA 2009, 106, 12115–12120, Erratum in Proc. Natl. Acad. Sci. USA 2023, 120, e2320128120. https://doi.org/10.1073/pnas.2320128120. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.S.; Carson, G.S.; Seo, H.C.; Hiraiwa, M.; Kishimoto, Y. Identification of prosaposin as a neurotrophic factor. Proc. Natl. Acad. Sci. USA 1994, 91, 9593–9596. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, C.F.; Corrêa-da-Silva, F.; Gonzatti, M.B.; Angelim, M.K.; Pretti, M.A.; Davanzo, G.G.; Castelucci, B.G.; Monteiro, L.B.; Castro, G.; Virgilio-da-Silva, J.V.; et al. Tissue-specific metabolic profile drives iNKT cell function during obesity and liver injury. Cell Rep. 2023, 42, 112035. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Yamamoto, Y.; Fujisawa, A.; Kashiba, M. Prosaposin is a novel coenzyme Q10-binding protein. J. Clin. Biochem. Nutr. 2024, 74, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Huang, X.; Li, S.; Sun, L.; Li, Y.; Li, H.; Zhou, Y.; Chu, Y.; Zhou, T. Identification of prosaposin as a novel interaction partner for Rhox5. J. Genet. Genom.Yi Chuan Xue Bao 2007, 34, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Ghassabeh, G.H.; De Baetselier, P.; Brys, L.; Noël, W.; Van Ginderachter, J.A.; Meerschaut, S.; Beschin, A.; Brombacher, F.; Raes, G. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood 2006, 108, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Misasi, R.; Garofalo, T.; Di Marzio, L.; Mattei, V.; Gizzi, C.; Hiraiwa, M.; Pavan, A.; Grazia Cifone, M.; Sorice, M. Prosaposin: A new player in cell death prevention of U937 monocytic cells. Exp. Cell Res. 2004, 298, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Misasi, R.; Sorice, M.; Di Marzio, L.; Campana, W.M.; Molinari, S.; Cifone, M.G.; Pavan, A.; Pontieri, G.M.; O’Brien, J.S. Prosaposin treatment induces PC12 entry in the S phase of the cell cycle and prevents apoptosis: Activation of ERKs and sphingosine kinase. FASEB J. 2001, 15, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ran, H.; Zamzow, M.; Kitatani, K.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Witte, D.P.; Hannun, Y.A.; Grabowski, G.A. Specific saposin C deficiency: CNS impairment and acid beta-glucosidase effects in the mouse. Hum. Mol. Genet. 2010, 19, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Ruz, C.; Barrero, F.J.; Pelegrina, J.; Bandrés-Ciga, S.; Vives, F.; Duran, R. Saposin C, Key Regulator in the Alpha-Synuclein Degradation Mediated by Lysosome. Int. J. Mol. Sci. 2022, 23, 12004. [Google Scholar] [CrossRef] [PubMed]
- Sharoar, M.G.; Palko, S.; Ge, Y.; Saido, T.C.; Yan, R. Accumulation of saposin in dystrophic neurites is linked to impaired lysosomal functions in Alzheimer’s disease brains. Mol. Neurodegener. 2021, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- van Leent, M.M.T.; Beldman, T.J.; Toner, Y.C.; Lameijer, M.A.; Rother, N.; Bekkering, S.; Teunissen, A.J.P.; Zhou, X.; van der Meel, R.; Malkus, J.; et al. Prosaposin mediates inflammation in atherosclerosis. Sci. Transl. Med. 2021, 13, abe1433. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, L.; Zou, W.; Xu, J.; Liu, H.; Wang, W.; Yun, X.; Gu, J. Prosaposin, a regulator of estrogen receptor alpha, promotes breast cancer growth. Cancer Sci. 2012, 103, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Lee, T.J.; Wang, R.; Sun, Y.; Delorme, N.; Hiraiwa, M.; Grabowski, G.A.; Culig, Z.; Minokadeh, A. Prosaposin is a novel androgen-regulated gene in prostate cancer cell line LNCaP. J. Cell. Biochem. 2007, 101, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhou, J.; Luo, P.; Gao, H.; Ma, Y.; Chen, Y.S.; Li, L.; Zou, D.; Zhang, Y.; Jing, Z. Prosaposin promotes the proliferation and tumorigenesis of glioma through toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway. EBioMedicine 2018, 37, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Yang, C.; Zou, D.; Liu, J.; Wang, S.; Liu, X.; Zhang, Y.; Zhang, Y. Pan-cancer analysis of PSAP identifies its expression and clinical relevance in gastric cancer. Pathol. Res. Pract. 2022, 238, 154027. [Google Scholar] [CrossRef] [PubMed]
- Robles, J.; Pintado-Berninches, L.; Boukich, I.; Escudero, B.; de Los Rios, V.; Bartolomé, R.A.; Jaén, M.; Martín-Regalado, Á.; Fernandez-Aceñero, M.J.; Imbaud, J.I.; et al. A prognostic six-gene expression risk-score derived from proteomic profiling of the metastatic colorectal cancer secretome. J. Pathol. Clin. Res. 2022, 8, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabuddhe, N.A.; Barbhuiya, M.A.; Bhunia, S.; Subbannayya, T.; Gowda, H.; Advani, J.; Shrivastav, B.R.; Navani, S.; Leal, P.; Roa, J.C.; et al. Identification of prosaposin and transgelin as potential biomarkers for gallbladder cancer using quantitative proteomics. Biochem. Biophys. Res. Commun. 2014, 446, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhong, X.; Li, J.; Xu, F. Circular RNA circVAPA Promotes Cell Proliferation in Hepatocellular Carcinoma. Hum. Gene Ther. Clin. Dev. 2019, 30, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, Y.; Takano, S.; Sogawa, K.; Tomizawa, S.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Ohtsuka, M. Prosaposin, tumor-secreted protein, promotes pancreatic cancer progression by decreasing tumor-infiltrating lymphocytes. Cancer Sci. 2022, 113, 2548–2559. [Google Scholar] [CrossRef] [PubMed]
- Lacerenza, S.; Ciregia, F.; Giusti, L.; Bonotti, A.; Greco, V.; Giannaccini, G.; D’Antongiovanni, V.; Fallahi, P.; Pieroni, L.; Cristaudo, A.; et al. Putative Biomarkers for Malignant Pleural Mesothelioma Suggested by Proteomic Analysis of Cell Secretome. Cancer Genom. Proteom. 2020, 17, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Deng, F.; Mao, Z.; Zhang, J.; Wang, H.; Wang, J.; Mu, J.; Deng, S.; Ma, D. The interaction of the second Kunitz-type domain (KD2) of TFPI-2 with a novel interaction partner, prosaposin, mediates the inhibition of the invasion and migration of human fibrosarcoma cells. Biochem. J. 2012, 441, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hiraiwa, M.; Martin, B.M.; Kishimoto, Y.; Conner, G.E.; Tsuji, S.; O’Brien, J.S. Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): Its mechanism and inhibition by ganglioside. Arch. Biochem. Biophys. 1997, 341, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hineno, T.; Sano, A.; Kondoh, K.; Ueno, S.; Kakimoto, Y.; Yoshida, K. Secretion of sphingolipid hydrolase activator precursor, prosaposin. Biochem. Biophys. Res. Commun. 1991, 176, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Carson, G.S.; Hiraiwa, M.; Grafe, M.; Kishimoto, Y.; O’Brien, J.S. Occurrence of prosaposin as a neuronal surface membrane component. J. Mol. Neurosci. 1994, 5, 59–67. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.S.; Carson, G.S.; Seo, H.C.; Hiraiwa, M.; Weiler, S.; Tomich, J.M.; Barranger, J.A.; Kahn, M.; Azuma, N.; Kishimoto, Y. Identification of the neurotrophic factor sequence of prosaposin. FASEB J. 1995, 9, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Hiraiwa, M.; Soeda, S.; Kishimoto, Y.; O’Brien, J.S. Binding and transport of gangliosides by prosaposin. Proc. Natl. Acad. Sci. USA 1992, 89, 11254–11258. [Google Scholar] [CrossRef] [PubMed]
- Misasi, R.; Sorice, M.; Carson, G.S.; Griggi, T.; Lenti, L.; Pontieri, G.M.; O’Brien, J.S. Prosaposin and prosaptide, a peptide from prosaposin, induce an increase in ganglioside content on NS20Y neuroblastoma cells. Glycoconj. J. 1996, 13, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Zurbruegg, M.; Li, T.; Paslawski, W.; Zhang, X.; Svenningsson, P. Prosaposin Reduces α-Synuclein in Cells and Saposin C Dislodges it from Glucosylceramide-enriched Lipid Membranes. J. Mol. Neurosci. 2022, 72, 2313–2325. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.C.; Giddens, M.M.; Schaefer, S.A.; Hall, R.A. GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. Proc. Natl. Acad. Sci. USA 2013, 110, 9529–9534. [Google Scholar] [CrossRef] [PubMed]
- Carvelli, L.; Libin, Y.; Morales, C.R. Prosaposin: A protein with differential sorting and multiple functions. Histol. Histopathol. 2015, 30, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Nabeka, H.; Yamamiya, K.; Khan, M.S.I.; Shimokawa, T.; Islam, F.; Doihara, T.; Wakisaka, H.; Kobayashi, N.; Hamada, F.; et al. The expression of prosaposin and its receptors, GRP37 and GPR37L1, are increased in the developing dorsal root ganglion. PLoS ONE 2021, 16, e0255958. [Google Scholar] [CrossRef] [PubMed]
- Morant-Ferrando, B.; Jimenez-Blasco, D.; Alonso-Batan, P.; Agulla, J.; Lapresa, R.; Garcia-Rodriguez, D.; Yunta-Sanchez, S.; Lopez-Fabuel, I.; Fernandez, E.; Carmeliet, P.; et al. Fatty acid oxidation organizes mitochondrial supercomplexes to sustain astrocytic ROS and cognition. Nat. Metab. 2023, 5, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Mosienko, V.; Vaccari Cardoso, B.; Prokudina, D.; Huentelman, M.; Teschemacher, A.G.; Kasparov, S. Glio- and neuro-protection by prosaposin is mediated by orphan G-protein coupled receptors GPR37L1 and GPR37. Glia 2018, 66, 2414–2426. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.; Townsend, K.L. The metabolic and functional roles of sensory nerves in adipose tissues. Nat. Metab. 2023, 5, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Gijzel, S.M.; Siersbæk, R.; Broekema, M.F.; de Haar, C.; Schipper, H.S.; Boes, M.; Mandrup, S.; Kalkhoven, E. CD1d-mediated presentation of endogenous lipid antigens by adipocytes requires microsomal triglyceride transfer protein. J. Biol. Chem. 2014, 289, 22128–22139. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.C.H.; Ansa, B.S.; Alvarez-Rivera, G.; Domínguez-Zorita, S.; Rodríguez-Pombo, P.; Pérez, B.; Calvo, E.; Paradela, A.; Miguez, D.G.; Cifuentes, A.; et al. An ETFDH-driven metabolon supports OXPHOS efficiency in skeletal muscle by regulating coenzyme Q homeostasis. Nat. Metab. 2024, 6, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Sugawara, K.; Okamoto, M.; Nakamura, A.; Tanaka, T.; Fujita, Y.; Ishiguro, K.; Yamazaki, H.; Okada, M.; Mikami, A.; et al. Reduced prosaposin levels in HepG2 cells with long-term coenzyme Q10 deficiency. J. Clin. Biochem. Nutr. 2022, 71, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Zhang, X.; Ly, K.; Kim, J.H.; Wan, Q.; Kim, J.; Lou, M.; Kain, L.; Teyton, L.; Winau, F. Hyperglycosylation of prosaposin in tumor dendritic cells drives immune escape. Science 2024, 383, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Griswold, M.D.; Roberts, K.; Bishop, P. Purification and characterization of a sulfated glycoprotein secreted by Sertoli cells. Biochemistry 1986, 25, 7265–7270. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Martin, B.M.; Yamamoto, Y.; Kretz, K.A.; O’Brien, J.S.; Kishimoto, Y. Saposin A: Second cerebrosidase activator protein. Proc. Natl. Acad. Sci. USA 1989, 86, 3389–3393. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.H.; Read, R.J.; Deane, J.E. Structure of human saposin A at lysosomal pH. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Ahn, V.E.; Leyko, P.; Alattia, J.R.; Chen, L.; Privé, G.G. Crystal structures of saposins A and C. Protein Sci. 2006, 15, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Popovic, K.; Holyoake, J.; Pomès, R.; Privé, G.G. Structure of saposin A lipoprotein discs. Proc. Natl. Acad. Sci. USA 2012, 109, 2908–2912. [Google Scholar] [CrossRef] [PubMed]
- Flayhan, A.; Mertens, H.D.T.; Ural-Blimke, Y.; Martinez Molledo, M.; Svergun, D.I.; Löw, C. Saposin Lipid Nanoparticles: A Highly Versatile and Modular Tool for Membrane Protein Research. Structure 2018, 26, 345–355.e5. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Custódio, T.F.; Jungnickel, K.E.J.; Löw, C. Salipro technology in membrane protein research. Curr. Opin. Struct. Biol. 2025, 93, 103050. [Google Scholar] [CrossRef] [PubMed]
- Kurgan, K.W.; Chen, B.; Brown, K.A.; Falco Cobra, P.; Ye, X.; Ge, Y.; Gellman, S.H. Stable Picodisc Assemblies from Saposin Proteins and Branched Detergents. Biochemistry 2021, 60, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Molodenskiy, D.S.; Svergun, D.I.; Mertens, H.D.T. MPBuilder: A PyMOL Plugin for Building and Refinement of Solubilized Membrane Proteins Against Small Angle X-ray Scattering Data. J. Mol. Biol. 2021, 433, 166888. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Richards, M.R.; Bagal, D.; Campuzano, I.D.; Kitova, E.N.; Xiong, Z.J.; Privé, G.G.; Klassen, J.S. Characterizing the Size and Composition of Saposin A Lipoprotein Picodiscs. Anal. Chem. 2016, 88, 9524–9531. [Google Scholar] [CrossRef] [PubMed]
- Harzer, K.; Paton, B.C.; Christomanou, H.; Chatelut, M.; Levade, T.; Hiraiwa, M.; O’Brien, J.S. Saposins (sap) A and C activate the degradation of galactosylceramide in living cells. FEBS Lett. 1997, 417, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Grabowski, G.A. Human acid beta-glucosidase. Use of inhibitory and activating monoclonal antibodies to investigate the enzyme’s catalytic mechanism and saposin A and C binding sites. J. Biol. Chem. 1991, 266, 15021–15027. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, L.; Wenger, D.A.; Matern, D.; Dahmoush, H.; Watiker, V.; Lee, C. Rare Saposin A deficiency: Novel variant and psychosine analysis. Mol. Genet. Metab. 2020, 129, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Vanier, M.T.; Saito, Y.; Tohyama, J.; Suzuki, K.; Suzuki, K. A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum. Mol. Genet. 2001, 10, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Locatelli-Hoops, S.; Remmel, N.; Klingenstein, R.; Breiden, B.; Rossocha, M.; Schoeniger, M.; Koenigs, C.; Saenger, W.; Sandhoff, K. Saposin A mobilizes lipids from low cholesterol and high bis(monoacylglycerol)phosphate-containing membranes: Patient variant Saposin A lacks lipid extraction capacity. J. Biol. Chem. 2006, 281, 32451–32460. [Google Scholar] [CrossRef] [PubMed]
- Mehl, E.; Jatzkewitz, H. A cerebrosidesulfatase from swine kidney. Hoppe-Seyler’s Z. Fur Physiol. Chem. 1964, 339, 260–276. [Google Scholar] [CrossRef]
- Fischer, G.; Jatzkewitz, H. The activator of cerebroside sulphatase. Purification from human liver and identification as a protein. Hoppe-Seyler’s Z. Fur Physiol. Chem. 1975, 356, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Inui, K.; Wenger, D.A. Biochemical, immunological, and structural studies on a sphingolipid activator protein (SAP-1). Arch. Biochem. Biophys. 1984, 233, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, S.; Conzelmann, E.; Sandhoff, K. Activator protein for the degradation of globotriaosylceramide by human alpha-galactosidase. J. Biol. Chem. 1983, 258, 12378–12385. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Kihara, H.; Serizawa, S.; Li, Y.T.; Fluharty, A.L.; Mayes, J.S.; Shapiro, L.J. Activator protein required for the enzymatic hydrolysis of cerebroside sulfate. Deficiency in urine of patients affected with cerebroside sulfatase activator deficiency and identity with activators for the enzymatic hydrolysis of GM1 ganglioside and globotriaosylceramide. J. Biol. Chem. 1985, 260, 1867–1871. [Google Scholar] [CrossRef] [PubMed]
- Ahn, V.E.; Faull, K.F.; Whitelegge, J.P.; Fluharty, A.L.; Privé, G.G. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA 2003, 100, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, A.; Mierzewska, H.; Bekiesińska-Figatowska, M.; Szczepanik, E.; Goszczańska-Ciuchta, A.; Bednarska-Makaruk, M. A homozygote for the c.459+1G>A mutation in the ARSA gene presents with cerebellar ataxia as the only first clinical sign of metachromatic leukodystrophy. J. Neurol. Sci. 2014, 338, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Stokeley, D.; Bemporad, D.; Gavaghan, D.; Sansom, M.S. Conformational dynamics of a lipid-interacting protein: MD simulations of saposin B. Biochemistry 2007, 46, 13573–13580. [Google Scholar] [CrossRef] [PubMed]
- Madaan, P.; Jauhari, P.; Chakrabarty, B.; Kumar, A.; Gulati, S. Saposin B-Deficient Metachromatic Leukodystrophy Mimicking Acute Flaccid Paralysis. Neuropediatrics 2019, 50, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Sonnino, S.; Tettamanti, G.; Li, Y.T. Characterization of a nonspecific activator protein for the enzymatic hydrolysis of glycolipids. J. Biol. Chem. 1988, 263, 6588–6591. [Google Scholar] [CrossRef] [PubMed]
- Kashiba, M.; Oizumi, M.; Suzuki, M.; Sawamura, Y.; Nagashima, K.; Yoshimura, S.; Yamamoto, Y. Prosaposin regulates coenzyme Q10 levels in HepG2 cells, especially those in mitochondria. J. Clin. Biochem. Nutr. 2014, 55, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Akil, O.; Sun, Y.; Vijayakumar, S.; Zhang, W.; Ku, T.; Lee, C.K.; Jones, S.; Grabowski, G.A.; Lustig, L.R. Spiral ganglion degeneration and hearing loss as a consequence of satellite cell death in saposin B-deficient mice. J. Neurosci. 2015, 35, 3263–3275. [Google Scholar] [CrossRef] [PubMed]
- Kretz, K.A.; Carson, G.S.; Morimoto, S.; Kishimoto, Y.; Fluharty, A.L.; O’Brien, J.S. Characterization of a mutation in a family with saposin B deficiency: A glycosylation site defect. Proc. Natl. Acad. Sci. USA 1990, 87, 2541–2544. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.W.; O’Brien, J.S. Gaucher’s disease: Deficiency of ‘acid’-glucosidase and reconstitution of enzyme activity in vitro. Proc. Natl. Acad. Sci. USA 1971, 68, 2810–2813. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.P.; Coyle, P.; Coffee, C.J.; Glew, R.H. Purification and properties of a heat-stable glucocerebrosidase activating factor from control and Gaucher spleen. J. Biol. Chem. 1977, 252, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Berent, B.L.; Radin, N.S. beta-Glucosidase activator protein from bovine spleen (“coglucosidase”). Arch. Biochem. Biophys. 1981, 208, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Martin, B.M.; Kishimoto, Y.; O’Brien, J.S. Saposin D: A sphingomyelinase activator. Biochem. Biophys. Res. Commun. 1988, 156, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Christomanou, H.; Kleinschmidt, T. Isolation of two forms of an activator protein for the enzymic sphingomyelin degradation from human Gaucher spleen. Biol. Chem. Hoppe-Seyler 1985, 366, 245–256. [Google Scholar] [CrossRef] [PubMed]
- De Alba, E.; Weiler, S.; Tjandra, N. Solution structure of human saposin C: pH-dependent interaction with phospholipid vesicles. Biochemistry 2003, 42, 14729–14740. [Google Scholar] [CrossRef] [PubMed]
- Preuss, K.D.; Hollak, C.E.M.; Fadle, N.; van Oers, M.; Regitz, E.; Pfreundschuh, M. Saposin C is a frequent target of paraproteins in Gaucher disease-associated MGUS/multiple myeloma. Br. J. Haematol. 2019, 184, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Tatti, M.; Motta, M.; Di Bartolomeo, S.; Scarpa, S.; Cianfanelli, V.; Cecconi, F.; Salvioli, R. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum. Mol. Genet. 2012, 21, 5159–5173. [Google Scholar] [CrossRef] [PubMed]
- Tatti, M.; Motta, M.; Di Bartolomeo, S.; Cianfanelli, V.; Salvioli, R. Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy 2013, 9, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.; Schultz-Heienbrok, R.; Behlke, J.; Remmel, N.; Alings, C.; Sandhoff, K.; Saenger, W.; Maier, T. Crystal structures of human saposins C andD: Implications for lipid recognition and membrane interactions. Structure 2008, 16, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Gebai, A.; Gorelik, A.; Nagar, B. Crystal structure of saposin D in an open conformation. J. Struct. Biol. 2018, 204, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Ciaffoni, F.; Tatti, M.; Salvioli, R.; Vaccaro, A.M. Interaction of saposin D with membranes: Effect of anionic phospholipids and sphingolipids. Biochem. J. 2003, 373, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Ciaffoni, F.; Salvioli, R.; Tatti, M.; Arancia, G.; Crateri, P.; Vaccaro, A.M. Saposin D solubilizes anionic phospholipid-containing membranes. J. Biol. Chem. 2001, 276, 31583–31589. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Kido, M.; Tadano-Aritomi, K.; Ishizuka, I.; Tominaga, K.; Toida, K.; Takeda, E.; Suzuki, K.; Kuroda, Y. Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Hum. Mol. Genet. 2004, 13, 2709–2723. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Subramanian, S.; Weinreb, N.J.; Kartha, R.V. Gaucher disease—More than just a rare lipid storage disease. J. Mol. Med. 2022, 100, 499–518. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Yamamoto, Y.; O’Brien, J.S.; Kishimoto, Y. Distribution of saposin proteins (sphingolipid activator proteins) in lysosomal storage and other diseases. Proc. Natl. Acad. Sci. USA 1990, 87, 3493–3497. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.M.; Ciaffoni, F.; Tatti, M.; Salvioli, R.; Barca, A.; Tognozzi, D.; Scerch, C. pH-dependent conformational properties of saposins and their interactions with phospholipid membranes. J. Biol. Chem. 1995, 270, 30576–30580. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.M.; Tatti, M.; Ciaffoni, F.; Salvioli, R.; Maras, B.; Barca, A. Function of saposin C in the reconstitution of glucosylceramidase by phosphatidylserine liposomes. FEBS Lett. 1993, 336, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.M.; Tatti, M.; Ciaffoni, F.; Salvioli, R.; Barca, A.; Scerch, C. Effect of saposins A and C on the enzymatic hydrolysis of liposomal glucosylceramide. J. Biol. Chem. 1997, 272, 16862–16867. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.M.; Salvioli, R.; Tatti, M.; Ciaffoni, F. Saposins and their interaction with lipids. Neurochem. Res. 1999, 24, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Salvioli, R.; Tatti, M.; Ciaffoni, F.; Vaccaro, A.M. Further studies on the reconstitution of glucosylceramidase activity by Sap C and anionic phospholipids. FEBS Lett. 2000, 472, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Yoneshige, A.; Suzuki, K.; Suzuki, K.; Matsuda, J. A mutation in the saposin C domain of the sphingolipid activator protein (Prosaposin) gene causes neurodegenerative disease in mice. J. Neurosci. Res. 2010, 88, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Quinn, B.; Witte, D.P.; Grabowski, G.A. Gaucher disease mouse models: Point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J. Lipid Res. 2005, 46, 2102–2113. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.M.; Motta, M.; Tatti, M.; Scarpa, S.; Masuelli, L.; Bhat, M.; Vanier, M.T.; Tylki-Szymanska, A.; Salvioli, R. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet. 2010, 19, 2987–2997. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhan, X.; Ye, J.; Han, L.; Qiu, W.; Gu, X.; Zhang, H. A rare form of Gaucher disease resulting from saposin C deficiency. Blood Cells Mol. Dis. 2018, 68, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Radha Rama Devi, A.; Kadali, S.; Radhika, A.; Singh, V.; Kumar, M.A.; Reddy, G.M.; Naushad, S.M. Acute Gaucher Disease-Like Condition in an Indian Infant with a Novel Biallelic Mutation in the Prosaposin Gene. J. Pediatr. Genet. 2019, 8, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.E.; Ali, A.; Al-Tenaiji, A.; Al-Jasmi, A.; Al-Jasmi, F. A Type 3 Gaucher-Like Disease Due To Saposin C Deficiency in Two Emirati Families Caused by a Novel Splice Site Variant in the PSAP Gene. J. Mol. Neurosci. 2022, 72, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Pavan, E.; Peruzzo, P.; Cattarossi, S.; Bergamin, N.; Bordugo, A.; Sechi, A.; Scarpa, M.; Biasizzo, J.; Colucci, F.; Dardis, A. Deficiency of Glucocerebrosidase Activity beyond Gaucher Disease: PSAP and LIMP-2 Dysfunctions. Int. J. Mol. Sci. 2024, 25, 6615. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymańska, A.; Czartoryska, B.; Vanier, M.T.; Poorthuis, B.J.; Groener, J.A.; Lugowska, A.; Millat, G.; Vaccaro, A.M.; Jurkiewicz, E. Non-neuronopathic Gaucher disease due to saposin C deficiency. Clin. Genet. 2007, 72, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Font, A.; Cormand, B.; Santamaria, R.; Vilageliu, L.; Grinberg, D.; Chabás, A. A mutation within the saposin D domain in a Gaucher disease patient with normal glucocerebrosidase activity. Hum. Genet. 2005, 117, 275–277. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kaya, I.; Shariatgorji, R.; Lundkvist, J.; Wahlberg, L.U.; Nilsson, A.; Mamula, D.; Kehr, J.; Zareba-Paslawska, J.; Biverstål, H.; et al. Prosaposin maintains lipid homeostasis in dopamine neurons and counteracts experimental parkinsonism in rodents. Nat. Commun. 2023, 14, 5804. [Google Scholar] [CrossRef] [PubMed]
- Mendsaikhan, A.; Tooyama, I.; Serrano, G.E.; Beach, T.G.; Walker, D.G. Loss of Lysosomal Proteins Progranulin and Prosaposin Associated with Increased Neurofibrillary Tangle Development in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2021, 80, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Stykel, M.G.; Siripala, S.V.; Soubeyrand, E.; Coackley, C.L.; Lu, P.; Camargo, S.; Thevasenan, S.; Figueroa, G.B.; So, R.W.L.; Stuart, E.; et al. G6PD deficiency triggers dopamine loss and the initiation of Parkinson’s disease pathogenesis. Cell Rep. 2025, 44, 115178. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Oji, Y.; Hatano, T.; Ueno, S.I.; Funayama, M.; Ishikawa, K.I.; Okuzumi, A.; Noda, S.; Sato, S.; Satake, W.; Toda, T.; et al. Variants in saposin D domain of prosaposin gene linked to Parkinson’s disease. Brain 2020, 143, 1190–1205. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Gu, X.J.; Ou, R.W.; Zhang, L.Y.; Hou, Y.B.; Liu, K.C.; Cao, B.; Wei, Q.Q.; Song, W.; Zhao, B.; et al. Genetic Analysis of Prosaposin, the Lysosomal Storage Disorder Gene in Parkinson’s Disease. Mol. Neurobiol. 2021, 58, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.C.; Chu, Y.T.; Su, Y.A.; Chen, M.L.; Wu, R.M. Prosaposin variants in sporadic, familial, and early-onset Parkinson’s disease: A Taiwanese case-control study and meta-analysis. Sci. Rep. 2024, 14, 2225. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Funayama, M.; Imai, Y.; Hatano, T. Pathogenesis of Parkinson’s disease: From hints from monogenic familial PD to biomarkers. J. Neural Transm. 2024, 131, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Tayebi, N.; Lopez, G.; Do, J.; Sidransky, E. Pro-cathepsin D, Prosaposin, and Progranulin: Lysosomal Networks in Parkinsonism. Trends Mol. Med. 2020, 26, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Gruschus, J.M.; Jiang, Z.; Yap, T.L.; Hill, S.A.; Grishaev, A.; Piszczek, G.; Sidransky, E.; Lee, J.C. Dissociation of glucocerebrosidase dimer in solution by its co-factor, saposin C. Biochem. Biophys. Res. Commun. 2015, 457, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Jeong, H.; Krainc, D. Lysosomal ceramides regulate cathepsin B-mediated processing of saposin C and glucocerebrosidase activity. Hum. Mol. Genet. 2022, 31, 2424–2437. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Shoji, M.; Imai, Y.; Inoue, H.; Kawarabayashi, T.; Matsubara, E.; Harigaya, Y.; Sasaki, A.; Takahashi, R.; Abe, K. Pael-R is accumulated in Lewy bodies of Parkinson’s disease. Ann. Neurol 2004, 55, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Lundius, E.G.; Vukojevic, V.; Hertz, E.; Stroth, N.; Cederlund, A.; Hiraiwa, M.; Terenius, L.; Svenningsson, P. GPR37 protein trafficking to the plasma membrane regulated by prosaposin and GM1 gangliosides promotes cell viability. J. Biol. Chem. 2014, 289, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Bogetofte, H.; Ryan, B.J.; Jensen, P.; Schmidt, S.I.; Vergoossen, D.L.E.; Barnkob, M.B.; Kiani, L.N.; Chughtai, U.; Heon-Roberts, R.; Caiazza, M.C.; et al. Post-translational proteomics platform identifies neurite outgrowth impairments in Parkinson’s disease GBA-N370S dopamine neurons. Cell Rep. 2023, 42, 112180. [Google Scholar] [CrossRef] [PubMed]
- Vilageliu, L.; Grinberg, D. Involvement of Gaucher Disease Mutations in Parkinson Disease. Curr. Protein Pept. Sci. 2017, 18, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Sidransky, E.; Lee, J.C. Saposin C protects glucocerebrosidase against α-synuclein inhibition. Biochemistry 2013, 52, 7161–7163. [Google Scholar] [CrossRef] [PubMed]
- Baden, P.; Perez, M.J.; Raji, H.; Bertoli, F.; Kalb, S.; Illescas, M.; Spanos, F.; Giuliano, C.; Calogero, A.M.; Oldrati, M.; et al. Glucocerebrosidase is imported into mitochondria and preserves complex I integrity and energy metabolism. Nat. Commun. 2023, 14, 1930. [Google Scholar] [CrossRef] [PubMed]
- Gehrlein, A.; Udayar, V.; Anastasi, N.; Morella, M.L.; Ruf, I.; Brugger, D.; von der Mark, S.; Thoma, R.; Rufer, A.; Heer, D.; et al. Targeting neuronal lysosomal dysfunction caused by β-glucocerebrosidase deficiency with an enzyme-based brain shuttle construct. Nat. Commun. 2023, 14, 2057. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.S.; Guo, M.; Zhao, J.X.; Zhang, H.; Wang, G.; Chen, W. Environmental enrichment in combination with Bifidobacterium breve HNXY26M4 intervention amplifies neuroprotective benefits in a mouse model of Alzheimer’s disease by modulating glutamine metabolism of the gut microbiome. Food Sci. Hum. Wellness 2024, 13, 982–992. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.G.; Wang, C.; Mazzarino, R.C.; Perez-Corredor, P.A.; Davtyan, H.; Blurton-Jones, M.; Lopera, F.; Arboleda-Velasquez, J.F.; Shi, Y. Microglial APOE3 Christchurch protects neurons from Tau pathology in a human iPSC-based model of Alzheimer’s disease. Cell Rep. 2024, 43, 114982. [Google Scholar] [CrossRef] [PubMed]
- Heywood, W.E.; Galimberti, D.; Bliss, E.; Sirka, E.; Paterson, R.W.; Magdalinou, N.K.; Carecchio, M.; Reid, E.; Heslegrave, A.; Fenoglio, C.; et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Zhou, W.; Vanmeter, A.; Heiby, M.; Magaki, S.; Ross, M.M.; Espina, V.; Schrag, M.; Dickson, C.; Liotta, L.A.; et al. The heme degradation pathway is a promising serum biomarker source for the early detection of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.; Tucker, B.; Nornes, S.; Ward, A.; Lardelli, M. Altering presenilin gene activity in zebrafish embryos causes changes in expression of genes with potential involvement in Alzheimer’s disease pathogenesis. J. Alzheimer’s Dis. 2009, 16, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.L.; Li, C.; Nabeka, H.; Shimokawa, T.; Wang, Z.Y.; Cao, Y.M.; Matsuda, S. An 18-mer Peptide Derived from Prosaposin Ameliorates the Effects of Aβ1–42 Neurotoxicity on Hippocampal Neurogenesis and Memory Deficit in Mice. J. Alzheimer’s Dis. 2016, 53, 1173–1192. [Google Scholar] [CrossRef] [PubMed]
- Paushter, D.H.; Du, H.; Feng, T.; Hu, F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 2018, 136, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mendsaikhan, A.; Tooyama, I.; Bellier, J.P.; Serrano, G.E.; Sue, L.I.; Lue, L.F.; Beach, T.G.; Walker, D.G. Characterization of lysosomal proteins Progranulin and Prosaposin and their interactions in Alzheimer’s disease and aged brains: Increased levels correlate with neuropathology. Acta Neuropathol. Commun. 2019, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.M.; Finch, N.A.; Almeida, M.; Perkerson, R.B.; van Blitterswijk, M.; Wojtas, A.; Cenik, B.; Rotondo, S.; Inskeep, V.; Almasy, L.; et al. Prosaposin is a regulator of progranulin levels and oligomerization. Nat. Commun. 2016, 7, 11992. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.T.; Chen, F.; Chen, J.H.; Wang, M.F. An overview of potential cardioprotective benefits of xanthophylls in atherosclerosis: An evidence-based review. Food Sci. Hum. Wellness 2024, 13, 1739–1755. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.M.; Herman, A.G. Apoptosis in atherosclerosis: Beneficial or detrimental? Cardiovasc. Res. 2000, 45, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Kockx, M.M. Apoptosis in atherosclerosis: Focus on oxidized lipids and inflammation. Curr. Opin. Lipidol. 2001, 12, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kapoor, D.; Jeong, S.J.; Fappi, A.; Stitham, J.; Shabrish, V.; Sergin, I.; Yousif, E.; Rodriguez-Velez, A.; Yeh, Y.S.; et al. Identification of a leucine-mediated threshold effect governing macrophage mTOR signalling and cardiovascular risk. Nat. Metab. 2024, 6, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Stroope, C.; Nettersheim, F.S.; Coon, B.; Finney, A.C.; Schwartz, M.A.; Ley, K.; Rom, O.; Yurdagul, A., Jr. Dysregulated cellular metabolism in atherosclerosis: Mediators and therapeutic opportunities. Nat. Metab. 2024, 6, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, U.K.; Singhal, A.; Subramanian, M. Dead cell and debris clearance in the atherosclerotic plaque: Mechanisms and therapeutic opportunities to promote inflammation resolution. Pharmacol. Res. 2021, 170, 105699. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Dhawan, U.K.; Hussain, M.T.; Singh, P.; Bhagat, K.K.; Singhal, A.; Austin-Williams, S.; Sengupta, S.; Subramanian, M. Efferocytes release extracellular vesicles to resolve inflammation and tissue injury via prosaposin-GPR37 signaling. Cell Rep. 2023, 42, 112808. [Google Scholar] [CrossRef] [PubMed]
- Haidar, B.; Kiss, R.S.; Sarov-Blat, L.; Brunet, R.; Harder, C.; McPherson, R.; Marcel, Y.L. Cathepsin D, a lysosomal protease, regulates ABCA1-mediated lipid efflux. J. Biol. Chem. 2006, 281, 39971–39981. [Google Scholar] [CrossRef] [PubMed]
- Greenlee, R.T.; Murray, T.; Bolden, S.; Wingo, P.A. Cancer statistics, 2000. CA Cancer J. Clin. 2000, 50, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.P. Regulation of signal transduction pathways by estrogen and progesterone. Annu. Rev. Physiol. 2005, 67, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Hilakivi-Clarke, L.; Cho, E.; Cabanes, A.; DeAssis, S.; Olivo, S.; Helferich, W.; Lippman, M.E.; Clarke, R. Dietary modulation of pregnancy estrogen levels and breast cancer risk among female rat offspring. Clin. Cancer Res. 2002, 8, 3601–3610. [Google Scholar] [PubMed]
- Meijer, D.; Jansen, M.P.; Look, M.P.; Ruigrok-Ritstier, K.; van Staveren, I.L.; Sieuwerts, A.M.; van Agthoven, T.; Foekens, J.A.; Dorssers, L.C.; Berns, E.M. TSC22D1 and PSAP predict clinical outcome of tamoxifen treatment in patients with recurrent breast cancer. Breast Cancer Res. Treat. 2009, 113, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Matha, V.; Lucas, A.; Huttler, S.; Sandhoff, K.; Garcia, M.; Rochefort, H. Procathepsin D interacts with prosaposin in cancer cells but its internalization is not mediated by LDL receptor-related protein. Exp. Cell Res. 2002, 277, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; de Bruijn, R.; van der Burg, E.; Drenth, A.P.; Wientjens, E.; Filipovic, T.; Bullock, E.; Brambillasca, C.S.; Pulver, E.M.; Nieuwland, M.; et al. CD26-negative and CD26-positive tissue-resident fibroblasts contribute to functionally distinct CAF subpopulations in breast cancer. Nat. Commun. 2023, 14, 183. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Inman, D.R.; Li, W.J.; Ponik, S.M.; Keely, P.J. Mechano-Signal Transduction in Mesenchymal Stem Cells Induces Prosaposin Secretion to Drive the Proliferation of Breast Cancer Cells. Cancer Res. 2017, 77, 6179–6189. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Ponik, S.M.; Haga, H. Mesenchymal stem cells in breast cancer: Response to chemical and mechanical stimuli. Oncoscience 2017, 4, 158–159. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Qiu, Q.; Wei, Y.; Mercatali, L.; Ibrahim, T.; Amadori, D.; Kang, Y. The microRNA-23b/27b/24 cluster promotes breast cancer lung metastasis by targeting metastasis-suppressive gene prosaposin. J. Biol. Chem. 2014, 289, 21888–21895. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Drapkin, R. Ovarian cancer pathogenesis: A model in evolution. J. Oncol. 2010, 2010, 932371. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Blois, A.; El Rayes, T.; Liu, J.F.; Hirsch, M.S.; Gravdal, K.; Palakurthi, S.; Bielenberg, D.R.; Akslen, L.A.; Drapkin, R.; et al. Development of a prosaposin-derived therapeutic cyclic peptide that targets ovarian cancer via the tumor microenvironment. Sci. Transl. Med. 2016, 8, 329ra34. [Google Scholar] [CrossRef] [PubMed]
- Hsing, A.W.; Chokkalingam, A.P. Prostate cancer epidemiology. Front. Biosci. 2006, 11, 1388–1413. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.M.; Srinivas, S.; Adra, N.; An, Y.; Barocas, D.; Bitting, R.; Bryce, A.; Chapin, B.; Cheng, H.H.; D’Amico, A.V.; et al. Prostate Cancer, Version 4.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 1067–1096. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Zhuang, Y.J.; Beroukhim, R.; Hsieh, C.L.; Hofer, M.D.; Zhau, H.E.; Hiraiwa, M.; Pattan, D.Y.; Ware, J.L.; Luftig, R.B.; et al. Amplification and overexpression of prosaposin in prostate cancer. Genes Chromosomes Cancer 2005, 44, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Delorme, N.; Liu, Z.; Liu, T.; Velasco-Gonzalez, C.; Garai, J.; Pullikuth, A.; Koochekpour, S. Prosaposin down-modulation decreases metastatic prostate cancer cell adhesion, migration, and invasion. Mol. Cancer 2010, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Hu, S.; Vellasco-Gonzalez, C.; Bernardo, R.; Azabdaftari, G.; Zhu, G.; Zhau, H.E.; Chung, L.W.; Vessella, R.L. Serum prosaposin levels are increased in patients with advanced prostate cancer. Prostate 2012, 72, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Lee, T.J.; Wang, R.; Culig, Z.; Delorme, N.; Caffey, S.; Marrero, L.; Aguirre, J. Prosaposin upregulates AR and PSA expression and activity in prostate cancer cells (LNCaP). Prostate 2007, 67, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Lee, T.J.; Sun, Y.; Hu, S.; Grabowski, G.A.; Liu, Z.; Garay, J. Prosaposin is an AR-target gene and its neurotrophic domain upregulates AR expression and activity in prostate stromal cells. J. Cell. Biochem. 2008, 104, 2272–2285. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Sartor, O.; Hiraiwa, M.; Lee, T.J.; Rayford, W.; Remmel, N.; Sandhoff, K.; Minokadeh, A.; Patten, D.Y. Saposin C stimulates growth and invasion, activates p42/44 and SAPK/JNK signaling pathways of MAPK and upregulates uPA/uPAR expression in prostate cancer and stromal cells. Asian J. Androl. 2005, 7, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.J.; Sartor, O.; Luftig, R.B.; Koochekpour, S. Saposin C promotes survival and prevents apoptosis via PI3K/Akt-dependent pathway in prostate cancer cells. Mol. Cancer 2004, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Alver, B.M.; Li, S.; Hlady, R.A.; Thompson, J.J.; Schroeder, M.A.; Lee, J.H.; Qiu, J.; Schwartz, P.H.; Sarkaria, J.N.; et al. Distinctive epigenomes characterize glioma stem cells and their response to differentiation cues. Genome Biol. 2018, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.H.; Wang, C.Y.; Hsieh, Y.T.; Fang, K.M.; Tzeng, S.F. Functional Role of Matrix gla Protein in Glioma Cell Migration. Mol. Neurobiol. 2018, 55, 4624–4636. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Sughrue, M.E. Glioblastoma: New therapeutic strategies to address cellular and genomic complexity. Oncotarget 2018, 9, 9540–9554. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhou, J.; Hou, D.; Luo, P.; Gao, H.; Ma, Y.; Chen, Y.S.; Li, L.; Zou, D.; Zhang, H.; et al. Prosaposin is a biomarker of mesenchymal glioblastoma and regulates mesenchymal transition through the TGF-β1/Smad signaling pathway. J. Pathol. 2019, 249, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, Y.; Cao, Z.; Chen, Z.; Pan, W. Identification of three immune subtypes characterized by distinct tumor immune microenvironment and therapeutic response in stomach adenocarcinoma. Gene 2022, 818, 146177. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, P.P.; Yin, L.; Wang, X.C.; Shan, Y.Y.; Yi, Y.L.; Zhou, Y.; Liu, B.F.; Wang, X.; Lue, X. Dietary Lactiplantibacillus plantarum KX041 attenuates colitis-associated tumorigenesis and modulates gut microbiota. Food Sci. Hum. Wellness 2023, 12, 1626–1636. [Google Scholar] [CrossRef]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Lazcano-Ponce, E.C.; Miquel, J.F.; Muñoz, N.; Herrero, R.; Ferrecio, C.; Wistuba, I.; Alonso de Ruiz, P.; Aristi Urista, G.; Nervi, F. Epidemiology and molecular pathology of gallbladder cancer. CA Cancer J. Clin. 2001, 51, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Elbert, A. Metabolic dysfunction-associated steatotic liver disease and urinary system cancers: Mere coincidence or reason for concern? Metab. Clin. Exp. 2025, 162, 156066. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Lonardo, A.; Stefan, N.; Targher, G. Metabolic dysfunction-associated steatotic liver disease and extrahepatic gastrointestinal cancers. Metab. Clin. Exp. 2024, 160, 156014. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Tacke, F. Diagnostics and omics technologies for the detection and prediction of metabolic dysfunction-associated steatotic liver disease-related malignancies. Metab. Clin. Exp. 2024, 161, 156015. [Google Scholar] [CrossRef] [PubMed]
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. An updated overview on hepatocellular carcinoma in patients with Metabolic dysfunction-Associated Steatotic Liver Disease: Trends, pathophysiology and risk-based surveillance. Metab. Clin. Exp. 2025, 162, 156080. [Google Scholar] [CrossRef] [PubMed]
- Ishteyaque, S.; Singh, G.; Yadav, K.S.; Verma, S.; Sharma, R.K.; Sen, S.; Srivastava, A.K.; Mitra, K.; Lahiri, A.; Bawankule, D.U.; et al. Cooperative STAT3-NFkB signaling modulates mitochondrial dysfunction and metabolic profiling in hepatocellular carcinoma. Metab. Clin. Exp. 2024, 152, 155771. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. reviews. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Zucali, P.A.; Ceresoli, G.L.; De Vincenzo, F.; Simonelli, M.; Lorenzi, E.; Gianoncelli, L.; Santoro, A. Advances in the biology of malignant pleural mesothelioma. Cancer Treat. Rev. 2011, 37, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Chand, H.S.; Du, X.; Ma, D.; Inzunza, H.D.; Kamei, S.; Foster, D.; Brodie, S.; Kisiel, W. The effect of human tissue factor pathway inhibitor-2 on the growth and metastasis of fibrosarcoma tumors in athymic mice. Blood 2004, 103, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Nabeshima, K.; Inoue, T.; Shimao, Y.; Sameshima, T. Matrix metalloproteinases in tumor invasion: Role for cell migration. Pathol. Int. 2002, 52, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Key Pathway/ Mechanism | Biomarker | Therapeutic Potential | Role in Tumor | Reference |
|---|---|---|---|---|---|
| Breast Cancer | ERα/MAPK signaling | Prognostic marker | Inhibition of miR-23b/27b/24/PSAP axis | Pro-tumor/Anti-tumor | [17,157,158] |
| Ovarian Cancer | TSP-1/CD36 apoptosis pathway | Unknown | Cyclic PSAP peptide | Anti-tumor | [160] |
| Prostate Cancer | AR signaling axis | Serum staging marker (TBC) | Blocking PSAP/AR axis | Pro-tumor | [18,167] |
| Gliomas | TLR4/NF-κB signaling | Unknown | Inhibition of the PSAP/TLR4/NF-κB axis via TLR4 inhibitors | Pro-tumor | [19] |
| Glioblastoma | TGF-β1/Smad- induced EMT | Unknown | Inhibiting PSAP/TGF-β/Smad axis | Pro-tumor | [175] |
| Gastric Cancer | PDCD1/TGFB1/CSF1R inhibition | Prognostic marker | Inhibiting PSAP/Immune checkpoint axis | Pro-tumor | [20] |
| Colorectal Cancer | dMMR/CIMP+/BRAF mutations | CMS4/CRIS-B subtype marker | Unknown | Pro-tumor | [21] |
| Gallbladder Cancer | Unknown | Early diagnostic marker (TBC) | Unknown | Pro-tumor | [22] |
| Hepatocellular carcinoma | circVAPA/miR-377-3p axis | Unknown | Inhibition of PSAP by miR-377-3p | Pro-tumor | [23] |
| Pancreatic ductal adenocarcinoma | CD8+ T-cell inhibition | Immune microenvironment marker (TBC) | Blocking PASP-induced immunosuppression | Pro-tumor | [24] |
| Malignant pleural mesothelioma | Oxidative stress defense | Co-diagnostic marker with QSOX1 (TBC) | Unknown | Pro-tumor | [25] |
| Fibrosarcoma | TFPI-2/PSAP/MMP2 pathway | Unknown | Inhibition of PSAP/MMP2 axis by TFPI-2 | Pro-tumor | [26] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Guo, L. Prosaposin: A Multifaceted Protein Orchestrating Biological Processes and Diseases. Cells 2025, 14, 1131. https://doi.org/10.3390/cells14151131
Li X, Guo L. Prosaposin: A Multifaceted Protein Orchestrating Biological Processes and Diseases. Cells. 2025; 14(15):1131. https://doi.org/10.3390/cells14151131
Chicago/Turabian StyleLi, Xin, and Liang Guo. 2025. "Prosaposin: A Multifaceted Protein Orchestrating Biological Processes and Diseases" Cells 14, no. 15: 1131. https://doi.org/10.3390/cells14151131
APA StyleLi, X., & Guo, L. (2025). Prosaposin: A Multifaceted Protein Orchestrating Biological Processes and Diseases. Cells, 14(15), 1131. https://doi.org/10.3390/cells14151131