
Histone Acetyltransferase MOF-Mediated AURKB K215 Acetylation Drives Breast Cancer Cell Proliferation via c-MYC Stabilization

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Culture
2.3. Plasmid Construction and Transfection
2.4. Expression of Recombinant Proteins in Escherichia Coli
2.5. siRNA/shRNA Knockdown
2.6. Immunoprecipitation (IP)
2.7. Immunofluorescence Staining
2.8. Reverse Transcription PCR
2.9. In Vitro KAT Assay
2.10. Flow Cytometry Analysis
2.11. EdU Assay
2.12. Cell Viability Assay
2.13. Colony Formation Assay
2.14. In Vivo Tumor Metastasis Experiments
2.15. Statistical Analysis
3. Results
3.1. A Reciprocal Interaction Between the MOF/MSL Complex and CPC in HEK293T Cells
3.2. MOF, MSL1, and AURKB May Collaborate During Early Mitosis
3.3. MOF/MSL Complex Mediates AURKB Acetylation, Stabilizing CPC Integrity in HEK293T Cells
3.4. MOF/MSL Complex-Mediated Acetylation of AURKB at K215 Maintains CPC Integrity and Kinase Activity
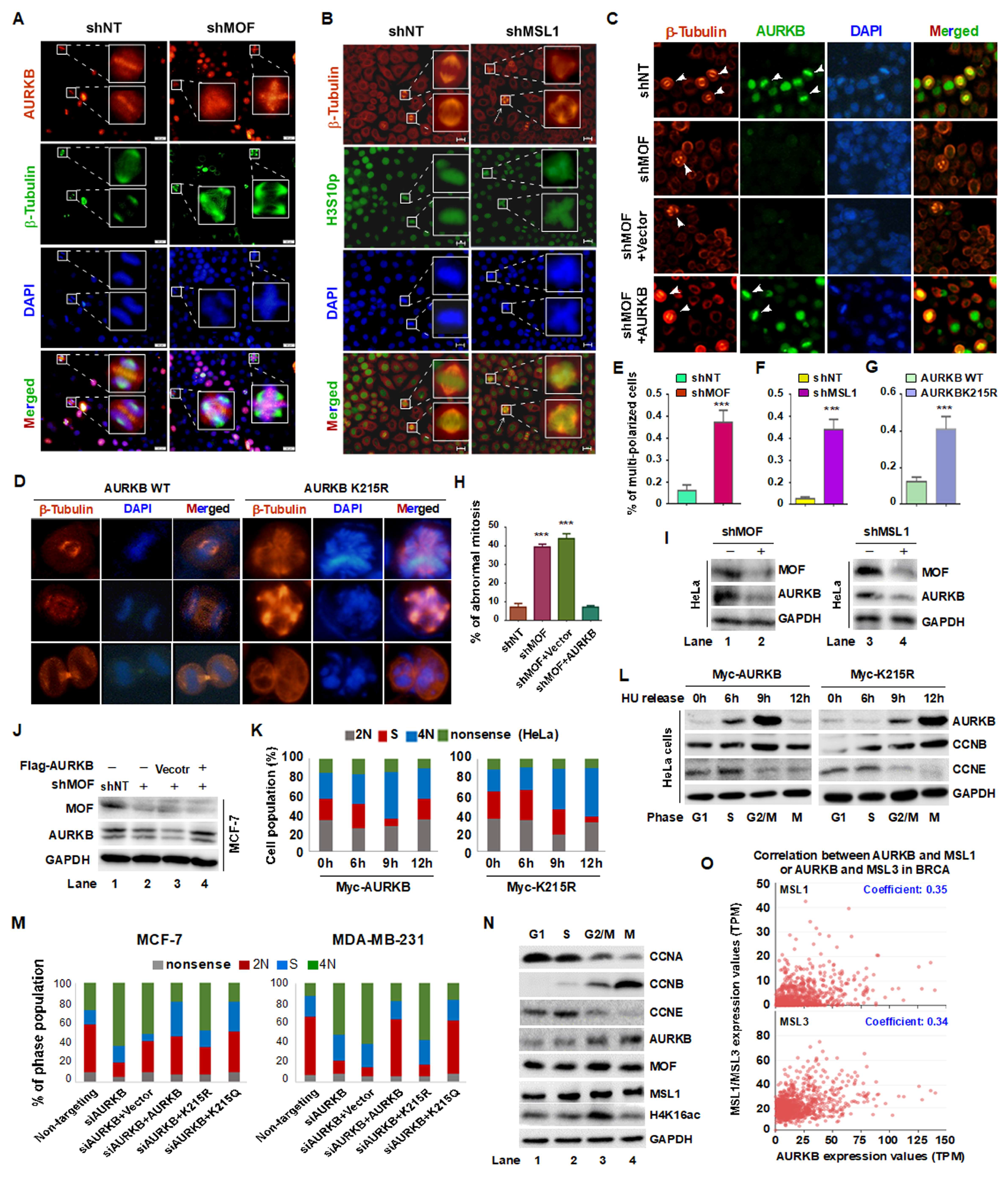
3.5. MOF/MSL1 Complex-Mediated Acetylation of AURKB at K215 Regulates G2/M Phase Progression in HeLa and MCF-7 Cells
3.6. MOF-Mediated Acetylation of AURKB at K215 Is Essential for Breast Cancer Cell Proliferation
3.7. Acetylation of AURKB at K215 Promotes Breast Cancer Cell Proliferation by Stabilizing c-MYC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AURKB | Aurora kinase B |
| INCENP | inner centromere protein |
| KATs | lysine acetyltransferases |
| CPC | chromosome passenger complex |
| H3S19pho | phosphorylation of histone H3S10 |
| MOF | males absent on the first |
| MSL | male-specific lethal |
| H4K16ac | acetylation of histone H4 at lysine16 |
| CHX | cycloheximide |
| PTMs | post-translational modifications |
| HATs | histone acetyltransferases |
| HDACs | histone deacetylases |
References
- Bolanos-Garcia, V.M. Aurora kinases. Int. J. Biochem. Cell Biol. 2005, 37, 1572–1577. [Google Scholar] [CrossRef]
- Niwa, H.; Abe, K.; Kunisada, T.; Yamamura, K. Cell-cycle-dependent expression of the STK-1 gene encoding a novel murine putative protein kinase. Gene 1996, 169, 197–201. [Google Scholar] [CrossRef]
- van der Horst, A.; Lens, S.M. Cell division: Control of the chromosomal passenger complex in time and space. Chromosoma 2014, 123, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Lampson, M.A.; Cheeseman, I.M. Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol. 2011, 21, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Welburn, J.P.; Vleugel, M.; Liu, D.; Yates, J.R., 3rd; Lampson, M.A.; Fukagawa, T.; Cheeseman, I.M. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol. Cell 2010, 38, 383–392. [Google Scholar] [CrossRef]
- Liu, D.; Vleugel, M.; Backer, C.B.; Hori, T.; Fukagawa, T.; Cheeseman, I.M.; Lampson, M.A. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol. 2010, 188, 809–820. [Google Scholar] [CrossRef]
- DeLuca, K.F.; Lens, S.M.; DeLuca, J.G. Temporal changes in Hec1 phosphorylation control kinetochore-microtubule attachment stability during mitosis. J. Cell Sci. 2011, 124, 622–634. [Google Scholar] [CrossRef]
- Sumara, I.; Quadroni, M.; Frei, C.; Olma, M.H.; Sumara, G.; Ricci, R.; Peter, M. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev. Cell 2007, 12, 887–900. [Google Scholar] [CrossRef]
- Adams, R.R.; Maiato, H.; Earnshaw, W.C.; Carmena, M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J. Cell Biol. 2001, 153, 865–880. [Google Scholar] [CrossRef]
- Monje-Casas, F.; Prabhu, V.R.; Lee, B.H.; Boselli, M.; Amon, A. Kinetochore orientation during meiosis is controlled by Aurora B and the monopolin complex. Cell 2007, 128, 477–490. [Google Scholar] [CrossRef]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef]
- Adams, R.R.; Eckley, D.M.; Vagnarelli, P.; Wheatley, S.P.; Gerloff, D.L.; Mackay, A.M.; Svingen, P.A.; Kaufmann, S.H.; Earnshaw, W.C. Human INCENP colocalizes with the Aurora-B/AIRK2 kinase on chromosomes and is overexpressed in tumour cells. Chromosoma 2001, 110, 65–74. [Google Scholar] [CrossRef]
- Belote, J.M.; Lucchesi, J.C. Male-specific lethal mutations of Drosophila melanogaster. Genetics 1980, 96, 165–186. [Google Scholar] [CrossRef]
- Rea, S.; Xouri, G.; Akhtar, A. Males absent on the first (MOF): From flies to humans. Oncogene 2007, 26, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Neal, K.C.; Pannuti, A.; Smith, E.R.; Lucchesi, J.C. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochim. Biophys. Acta 2000, 149, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Cayrou, C.; Huang, R.; Lane, W.S.; Côté, J.; Lucchesi, J.C. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell Biol. 2005, 25, 9175–9188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Su, J.; Wang, F.; Liu, D.; Ding, J.; Yang, Y.; Conaway, J.W.; Conaway, R.C.; Cao, L.; Wu, D.; et al. Crosstalk between NSL histone acetyltransferase and MLL/SET complexes: NSL complex functions in promoting histone H3K4 di-methylation activity by MLL/SET complexes. PLoS Genet. 2013, 9, e1003940. [Google Scholar] [CrossRef]
- Kapoor-Vazirani, P.; Kagey, J.D.; Powell, D.R.; Vertino, P.M. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008, 68, 6810–6821. [Google Scholar] [CrossRef]
- Singh, M.; Bacolla, A.; Chaudhary, S.; Hunt, C.R.; Pandita, S.; Chauhan, R.; Gupta, A.; Tainer, J.A.; Pandita, T.K. Histone Acetyltransferase MOF Orchestrates Outcomes at the Crossroad of Oncogenesis, DNA Damage Response, Proliferation, and Stem Cell Development. Mol. Cell Biol. 2020, 40, e00232-20. [Google Scholar] [CrossRef]
- Zhong, J.; Li, X.; Cai, W.; Wang, Y.; Dong, S.; Yang, J.; Zhang, J.; Wu, N.; Li, Y.; Mao, F.; et al. TET1 modulates H4K16 acetylation by controlling auto-acetylation of hMOF to affect gene regulation and DNA repair function. Nucleic Acids Res. 2017, 45, 672–684. [Google Scholar] [CrossRef]
- Li, X.; Li, L.; Pandey, R.; Byun, J.S.; Gardner, K.; Qin, Z.; Dou, Y. The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell 2012, 11, 163–178. [Google Scholar] [CrossRef]
- Chen, Z.; Ye, X.; Tang, N.; Shen, S.; Li, Z.; Niu, X.; Lu, S.; Xu, L. The histone acetylranseferase hMOF acetylates Nrf2 and regulates anti-drug responses in human non-small cell lung cancer. Br. J. Pharmacol. 2014, 171, 3196–3211. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Xu, S.; Jin, Y.; Yu, J.; Dai, S.; Shi, X.J.; Guo, R. hMOF induces cisplatin resistance of ovarian cancer by regulating the stability and expression of MDM2. Cell Death Discov. 2023, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.B.; Glasser, J.R.; Coon, T.A.; Mallampalli, R.K. Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates Aurora B to inhibit tumorigenesis. Cell Death Dis. 2013, 4, e759. [Google Scholar] [CrossRef] [PubMed]
- Krupina, K.; Kleiss, C.; Metzger, T.; Fournane, S.; Schmucker, S.; Hofmann, K.; Fischer, B.; Paul, N.; Porter, I.M.; Raffelsberger, W.; et al. Ubiquitin Receptor Protein UBASH3B Drives Aurora B Recruitment to Mitotic Microtubules. Dev. Cell. 2016, 36, 63–78. [Google Scholar] [CrossRef]
- Xu, J.L.; Yuan, Y.J.; Lv, J.; Qi, D.; Wu, M.D.; Lan, J.; Liu, S.N.; Yang, Y.; Zhai, J.; Jiang, H.M. Inhibition of BRD4 triggers cellular senescence through suppressing aurora kinases in oesophageal cancer cells. J. Cell Mol. Med. 2020, 24, 13036–13045. [Google Scholar] [CrossRef]
- Wang, C.; Chen, J.; Cao, W.; Sun, L.; Sun, H.; Liu, Y. Aurora-B and HDAC synergistically regulate survival and proliferation of lymphoma cell via AKT, mTOR and Notch pathways. Eur. J. Pharmacol. 2016, 779, 1–7. [Google Scholar] [CrossRef]
- Mo, F.; Zhuang, X.; Liu, X.; Yao, P.Y.; Qin, B.; Su, Z.; Zang, J.; Wang, Z.; Zhang, J.; Dou, Z.; et al. Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat. Chem. Biol. 2016, 12, 226–232. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, B.; Cai, C.; Chen, Y.; Miao, Y.; Chu, J.; Sui, Y.; Li, F.; Chen, W.; Cai, Y.; et al. The Males Absent on the First (MOF) Mediated Acetylation Alters the Protein Stability and Transcriptional Activity of YY1 in HCT116 Cells. Int. J. Mol. Sci. 2023, 24, 8719. [Google Scholar] [CrossRef]
- Wei, T.; Liu, H.; Zhu, H.; Chen, W.; Wu, T.; Bai, Y.; Zhang, X.; Miao, Y.; Wang, F.; Cai, Y.; et al. Two distinct males absent on the first (MOF)-containing histone acetyltransferases are involved in the epithelial-mesenchymal transition in different ways in human cells. Cell Mol. Life Sci. 2022, 79, 238. [Google Scholar] [CrossRef] [PubMed]
- Valerio, D.G.; Xu, H.; Eisold, M.E.; Woolthuis, C.M.; Pandita, T.K.; Armstrong, S.A. Histone acetyltransferase activity of MOF is required for adult but not early fetal hematopoiesis in mice. Blood J. Am. Soc. Hematol. 2017, 129, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Rohrberg, J.; Van de Mark, D.; Amouzgar, M.; Lee, J.V.; Taileb, M.; Corella, A.; Kilinc, S.; Williams, J.; Jokisch, M.L.; Camarda, R.; et al. MYC Dysregulates Mitosis, Revealing Cancer Vulnerabilities. Cell Rep. 2020, 30, 3368–3382. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, J.; Yue, M.; Cai, X.; Wang, T.; Wu, C.; Su, H.; Wang, Y.; Han, M.; Zhang, Y.; et al. Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-cell Leukemogenesis. Cancer Cell 2020, 37, 200–215. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Zhang, Y.; Cheng, Y.; Li, Y. RNAi screening identifies KAT8 as a key molecule important for cancer cell survival. Int. J. Clin. Exp. Pathol. 2013, 6, 870–877. [Google Scholar]
- Tanaka, K.; Helena, A.Y.; Yang, S.; Han, S.; Selcuklu, S.D.; Kim, K.; Ramani, S.; Ganesan, Y.T.; Moyer, A.; Sinha, S.; et al. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell 2021, 39, 1245–1261. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, F.; Ward, T.; Yan, F.; Wu, Q.; Wang, Z.; McGlothen, T.; Peng, W.; You, T.; Sun, M.; et al. Phosphorylation of HsMis13 by Aurora B kinase is essential for assembly of functional kinetochore. J. Biol. Chem. 2008, 283, 26726–26736. [Google Scholar] [CrossRef]
- Cimini, D.; Wan, X.; Hirel, C.B.; Salmon, E.D. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr. Biol. 2006, 16, 1711–1718. [Google Scholar] [CrossRef]
- Alushin, G.M.; Musinipally, V.; Matson, D.; Tooley, J.; Stukenberg, P.T.; Nogales, E. Multimodal microtubule binding by the Ndc80 kinetochore complex. Nat. Struct. Mol. Biol. 2012, 19, 1161–1167. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Gestaut, D.R.; Tien, J.F.; Vollmar, B.S.; Gonen, T.; Asbury, C.L.; Davis, T.N. The Ndc80 kinetochore complex directly modulates microtubule dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 16113–16118. [Google Scholar] [CrossRef]
- Muir, K.W.; Batters, C.; Dendooven, T.; Yang, J.; Zhang, Z.; Burt, A.; Barford, D. Structural mechanism of outer kinetochore Dam1-Ndc80 complex assembly on microtubules. Science 2023, 382, 1184–1190. [Google Scholar] [CrossRef]
- Ahmed, A.; Shamsi, A.; Mohammad, T.; Hasan, G.M.; Islam, A.; Hassan, M.I. Aurora B kinase: A potential drug target for cancer therapy. J. Cancer Res. Clin. Oncol. 2021, 147, 2187–2198. [Google Scholar] [CrossRef]
- Liu, M.; Li, Y.; Zhang, C.; Zhang, Q. Role of aurora kinase B in regulating resistance to paclitaxel in breast cancer cells. Hum. Cell 2022, 35, 678–693. [Google Scholar] [CrossRef]
- Al-Khafaji, A.S.; Davies, M.P.; Risk, J.M.; Marcus, M.W.; Koffa, M.; Gosney, J.R.; Shaw, R.J.; Field, J.K.; Liloglou, T. Aurora B expression modulates paclitaxel response in non-small cell lung cancer. Br. J. Cancer 2017, 116, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Hartsink-Segers, S.A.; Zwaan, C.M.; Exalto, C.; Luijendijk, M.W.; Calvert, V.S.; Petricoin, E.F.; Evans, W.E.; Reinhardt, D.; de Haas, V.; Hedtjärn, M.; et al. Aurora kinases in childhood acute leukemia: The promise of aurora B as therapeutic target. Leukemia 2013, 27, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.Z.; Jeng, Y.M.; Hu, F.C.; Pan, H.W.; Tsao, H.W.; Lai, P.L.; Lee, P.H.; Cheng, A.L.; Hsu, H.C. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer 2010, 10, 461. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Lourenco, C.; Kalkat, M.; Houlahan, K.E.; De Melo, J.; Longo, J.; Done, S.J.; Boutros, P.C.; Penn, L.Z. Modelling the MYC-driven normal-to-tumour switch in breast cancer. Dis. Model. Mech. 2019, 12, dmm038083. [Google Scholar] [CrossRef]
- Li, M.; Li, A.; Zhou, S.; Lv, H.; Yang, W. SPAG5 upregulation contributes to enhanced c-MYC transcriptional activity via interaction with c-MYC binding protein in triple-negative breast cancer. J. Hematol. Oncol. 2019, 12, 14. [Google Scholar] [CrossRef]
- Gao, F.Y.; Li, X.T.; Xu, K.; Wang, R.T.; Guan, X.X. c-MYC mediates the crosstalk between breast cancer cells and tumor microenvironment. Cell Commun. Signal. 2023, 21, 28. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, Y.; Zhang, N.; Li, F.; Wang, F.; Chen, Y.; Li, F.; Cui, X.; Zhao, Q.; Cai, Y.; Jin, J. Histone Acetyltransferase MOF-Mediated AURKB K215 Acetylation Drives Breast Cancer Cell Proliferation via c-MYC Stabilization. Cells 2025, 14, 1100. https://doi.org/10.3390/cells14141100
Miao Y, Zhang N, Li F, Wang F, Chen Y, Li F, Cui X, Zhao Q, Cai Y, Jin J. Histone Acetyltransferase MOF-Mediated AURKB K215 Acetylation Drives Breast Cancer Cell Proliferation via c-MYC Stabilization. Cells. 2025; 14(14):1100. https://doi.org/10.3390/cells14141100
Chicago/Turabian StyleMiao, Yujuan, Na Zhang, Fuqing Li, Fei Wang, Yuyang Chen, Fuqiang Li, Xueli Cui, Qingzhi Zhao, Yong Cai, and Jingji Jin. 2025. "Histone Acetyltransferase MOF-Mediated AURKB K215 Acetylation Drives Breast Cancer Cell Proliferation via c-MYC Stabilization" Cells 14, no. 14: 1100. https://doi.org/10.3390/cells14141100
APA StyleMiao, Y., Zhang, N., Li, F., Wang, F., Chen, Y., Li, F., Cui, X., Zhao, Q., Cai, Y., & Jin, J. (2025). Histone Acetyltransferase MOF-Mediated AURKB K215 Acetylation Drives Breast Cancer Cell Proliferation via c-MYC Stabilization. Cells, 14(14), 1100. https://doi.org/10.3390/cells14141100