
Dual Roles of Hypoxia-Inducible Factor 1 in Acute Lung Injury: Tissue-Specific Mechanisms and Therapeutic Modulation


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
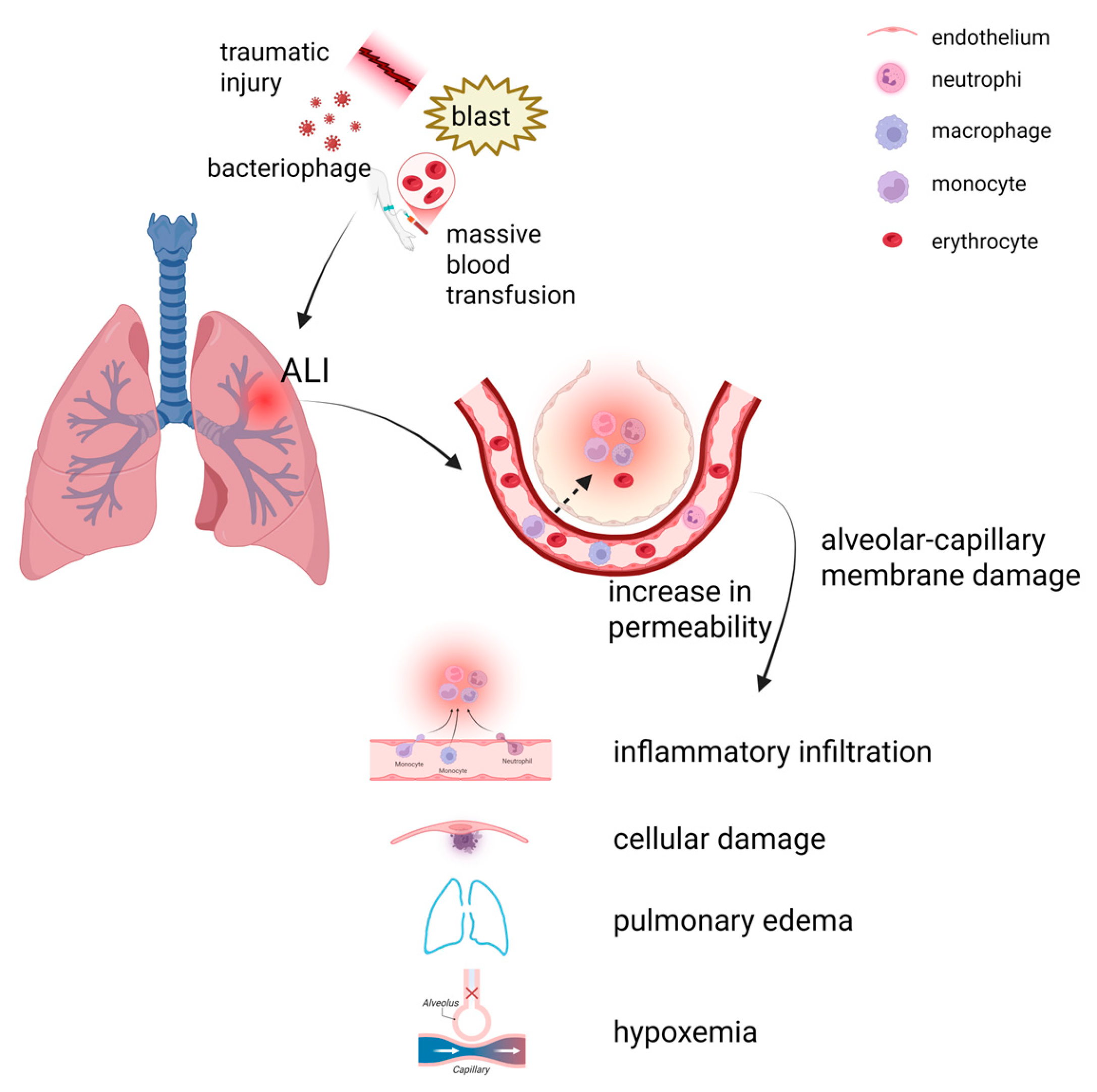
2. Pathophysiology of ALI
2.1. Direct Factors
2.2. Indirect Factors
2.3. Endothelial and Epithelial Cell Apoptosis, and Neutrophil Recruitment
2.4. Polarization of Macrophages
2.5. Vascular Leakage and Hypoxemia
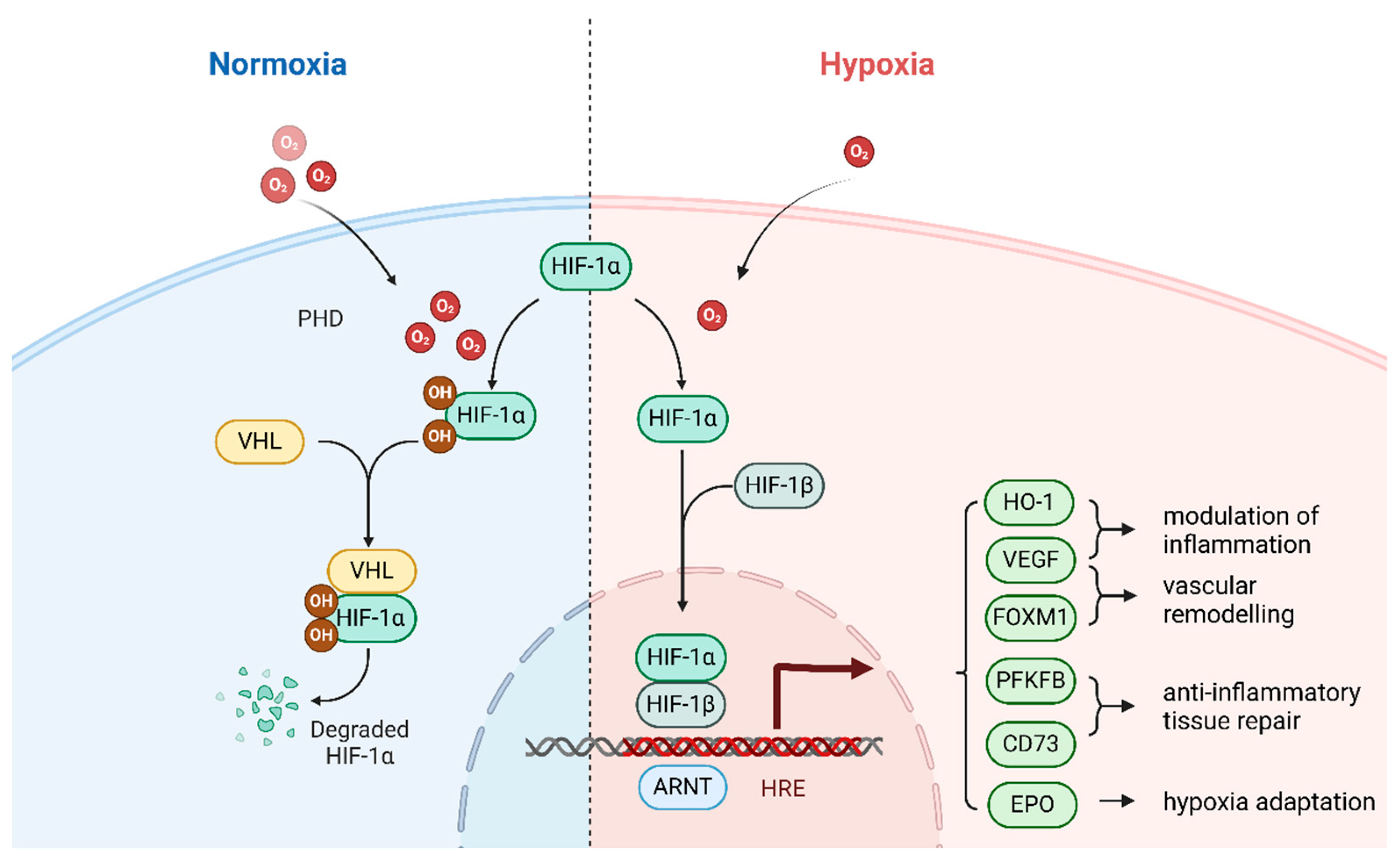
3. Hypoxia-Inducible Factor (HIF)-1
3.1. Activation Mechanisms of HIFs
3.2. Structure of HIF-1
3.3. Regulatory Mechanisms of HIF-1
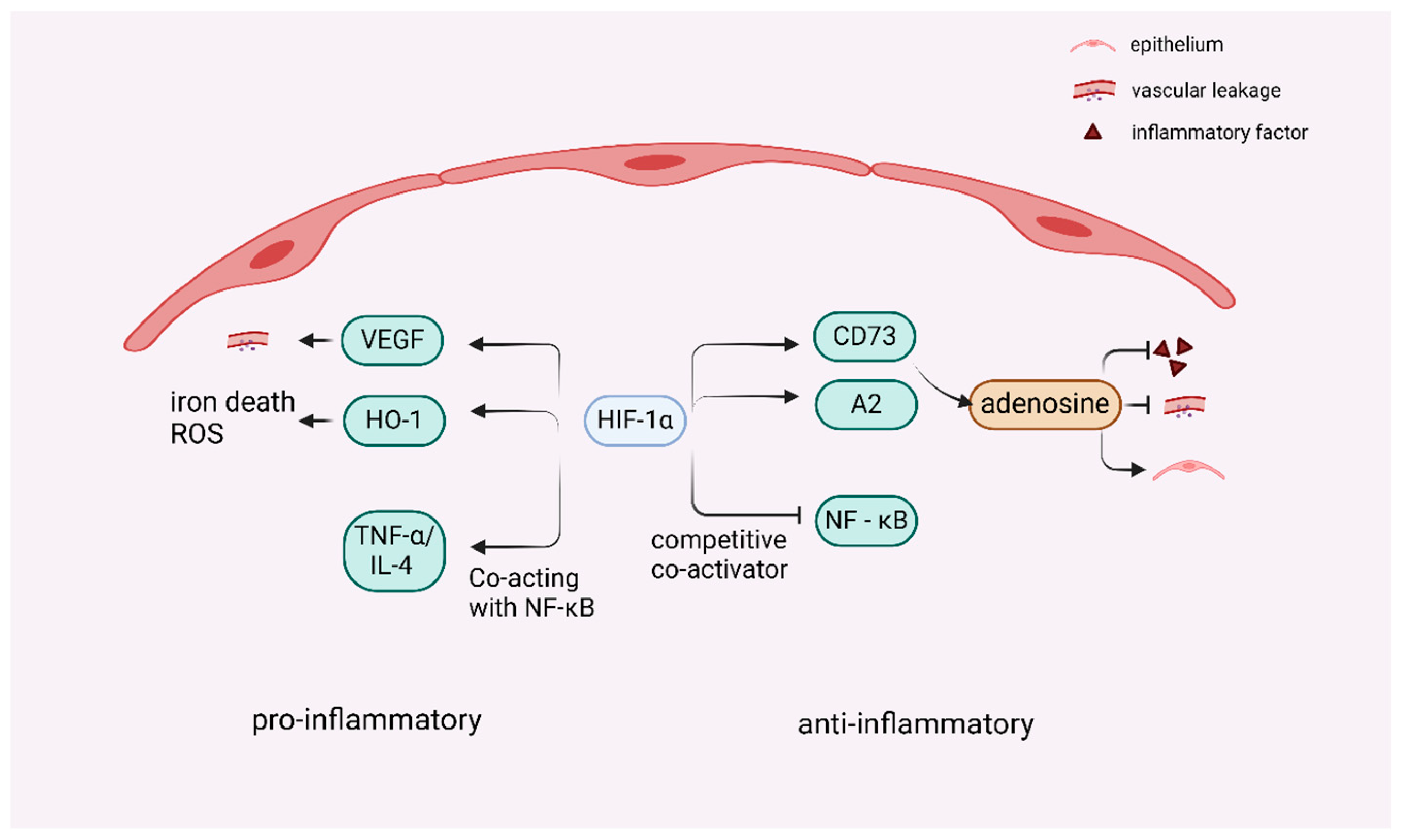
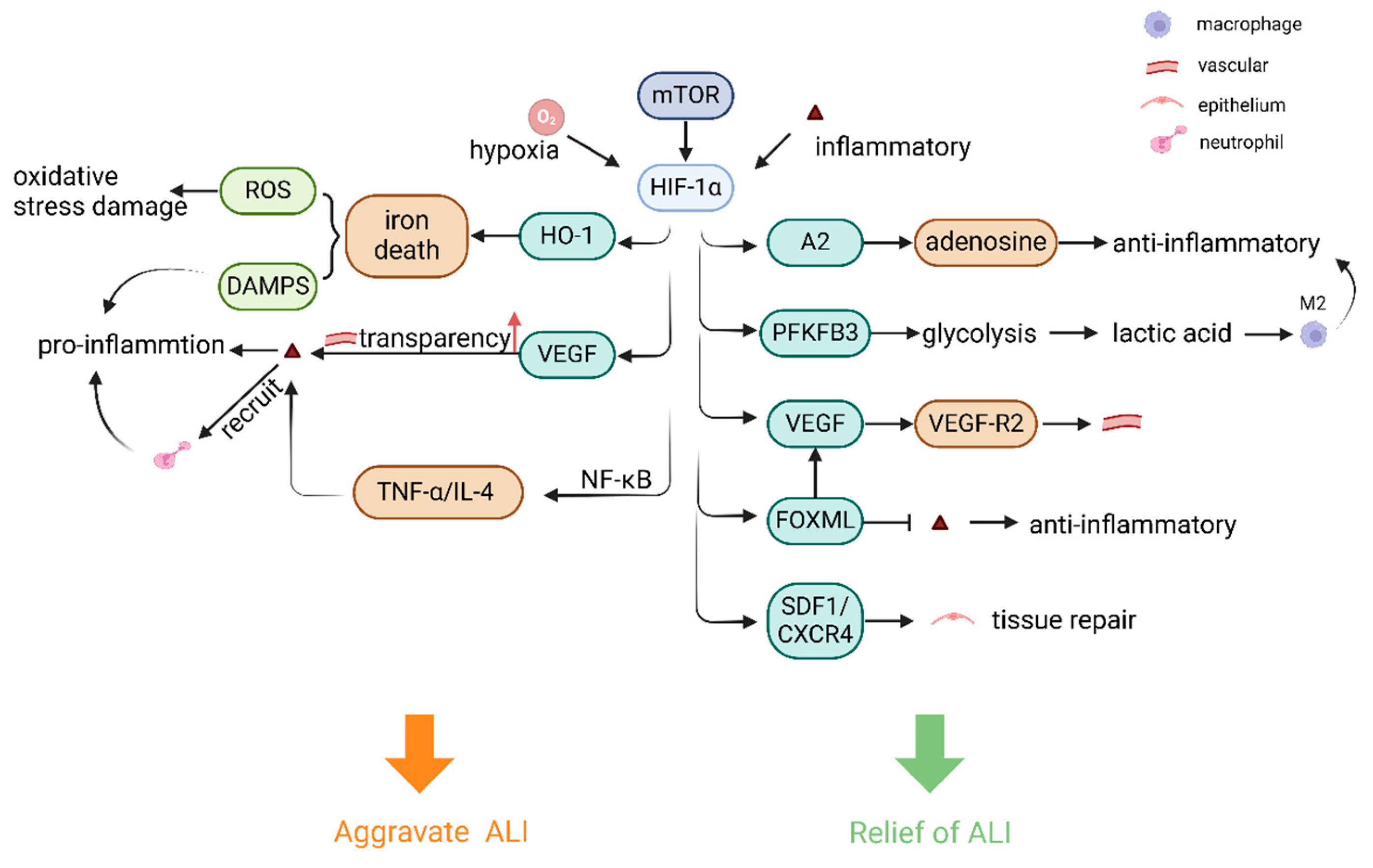
4. Regulation of HIF-1 in ALI
4.1. Dual Role of HIF-1 in ALI-Associated Inflammation
4.2. Regulation of Pulmonary Endothelial Cells and Vascular Remodeling by HIF-1 in ALI
4.3. Regulation of Lung Epithelial Cells and Lung Macrophages by HIF-1 in ALI
4.4. Regulation of Cell and Lung Tissue Repair by HIF-1 in ALI
4.5. Potential Influencing Factors and Drug Study of Targeting HIF-1 for ALI Treatment
5. Discussion
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALI | Acute lung injury |
| HIF | Hypoxia-inducible factor |
| ARDS | Acute respiratory distress syndrome |
| LECs | Lymphatic endothelial cells |
| AECs | Alveolar epithelial cells |
| GM-CSF | Granulocyte-macrophage stimulator |
| TBI | Traumatic brain injury |
| BRMs | Biological response modifiers |
| DAMPs | Damage-associated molecules |
| PRRs | Pattern recognition receptors |
| ATI cell | Alveolar type I epithelial cell |
| ATII cell | Alveolar type II epithelial cell |
| TNF-α | Tumor necrosis factor-alpha |
| IL-6 | Interleukin 6 |
| IL-1β | Interleukin-1β |
| IL-12 | Interleukin 12 |
| IL-10 | Interleukin 10 |
| AMs | Alveolar macrophages |
| IFN-γ | Interferon-γ |
| LPS | Lipopolysaccharide |
| iNOS | Inducible nitric oxide synthase |
| ROS | Reactive oxygen species |
| V/Q | ventilation-perfusion |
| PHD | Prolyl hydroxylase |
| VHL | Von Hippel-Lindau |
| HRE | Hypoxia response element |
| TADs | Transcriptional activation domains |
| VEGF | Vascular endothelial growth factor |
| PDGF | Platelet-derived growth factor |
| PI3K | Phosphatidylinositol 3-kinase |
| Akt | Protein kinase B |
| mTOR | mechanistic Target Of Rapamycin |
| A2AR | Adenosine A2A receptor |
| cAMP | cyclic AMP |
| A2BR | Adenosine A2B receptor |
| CD73 | Cluster of differentiation 73, ecto-5′-nucleotidase |
| HO-1 | Hemoglobin oxygenase-1 |
| ARA | Araloside A |
| FOXM1 | Forkhead box protein M1 |
| PAECs | Pulmonary artery endothelial cells |
| VEGFR2 | Endothelial growth factor receptor 2 |
| PFKFB | Phosphofructokinase-2/fructose-2,6-bisphosphatase |
| Arg-1 | Arginase-1 |
| SDF | Stromal cell derived factor |
| CXCR4 | C-X-C chemokine receptor type 4 |
| PF | Pulmonary fibrosis |
| EMT | Epithelial–mesenchymal transition |
| TGF-β1 | Transforming growth factor beta 1 |
| CCT6A | A member of the chaperone-containing TCP1 complex (CCT) |
| H2O2 | Hydrogen peroxide |
References
- Lv, H.; Liu, Q.; Wen, Z.; Feng, H.; Deng, X.; Ci, X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox Biol. 2017, 12, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhang, C.; Lv, N.; Liang, Z.; Ma, T.; Cheng, H.; Xia, Y.; Shi, L. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. Theranostics 2022, 12, 2928–2947. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Deng, S.-h.; Li, J.; Li, L.; Zhang, F.; Zou, Y.; Wu, D.-m.; Xu, Y. Obacunone alleviates ferroptosis during lipopolysaccharide-induced acute lung injury by upregulating Nrf2-dependent antioxidant responses. Cell. Mol. Biol. Lett. 2022, 27, 29. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zou, S.; Wang, B.; Liu, H. Targeting immunometabolism against acute lung injury. Clin. Immunol. 2023, 249, 109289. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Z. The role of macrophages polarization in sepsis-induced acute lung injury. Front. Immunol. 2023, 14, 1209438. [Google Scholar] [CrossRef]
- Fröhlich, S.; Boylan, J.; McLoughlin, P. Hypoxia-Induced Inflammation in the Lung. Am. J. Respir. Cell Mol. Biol. 2013, 48, 271–279. [Google Scholar] [CrossRef]
- Evans, C.E. Hypoxia-Inducible Factor Signaling in Inflammatory Lung Injury and Repair. Cells 2022, 11, 183. [Google Scholar] [CrossRef]
- Suresh, M.V.; Aggarwal, V.; Raghavendran, K. The Intersection of Pulmonary Vascular Disease and Hypoxia-Inducible Factors. Interv. Cardiol. Clin. 2023, 12, 443–452. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, D.; Liu, J.; Wang, M.; Duan, L.-J.; Liu, M.; Meng, H.; Zhuang, Y.; Wang, H.; Wang, Y.; et al. Prolonged hypoxia alleviates prolyl hydroxylation-mediated suppression of RIPK1 to promote necroptosis and inflammation. Nat. Cell Biol. 2023, 25, 950–962. [Google Scholar] [CrossRef]
- The Recovery Collaborative Group. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Pan, T.; Tuoerxun, T.; Chen, X.; Yang, C.-J.; Jiang, C.-Y.; Zhu, Y.-F.; Li, Z.-S.; Jiang, X.-Y.; Zhang, H.-T.; Zhang, H.; et al. The neutrophil elastase inhibitor, sivelestat, attenuates acute lung injury in patients with cardiopulmonary bypass. Front. Immunol. 2023, 14, 1082830. [Google Scholar] [CrossRef] [PubMed]
- Paine, R.I.; Standiford, T.J.; Dechert, R.E.; Moss, M.; Martin, G.S.; Rosenberg, A.L.; Thannickal, V.J.; Burnham, E.L.; Brown, M.B.; Hyzy, R.C. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit. Care Med. 2012, 40, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Kor, D.J. Statins and acute lung injury: Holy Grail or the next to fail? Crit. Care Med. 2012, 40, 1661–1663. [Google Scholar] [CrossRef] [PubMed]
- Ghadimi, K.; Cappiello, J.L.; Wright, M.C.; Levy, J.H.; Bryner, B.S.; DeVore, A.D.; Schroder, J.N.; Patel, C.B.; Rajagopal, S.; Shah, S.H.; et al. Inhaled Epoprostenol Compared with Nitric Oxide for Right Ventricular Support After Major Cardiac Surgery. Circulation 2023, 148, 1316–1329. [Google Scholar] [CrossRef]
- Peter, J.V.; John, P.; Graham, P.L.; Moran, J.L.; George, I.A.; Bersten, A. Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: Meta-analysis. BMJ 2008, 336, 1006–1009. [Google Scholar] [CrossRef]
- Abani, O.; Abbas, A.; Abbas, F.; Abbas, J.; Abbas, K.; Abbas, M.; Abbasi, S.; Abbass, H.; Abbott, A.; Abdallah, N.; et al. Higher dose corticosteroids in patients admitted to hospital with COVID-19 who are hypoxic but not requiring ventilatory support (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2023, 401, 1499–1507. [Google Scholar] [CrossRef]
- Iwata, K.; Doi, A.; Ohji, G.; Oka, H.; Oba, Y.; Takimoto, K.; Igarashi, W.; Gremillion, D.H.; Shimada, T. Effect of Neutrophil Elastase Inhibitor (Sivelestat Sodium) in the Treatment of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS): A Systematic Review and Meta-Analysis. Intern. Med. 2010, 49, 2423–2432. [Google Scholar] [CrossRef]
- Moore, B.B.; Coffey, M.J.; Christensen, P.; Sitterding, S.; Ngan, R.; Wilke, C.A.; McDonald, R.; Phare, S.M.; Peters-Golden, M.; Paine, R., III; et al. GM-CSF Regulates Bleomycin-Induced Pulmonary Fibrosis Via a Prostaglandin-Dependent Mechanism. J. Immunol. 2000, 165, 4032–4039. [Google Scholar] [CrossRef]
- The National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. Rosuvastatin for Sepsis-Associated Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2014, 370, 2191–2200. [Google Scholar] [CrossRef]
- Zhou, J.; Peng, Z.; Wang, J. Trelagliptin Alleviates Lipopolysaccharide (LPS)-Induced Inflammation and Oxidative Stress in Acute Lung Injury Mice. Inflammation 2021, 44, 1507–1517. [Google Scholar] [CrossRef]
- Grieco, D.L.; Menga, L.S.; Eleuteri, D.; Antonelli, M. Patient self-inflicted lung injury: Implications for acute hypoxemic respiratory failure and ARDS patients on non-invasive support. Minerva Anestesiol. 2019, 85, 1014–1023. [Google Scholar] [CrossRef]
- Nieman, G.F.; Gatto, L.A.; Habashi, N.M. Impact of mechanical ventilation on the pathophysiology of progressive acute lung injury. J. Appl. Physiol. 2015, 119, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Lin, H.; Zhang, A.; Cao, C.; Hu, X.; Zhang, J.; Wang, L.; Pan, X.; Zhu, Y.; Qian, F.; et al. N-phenethyl-5-phenylpicolinamide alleviates inflammation in acute lung injury by inhibiting HIF-1α/glycolysis/ASIC1a pathway. Life Sci. 2022, 309, 120987. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Butt, Y.; Kurdowska, A.; Allen, T.C. Acute Lung Injury: A Clinical and Molecular Review. Arch. Pathol. Lab. Med. 2016, 140, 345–350. [Google Scholar] [CrossRef]
- He, Y.-Q.; Zhou, C.-C.; Yu, L.-Y.; Wang, L.; Deng, J.-l.; Tao, Y.-L.; Zhang, F.; Chen, W.-S. Natural product derived phytochemicals in managing acute lung injury by multiple mechanisms. Pharmacol. Res. 2021, 163, 105224. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, S.; Yang, Y.; Yao, J.-Q.; Tang, W.-F.; Lyon, C.J.; Hu, T.Y.; Wan, M.-H. Extracellular vesicles in the pathogenesis and treatment of acute lung injury. Mil. Med. Res. 2022, 9, 61. [Google Scholar] [CrossRef]
- Long, M.E.; Mallampalli, R.K.; Horowitz, J.C. Pathogenesis of pneumonia and acute lung injury. Clin. Sci. 2022, 136, 747–769. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Na, E.; Allen, E.; Baird, L.A.; Odom, C.V.; Korkmaz, F.T.; Shenoy, A.T.; Matschulat, A.M.; Jones, M.R.; Kotton, D.N.; Mizgerd, J.P.; et al. Epithelial LIF signaling limits apoptosis and lung injury during bacterial pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L550–L563. [Google Scholar] [CrossRef]
- Zhao, J.; Zhen, N.; Zhou, Q.; Lou, J.; Cui, W.; Zhang, G.; Tian, B. NETs Promote Inflammatory Injury by Activating cGAS-STING Pathway in Acute Lung Injury. Int. J. Mol. Sci. 2023, 24, 5125. [Google Scholar] [CrossRef] [PubMed]
- Störmann, P.; Lustenberger, T.; Relja, B.; Marzi, I.; Wutzler, S. Role of biomarkers in acute traumatic lung injury. Injury 2017, 48, 2400–2406. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, G.; Li, Z.; Chen, R.; Wu, H.; Li, H.; Shen, C.; Deng, M.; Hao, Z.; Wu, S.; et al. Accurate identification of traumatic lung injury (TLI) by ATR-FTIR spectroscopy combined with chemometrics. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 288, 122186. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Jung, H.; Youn, D.H.; Kim, T.Y.; Han, S.W.; Kim, B.J.; Lee, J.J.; Jeon, J.P. Mild Traumatic Brain Injury and Subsequent Acute Pulmonary Inflammatory Response. J. Korean Neurosurg. Soc. 2022, 65, 680–687. [Google Scholar] [CrossRef]
- Zhang, C.-N.; Li, F.-J.; Zhao, Z.-L.; Zhang, J.-N. The role of extracellular vesicles in traumatic brain injury-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L885–L891. [Google Scholar] [CrossRef]
- Tung, J.-P.; Chiaretti, S.; Dean, M.M.; Sultana, A.J.; Reade, M.C.; Fung, Y.L. Transfusion-related acute lung injury (TRALI): Potential pathways of development, strategies for prevention and treatment, and future research directions. Blood Rev. 2022, 53, 100926. [Google Scholar] [CrossRef]
- Yu, Y.; Lian, Z. Update on transfusion-related acute lung injury: An overview of its pathogenesis and management. Front. Immunol. 2023, 14, 1175387. [Google Scholar] [CrossRef]
- Li, X.; Jamal, M.; Guo, P.; Jin, Z.; Zheng, F.; Song, X.; Zhan, J.; Wu, H. Irisin alleviates pulmonary epithelial barrier dysfunction in sepsis-induced acute lung injury via activation of AMPK/SIRT1 pathways. Biomed. Pharmacother. 2019, 118, 109363. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Mutlu, G.M. Alveolar Epithelial Cells Burn Fat to Survive Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2019, 60, 135–136. [Google Scholar] [CrossRef]
- Qiao, X.; Yin, J.; Zheng, Z.; Li, L.; Feng, X. Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: Pathogenesis and therapeutic implications. Cell Commun. Signal. 2024, 22, 241. [Google Scholar] [CrossRef]
- Orfanos, S.E.; Mavrommati, I.; Korovesi, I.; Roussos, C. Pulmonary endothelium in acute lung injury: From basic science to the critically ill. Intensive Care Med. 2004, 30, 1702–1714. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-H.; Jiang, H.-L.; Tao, J.-H.; Zhang, C.-Y.; Xiong, J.-B.; Yang, J.-T.; Liu, Y.-B.; Zhong, W.-J.; Guan, X.-X.; Duan, J.-X.; et al. Mitochondrial citrate accumulation drives alveolar epithelial cell necroptosis in lipopolysaccharide-induced acute lung injury. Exp. Mol. Med. 2022, 54, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Yazicioglu, T.; Mühlfeld, C.; Autilio, C.; Huang, C.-K.; Bär, C.; Dittrich-Breiholz, O.; Thum, T.; Pérez-Gil, J.; Schmiedl, A.; Brandenberger, C. Aging impairs alveolar epithelial type II cell function in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L755–L769. [Google Scholar] [CrossRef] [PubMed]
- Grommes, J.; Soehnlein, O. Contribution of Neutrophils to Acute Lung Injury. Mol. Med. 2010, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Burkard, P.; Schonhart, C.; Vögtle, T.; Köhler, D.; Tang, L.; Johnson, D.; Hemmen, K.; Heinze, K.G.; Zarbock, A.; Hermanns, H.M.; et al. A key role for platelet GPVI in neutrophil recruitment, migration, and NETosis in the early stages of acute lung injury. Blood 2023, 142, 1463–1477. [Google Scholar] [CrossRef]
- Liu, P.-Y.; Chen, C.-Y.; Lin, Y.-L.; Lin, C.-M.; Tsai, W.-C.; Tsai, Y.-L.; Lin, G.-J.; Chen, Y.-G.; Wang, S.-Y.; Sun, R.-N.; et al. RNF128 regulates neutrophil infiltration and myeloperoxidase functions to prevent acute lung injury. Cell Death Dis. 2023, 14, 369. [Google Scholar] [CrossRef]
- Rehring, J.F.; Bui, T.M.; Galán-Enríquez, C.S.; Urbanczyk, J.M.; Ren, X.; Wiesolek, H.L.; Sullivan, D.P.; Sumagin, R. Released Myeloperoxidase Attenuates Neutrophil Migration and Accumulation in Inflamed Tissue. Front. Immunol. 2021, 12, 654259. [Google Scholar] [CrossRef]
- Chen, Q.; Shao, X.; He, Y.; Lu, E.; Zhu, L.; Tang, W. Norisoboldine Attenuates Sepsis-Induced Acute Lung Injury by Modulating Macrophage Polarization via PKM2/HIF-1α/PGC-1α Pathway. Biol. Pharm. Bull. 2021, 44, 1536–1547. [Google Scholar] [CrossRef]
- Kulshrestha, R.; Dhanda, H.; Pandey, A.; Singh, A.; Kumar, R. Immunopathogenesis and therapeutic potential of macrophage influx in diffuse parenchymal lung diseases. Expert. Rev. Respir. Med. 2020, 14, 917–928. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1—M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Johnston, L.K.; Rims, C.R.; Gill, S.E.; McGuire, J.K.; Manicone, A.M. Pulmonary Macrophage Subpopulations in the Induction and Resolution of Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 417–426. [Google Scholar] [CrossRef]
- Wang, L.; Wang, D.; Zhang, T.; Ma, Y.; Tong, X.; Fan, H. The role of immunometabolism in macrophage polarization and its impact on acute lung injury/acute respiratory distress syndrome. Front. Immunol. 2023, 14, 1117548. [Google Scholar] [CrossRef]
- Wang, R.; Ma, S.; Yang, J.; Luo, K.; Qian, Q.; Pan, J.; Liang, K.; Wang, Y.; Gao, Y.; Li, M. Sodium Hydrosulfide Protects Rats from Hypobaric-Hypoxia-Induced Acute Lung Injury. Int. J. Mol. Sci. 2024, 25, 10734. [Google Scholar] [CrossRef] [PubMed]
- Jahani, M.; Dokaneheifard, S.; Mansouri, K. Hypoxia: A key feature of COVID-19 launching activation of HIF-1 and cytokine storm. J. Inflamm. 2020, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhao, Z.; Chao, Y.; Chen, D.; Chen, H.; Zhang, R.; Liu, S.; Xie, J.; Yang, Y.; Qiu, H.; et al. Effects of early versus delayed application of prone position on ventilation–perfusion mismatch in patients with acute respiratory distress syndrome: A prospective observational study. Crit. Care 2023, 27, 462. [Google Scholar] [CrossRef] [PubMed]
- Mirchandani, A.S.; Jenkins, S.J.; Bain, C.C.; Sanchez-Garcia, M.A.; Lawson, H.; Coelho, P.; Murphy, F.; Griffith, D.M.; Zhang, A.; Morrison, T.; et al. Hypoxia shapes the immune landscape in lung injury and promotes the persistence of inflammation. Nat. Immunol. 2022, 23, 927–939. [Google Scholar] [CrossRef]
- Pu, X.; Li, F.; Lin, X.; Wang, R.; Chen, Z. Oxidative stress and expression of inflammatory factors in lung tissue of acute mountain sickness rats. Mol. Med. Rep. 2021, 25, 49. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 Transcription Factors—Similar but Not Identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Cowman, S.J.; Koh, M.Y. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer 2022, 8, 28–42. [Google Scholar] [CrossRef]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef]
- Yfantis, A.; Mylonis, I.; Chachami, G.; Nikolaidis, M.; Amoutzias, G.D.; Paraskeva, E.; Simos, G. Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators. Cells 2023, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-C.; Yang, Y.-H.; Liao, C.-C.; Lee, H.-C. Xanthoxylin Attenuates Lipopolysaccharide-Induced Lung Injury through Modulation of Akt/HIF-1α/NF-κB and Nrf2 Pathways. Int. J. Mol. Sci. 2024, 25, 8742. [Google Scholar] [CrossRef] [PubMed]
- Koval, M.; Tang, M.; Tian, Y.; Li, D.; Lv, J.; Li, Q.; Kuang, C.; Hu, P.; Wang, Y.; Wang, J.; et al. TNF-α Mediated Increase of HIF-1α Inhibits VASP Expression, Which Reduces Alveolar-Capillary Barrier Function during Acute Lung Injury (ALI). PLoS ONE 2014, 9, e102967. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Suresh, M.V.; Aktay, S.; Yalamanchili, G.; Solanki, S.; Sathyarajan, D.T.; Arnipalli, M.S.; Pennathur, S.; Raghavendran, K. Role of succinate in airway epithelial cell regulation following traumatic lung injury. JCI Insight 2023, 8, 166860. [Google Scholar] [CrossRef]
- Xin, Y.; Zhao, L.; Peng, R. HIF-1 signaling: An emerging mechanism for mitochondrial dynamics. J. Physiol. Biochem. 2023, 79, 489–500. [Google Scholar] [CrossRef]
- Yang, C.; Zhong, Z.-F.; Wang, S.-P.; Vong, C.-T.; Yu, B.; Wang, Y.-T. HIF-1: Structure, biology and natural modulators. Chin. J. Nat. Med. 2021, 19, 521–527. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, X.; Lingappan, K. Role of HIF-1α-miR30a-Snai1 Axis in Neonatal Hyperoxic Lung Injury. Oxidative Med. Cell. Longev. 2019, 2019, 8327486. [Google Scholar] [CrossRef] [PubMed]
- Devraj, G.; Beerlage, C.; Brüne, B.; Kempf, V.A.J. Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect. 2017, 19, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Ruan, H.; Zhang, Q.; Zhang, Y.-P.; Li, S.-S.; Ran, X. Unraveling the role of HIF-1α in sepsis: From pathophysiology to potential therapeutics—A narrative review. Crit. Care 2024, 28, 100. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiong, W.; Li, C.; Zhao, R.; Lu, H.; Song, S.; Zhou, Y.; Hu, Y.; Shi, B.; Ge, J. Hypoxia-induced signaling in the cardiovascular system: Pathogenesis and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 431. [Google Scholar] [CrossRef]
- Tang, J.; Xu, L.; Zeng, Y.; Gong, F. Effect of gut microbiota on LPS-induced acute lung injury by regulating the TLR4/NF-kB signaling pathway. Int. Immunopharmacol. 2021, 91, 107272. [Google Scholar] [CrossRef]
- Luo, L.; Huang, F.; Zhong, S.; Ding, R.; Su, J.; Li, X. Astaxanthin attenuates ferroptosis via Keap1-Nrf2/HO-1 signaling pathways in LPS-induced acute lung injury. Life Sci. 2022, 311, 121091. [Google Scholar] [CrossRef]
- Liu, Y.; Shang, L.; Zhou, J.; Pan, G.; Zhou, F.; Yang, S. Emodin Attenuates LPS-Induced Acute Lung Injury by Inhibiting NLRP3 Inflammasome-Dependent Pyroptosis Signaling Pathway in vitro and in vivo. Inflammation 2021, 45, 753–767. [Google Scholar] [CrossRef]
- Deng, H.; Chen, Y.; Li, P.; Hang, Q.; Zhang, P.; Jin, Y.; Chen, M. PI3K/AKT/mTOR pathway, hypoxia, and glucose metabolism: Potential targets to overcome radioresistance in small cell lung cancer. Cancer Pathog. Ther. 2023, 1, 56–66. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef]
- Liu, Y.; Xiang, D.; Zhang, H.; Yao, H.; Wang, Y.; Barreto, E. Hypoxia-Inducible Factor-1: A Potential Target to Treat Acute Lung Injury. Oxidative Med. Cell. Longev. 2020, 2020, 8871476. [Google Scholar] [CrossRef]
- Eckle, T.; Brodsky, K.; Bonney, M.; Packard, T.; Han, J.; Borchers, C.H.; Mariani, T.J.; Kominsky, D.J.; Mittelbronn, M.; Eltzschig, H.K. HIF1A Reduces Acute Lung Injury by Optimizing Carbohydrate Metabolism in the Alveolar Epithelium. PLOS Biol. 2013, 11, e1001665. [Google Scholar] [CrossRef]
- Millar, M.W.; Fazal, F.; Rahman, A. Therapeutic Targeting of NF-κB in Acute Lung Injury: A Double-Edged Sword. Cells 2022, 11, 3317. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Kewley, E.M.; Brodsky, K.S.; Tak, E.; Bonney, S.; Gobel, M.; Anderson, D.; Glover, L.E.; Riegel, A.K.; Colgan, S.P.; et al. Identification of Hypoxia-Inducible Factor HIF-1A as Transcriptional Regulator of the A2B Adenosine Receptor during Acute Lung Injury. J. Immunol. 2014, 192, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Morote-Garcia, J.C.; Rosenberger, P.; Kuhlicke, J.; Eltzschig, H.K. HIF-1–dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 2008, 111, 5571–5580. [Google Scholar] [CrossRef] [PubMed]
- Vohwinkel, C.U.; Coit, E.J.; Burns, N.; Elajaili, H.; Hernandez-Saavedra, D.; Yuan, X.; Eckle, T.; Nozik, E.; Tuder, R.M.; Eltzschig, H.K. Targeting alveolar-specific succinate dehydrogenase A attenuates pulmonary inflammation during acute lung injury. FASEB J. 2021, 35, e21468. [Google Scholar] [CrossRef]
- Fang, Y.; Xiao, C.; Wang, L.; Wang, Y.; Zeng, J.; Liang, Y.; Huang, R.; Shi, Y.; Wu, S.; Du, X.; et al. Synergistic Enhancement of Isoforskolin and Dexamethasone Against Sepsis and Acute Lung Injury Mouse Models. J. Inflamm. Res. 2023, 16, 5989–6001. [Google Scholar] [CrossRef]
- Halpin-Veszeleiova, K.; Hatfield, S.M. Therapeutic Targeting of Hypoxia-A2-Adenosinergic Pathway in COVID-19 Patients. Physiology 2022, 37, 46–52. [Google Scholar] [CrossRef]
- Cai, J.; Wang, Y.-l.; Sheng, X.-d.; Zhang, L.; Lv, X. Shufeng Jiedu capsule inhibits inflammation and apoptosis by activating A2AAR and inhibiting NF-κB to alleviate LPS-induced ALI. J. Ethnopharmacol. 2022, 298, 115661. [Google Scholar] [CrossRef]
- Hatfield, S.; Belikoff, B.; Lukashev, D.; Sitkovsky, M.; Ohta, A. The antihypoxia–adenosinergic pathogenesis as a result of collateral damage by overactive immune cells. J. Leukoc. Biol. 2009, 86, 545–548. [Google Scholar] [CrossRef]
- Li, X.; Berg, N.K.; Mills, T.; Zhang, K.; Eltzschig, H.K.; Yuan, X. Adenosine at the Interphase of Hypoxia and Inflammation in Lung Injury. Front. Immunol. 2021, 11, 604944. [Google Scholar] [CrossRef]
- Li, X.; Shan, C.; Wu, Z.; Yu, H.; Yang, A.; Tan, B. Emodin alleviated pulmonary inflammation in rats with LPS-induced acute lung injury through inhibiting the mTOR/HIF-1α/VEGF signaling pathway. Inflamm. Res. 2020, 69, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Song, S.; Wang, Y.; Wu, K.; Liang, G.; Wang, A.; Xu, X. Esketamine alleviates ferroptosis-mediated acute lung injury by modulating the HIF-1α/HO-1 pathway. Int. Immunopharmacol. 2024, 142, 113065. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Meng, X.; Yang, W.; Wang, J.; Zhang, J.; Tian, R.; Wang, R.; Su, Q.; Jin, W. HIF-1α promotes paraquat induced acute lung injury and implicates a role NF-κB and Rac2 activity. Toxicology 2023, 483, 153388. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hu, R.; Sun, L.; Chai, D.; Cao, Z.; Li, Q. Critical Role of Toll-Like Receptor 4 in Hypoxia-Inducible Factor 1α Activation During Trauma/Hemorrhagic Shock–Induced Acute Lung Injury After Lymph Infusion in Mice. Shock 2014, 42, 271–278. [Google Scholar] [CrossRef]
- Shang, L.; Zhang, M.; Li, J.; Zhou, F.; Wang, S.; Chen, L.; Yang, S. Dachengqi decoction alleviates acute lung injury by suppressing HIF-1α-mediated glycolysis. J. Ethnopharmacol. 2024, 321, 117410. [Google Scholar] [CrossRef]
- Ma, J.; Wang, J.; Wang, J.; Zhou, J.; Jiang, C.; Chen, W.; Zhang, X.; Pan, J.; Zhu, J.; Chen, M. Araloside A alleviates sepsis-induced acute lung injury via PHD2/HIF-1α in macrophages. Phytomedicine 2024, 135, 156089. [Google Scholar] [CrossRef]
- Suresh, M.V.; Yalamanchili, G.; Rao, T.C.; Aktay, S.; Kralovich, A.; Shah, Y.M.; Raghavendran, K. Hypoxia-inducible factor (HIF)-1α-induced regulation of lung injury in pulmonary aspiration is mediated through NF-kB. FASEB BioAdvances 2022, 4, 309–328. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2015, 283, 413–424. [Google Scholar] [CrossRef]
- Zhou, Q.; Gong, X.; Kuang, G.; Jiang, R.; Xie, T.; Tie, H.; Chen, X.; Li, K.; Wan, J.; Wang, B. Ferulic Acid Protected from Kidney Ischemia Reperfusion Injury in Mice: Possible Mechanism Through Increasing Adenosine Generation via HIF-1α. Inflammation 2018, 41, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, P.; Goodwin, A.J.; Cook, J.A.; Halushka, P.V.; Chang, E.; Zingarelli, B.; Fan, H. Exosomes from endothelial progenitor cells improve outcomes of the lipopolysaccharide-induced acute lung injury. Crit. Care 2019, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Borek, I.; Birnhuber, A.; Voelkel, N.F.; Marsh, L.M.; Kwapiszewska, G. The vascular perspective on acute and chronic lung disease. J. Clin. Investig. 2023, 133, e170502. [Google Scholar] [CrossRef] [PubMed]
- Rafat, N.; Tönshoff, B.; Bierhaus, A.; Beck, G.C. Endothelial Progenitor Cells in Regeneration after Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2013, 48, 399–405. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, X.; Zhao, D.X.; Yin, J.; Hu, G.; Evans, C.E.; Zhao, Y.-Y. Endothelial Hypoxia-Inducible Factor-1α Is Required for Vascular Repair and Resolution of Inflammatory Lung Injury through Forkhead Box Protein M1. Am. J. Pathol. 2019, 189, 1664–1679. [Google Scholar] [CrossRef]
- Ding, Z.; Zhong, R.; Yang, Y.; Xia, T.; Wang, W.; Wang, Y.; Xing, N.; Luo, Y.; Li, S.; Shang, L.; et al. Systems pharmacology reveals the mechanism of activity of Ge-Gen-Qin-Lian decoction against LPS-induced acute lung injury: A novel strategy for exploring active components and effective mechanism of TCM formulae. Pharmacol. Res. 2020, 156, 104759. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Miyatake, S.-I.; Fukumoto, M.; Furuse, M.; Hiramatsu, R.; Kawabata, S.; Kuroiwa, T.; Tsuji, M.; Fukumoto, M.; Ono, K. The distribution of vascular endothelial growth factor-producing cells in clinical radiation necrosis of the brain: Pathological consideration of their potential roles. J. Neuro-Oncol. 2011, 105, 423–431. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2020, 599, 23–37. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, J.; Zhang, R.; Zhang, M.; Cui, H.; Wang, L.; Cui, Y.; Wang, W.; Sun, Y.; Wang, C. Targeting Endothelial ENO1 (Alpha-Enolase)-PI3K-Akt-mTOR Axis Alleviates Hypoxic Pulmonary Hypertension. Hypertension 2023, 80, 1035–1047. [Google Scholar] [CrossRef]
- Fan, J.; Lv, H.; Li, J.; Che, Y.; Xu, B.; Tao, Z.; Jiang, W. Roles of Nrf2/HO-1 and HIF-1α/VEGF in lung tissue injury and repair following cerebral ischemia/reperfusion injury. J. Cell. Physiol. 2019, 234, 7695–7707. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Kadota, T.; Kaneko, R.; Hirano, Y.; Fujimoto, S.; Watanabe, N.; Kizawa, R.; Ohtsuka, T.; Kuwano, K.; Ochiya, T.; et al. Mitigation of acute lung injury by human bronchial epithelial cell-derived extracellular vesicles via ANXA1-mediated FPR signaling. Commun. Biol. 2024, 7, 514. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Yuan, Y.; He, Y.; Wasti, B.; Duan, W.; Chen, Z.; Li, D.; Sun, W.; Zeng, Q.; Ma, L.; et al. Inhibition of METTL3 alleviated LPS-induced alveolar epithelial cell apoptosis and acute lung injury via restoring neprilysin expression. Life Sci. 2023, 333, 122148. [Google Scholar] [CrossRef] [PubMed]
- Vohwinkel, C.U.; Burns, N.; Coit, E.; Yuan, X.; Vladar, E.K.; Sul, C.; Schmidt, E.P.; Carmeliet, P.; Stenmark, K.; Nozik, E.S.; et al. HIF1A-dependent induction of alveolar epithelial PFKFB3 dampens acute lung injury. JCI Insight 2022, 7, 157855. [Google Scholar] [CrossRef]
- Feng, Z.; Jing, Z.; Li, Q.; Chu, L.; Jiang, Y.; Zhang, X.; Yan, L.; Liu, Y.; Jiang, J.; Xu, P.; et al. Exosomal STIMATE derived from type II alveolar epithelial cells controls metabolic reprogramming of tissue-resident alveolar macrophages. Theranostics 2023, 13, 991–1009. [Google Scholar] [CrossRef]
- Roy, R.M.; Allawzi, A.; Burns, N.; Sul, C.; Rubio, V.; Graham, J.; Stenmark, K.; Nozik, E.S.; Tuder, R.M.; Vohwinkel, C.U. Lactate produced by alveolar type II cells suppresses inflammatory alveolar macrophages in acute lung injury. FASEB J. 2023, 37, e23316. [Google Scholar] [CrossRef]
- Evans, C.E.; Peng, Y.; Zhu, M.M.; Dai, Z.; Zhang, X.; Zhao, Y.-Y. Rabeprazole Promotes Vascular Repair and Resolution of Sepsis-Induced Inflammatory Lung Injury through HIF-1α. Cells 2022, 11, 1425. [Google Scholar] [CrossRef]
- Yeh, C.H.; Cho, W.; So, E.C.; Chu, C.C.; Lin, M.C.; Wang, J.J.; Hsing, C.H. Propofol inhibits lipopolysaccharide-induced lung epithelial cell injury by reducing hypoxia-inducible factor-1α expression. Br. J. Anaesth. 2011, 106, 590–599. [Google Scholar] [CrossRef]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 14959. [Google Scholar] [CrossRef]
- Du, J.; Zhu, Y.; Meng, X.; Xie, H.; Wang, J.; Zhou, Z.; Wang, R. Atorvastatin attenuates paraquat poisoning-induced epithelial-mesenchymal transition via downregulating hypoxia-inducible factor-1 alpha. Life Sci. 2018, 213, 126–133. [Google Scholar] [CrossRef]
- Yan, P.; Yang, K.; Xu, M.; Zhu, M.; Duan, Y.; Li, W.; Liu, L.; Liang, C.; Li, Z.; Pan, X.; et al. CCT6A alleviates pulmonary fibrosis by inhibiting HIF-1α-mediated lactate production. J. Mol. Cell Biol. 2024, 16, mjae021. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef] [PubMed]
- Galani, V.; Tatsaki, E.; Bai, M.; Kitsoulis, P.; Lekka, M.; Nakos, G.; Kanavaros, P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): An up-to-date cell-specific review. Pathol. Res. Pract. 2010, 206, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.N.; Yang, M.; Wu, B.; Huo, Y.; Xu, W. The long non-coding RNA mir155hg promotes NLRP3-inflammasome activation and oxidative stress response in acute lung injury by targeting miR-450b-5p to regulate HIF-1α. Free Radic. Biol. Med. 2024, 222, 638–649. [Google Scholar] [CrossRef]
- Ålgars, A.; Karikoski, M.; Yegutkin, G.G.; Stoitzner, P.; Niemelä, J.; Salmi, M.; Jalkanen, S. Different role of CD73 in leukocyte trafficking via blood and lymph vessels. Blood 2011, 117, 4387–4393. [Google Scholar] [CrossRef]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- McClendon, J.; Jansing, N.L.; Redente, E.F.; Gandjeva, A.; Ito, Y.; Colgan, S.P.; Ahmad, A.; Riches, D.W.H.; Chapman, H.A.; Mason, R.J.; et al. Hypoxia-Inducible Factor 1α Signaling Promotes Repair of the Alveolar Epithelium after Acute Lung Injury. Am. J. Pathol. 2017, 187, 1772–1786. [Google Scholar] [CrossRef]
- Luo, L.; Shaver, C.M.; Zhao, Z.; Koyama, T.; Calfee, C.S.; Bastarache, J.A.; Ware, L.B. Clinical Predictors of Hospital Mortality Differ Between Direct and Indirect ARDS. Chest 2017, 151, 755–763. [Google Scholar] [CrossRef]
- Calfee, C.S.; Janz, D.R.; Bernard, G.R.; May, A.K.; Kangelaris, K.N.; Matthay, M.A.; Ware, L.B. Distinct Molecular Phenotypes of Direct vs Indirect ARDS in Single-Center and Multicenter Studies. Chest 2015, 147, 1539–1548. [Google Scholar] [CrossRef]
- Matthay, M.A.; Arabi, Y.M.; Siegel, E.R.; Ware, L.B.; Bos, L.D.J.; Sinha, P.; Beitler, J.R.; Wick, K.D.; Curley, M.A.Q.; Constantin, J.-M.; et al. Phenotypes and personalized medicine in the acute respiratory distress syndrome. Intensive Care Med. 2020, 46, 2136–2152. [Google Scholar] [CrossRef]
- Beitler, J.R.; Goligher, E.C.; Schmidt, M.; Spieth, P.M.; Zanella, A.; Martin-Loeches, I.; Calfee, C.S.; Cavalcanti, A.B.; The ARDSne(x)t Investigators. Personalized medicine for ARDS: The 2035 research agenda. Intensive Care Med. 2016, 42, 756–767. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, S.; Zuo, A.; Zhang, J.; Wen, W.; Jiang, W.; Chen, H.; Liang, D.; Sun, J.; Wang, M. HIF-1α/JMJD1A signaling regulates inflammation and oxidative stress following hyperglycemia and hypoxia-induced vascular cell injury. Cell. Mol. Biol. Lett. 2021, 26, 40. [Google Scholar] [CrossRef] [PubMed]
- Gautam, Y.; Afanador, Y.; Abebe, T.; López, J.E.; Mersha, T.B. Genome-wide analysis revealed sex-specific gene expression in asthmatics. Hum. Mol. Genet. 2019, 28, 2600–2614. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, K.S.; Park, S.J.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Hong, S.H.; Koh, G.Y.; Lee, Y.C. Angiopoietin-1 variant, COMP-Ang1 attenuates hydrogen peroxide-induced acute lung injury. Exp. Mol. Med. 2008, 40, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Tojo, K.; Tamada, N.; Nagamine, Y.; Yazawa, T.; Ota, S.; Goto, T. Enhancement of glycolysis by inhibition of oxygen-sensing prolyl hydroxylases protects alveolar epithelial cells from acute lung injury. FASEB J. 2018, 32, 2258–2268. [Google Scholar] [CrossRef]
- Wing, P.A.C.; Keeley, T.P.; Zhuang, X.; Lee, J.Y.; Prange-Barczynska, M.; Tsukuda, S.; Morgan, S.B.; Harding, A.C.; Argles, I.L.A.; Kurlekar, S.; et al. Hypoxic and pharmacological activation of HIF inhibits SARS-CoV-2 infection of lung epithelial cells. Cell Rep. 2021, 35, 109020. [Google Scholar] [CrossRef]
- Lv, Y.-W.; Du, Y.; Ma, S.-S.; Shi, Y.-C.; Xu, H.-C.; Deng, L.; Chen, X.-Y. Proanthocyanidins attenuates ferroptosis against influenza-induced acute lung injury in mice by reducing IFN-γ. Life Sci. 2023, 314, 121279. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.; Wang, Y.; Wang, J.; Sang, A.; Song, X.; Li, X. YAP1 alleviates sepsis-induced acute lung injury via inhibiting ferritinophagy-mediated ferroptosis. Front. Immunol. 2022, 13, 884362. [Google Scholar] [CrossRef]
- Nguyen, N.; Xu, S.; Lam, T.Y.W.; Liao, W.; Wong, W.S.F.; Ge, R. ISM1 suppresses LPS-induced acute lung injury and post-injury lung fibrosis in mice. Mol. Med. 2022, 28, 72. [Google Scholar] [CrossRef]
- De Ponti, C.; Carini, R.; Alchera, E.; Nitti, M.P.; Locati, M.; Albano, E.; Cairo, G.; Tacchini, L. Adenosine A2a receptor-mediated, normoxic induction of HIF-1 through PKC and PI-3K-dependent pathways in macrophages. J. Leukoc. Biol. 2007, 82, 392–402. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Gong, L.; Zhang, Y.; Dong, S.; Shi, J.; Li, C.; Li, Y.; Zhang, Y.; Li, H. Heme oxygenase-1(HO-1) regulates Golgi stress and attenuates endotoxin-induced acute lung injury through hypoxia inducible factor-1α (HIF-1α)/HO-1 signaling pathway. Free Radic. Biol. Med. 2021, 165, 243–253. [Google Scholar] [CrossRef]
- Shi, J.; Yu, T.; Song, K.; Du, S.; He, S.; Hu, X.; Li, X.; Li, H.; Dong, S.; Zhang, Y.; et al. Dexmedetomidine ameliorates endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the HIF-1a/HO-1 signaling pathway. Redox Biol. 2021, 41, 101954. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Wu, G.; Han, S.; Li, Z.; Jia, Y.; Bai, L.; Li, X.; Wang, K.; Yang, F.; Zhang, J.; et al. Hypoxia-inducible factor prolyl-hydroxylase inhibitor roxadustat (FG-4592) alleviates sepsis-induced acute lung injury. Respir. Physiol. Neurobiol. 2020, 281, 103506. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, J.; Zhao, T.; Sun, M.; Xu, M.; Che, S.; Pan, Z.; Wu, C.; Shen, L. Polystyrenenanoplastics lead to ferroptosis in the lungs. J. Adv. Res. 2024, 56, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Liu, Z.; Li, M.; Guo, J.; Chen, L.; Ding, L.; Ding, X.; Zhou, T.; Zhang, J. Polysaccharide from Potentilla anserina L ameliorate pulmonary edema induced by hypobaric hypoxia in rats. Biomed. Pharmacother. 2021, 139, 111669. [Google Scholar] [CrossRef]
- Armstrong, S.M.; Mubareka, S.; Lee, W.L. The lung microvascular endothelium as a therapeutic target in severe influenza. Antivir. Res. 2013, 99, 113–118. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, J.; Liu, P.; Zhang, R.-J.; Li, J.-D.; Bi, Y.-H.; Li, Y. Regulatory role of ncRNAs in pulmonary epithelial and endothelial barriers: Molecular therapy clues of influenza-induced acute lung injury. Pharmacol. Res. 2022, 185, 106509. [Google Scholar] [CrossRef]
- Mirza, M.K.; Sun, Y.; Zhao, Y.D.; Potula, H.-H.S.; Frey, R.S.; Vogel, S.M.; Malik, A.B.; Zhao, Y.-Y. FoxM1 regulates re-annealing of endothelial adherens junctions through transcriptional control of β-catenin expression. J. Exp. Med. 2010, 207, 1675–1685. [Google Scholar] [CrossRef]
- Luo, Z.; Tian, M.; Yang, G.; Tan, Q.; Chen, Y.; Li, G.; Zhang, Q.; Li, Y.; Wan, P.; Wu, J. Hypoxia signaling in human health and diseases: Implications and prospects for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 218. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef]
- LaCanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef]
- Bayry, J.; Errea, A.; Cayet, D.; Marchetti, P.; Tang, C.; Kluza, J.; Offermanns, S.; Sirard, J.-C.; Rumbo, M. Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PLoS ONE 2016, 11, e0163694. [Google Scholar] [CrossRef]
- Mould, K.J.; Barthel, L.; Mohning, M.P.; Thomas, S.M.; McCubbrey, A.L.; Danhorn, T.; Leach, S.M.; Fingerlin, T.E.; O’Connor, B.P.; Reisz, J.A.; et al. Cell Origin Dictates Programming of Resident versus Recruited Macrophages during Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2017, 57, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Ruan, W.; Bobrow, B.; Carmeliet, P.; Eltzschig, H.K. Targeting hypoxia-inducible factors: Therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 2024, 23, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Guo, Y.; Wang, Q.; Ma, L.; Zhang, Q.; Zhang, Y.; Geng, Y.; Jin, T.; Guo, J.; Yang, R.; et al. YAP1 inhibits the senescence of alveolar epithelial cells by targeting Prdx3 to alleviate pulmonary fibrosis. Exp. Mol. Med. 2024, 56, 1643–1654. [Google Scholar] [CrossRef]
- Tanaka, K.; Fujita, T.; Umezawa, H.; Namiki, K.; Yoshioka, K.; Hagihara, M.; Sudo, T.; Kimura, S.; Tatsumi, K.; Kasuya, Y. Therapeutic effect of lung mixed culture-derived epithelial cells on lung fibrosis. Lab. Investig. 2014, 94, 1247–1259. [Google Scholar] [CrossRef]
- Liu, H.; Xue, W.; Ge, G.; Luo, X.; Li, Y.; Xiang, H.; Ding, X.; Tian, P.; Tian, X. Hypoxic preconditioning advances CXCR4 and CXCR7 expression by activating HIF-1α in MSCs. Biochem. Biophys. Res. Commun. 2010, 401, 509–515. [Google Scholar] [CrossRef]
- Hoegl, S.; Brodsky, K.S.; Blackburn, M.R.; Karmouty-Quintana, H.; Zwissler, B.; Eltzschig, H.K. Alveolar Epithelial A2B Adenosine Receptors in Pulmonary Protection during Acute Lung Injury. J. Immunol. 2015, 195, 1815–1824. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Figarella, K.; Kim, J.; Ruan, W.; Mills, T.; Eltzschig, H.K.; Yuan, X. Hypoxia-adenosine axis as therapeutic targets for acute respiratory distress syndrome. Front. Immunol. 2024, 15, 1328565. [Google Scholar] [CrossRef]
- Ruan, W.; Eltzschig, H.K.; Yuan, X. Hypoxia-stabilized RIPK1 promotes cell death. Nat. Cell Biol. 2023, 25, 921–922. [Google Scholar] [CrossRef]
- Eckle, T.; Grenz, A.; Laucher, S.; Eltzschig, H.K. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J. Clin. Investig. 2008, 118, 3301–3315. [Google Scholar] [CrossRef]
- Le, T.-T.T.; Berg, N.K.; Harting, M.T.; Li, X.; Eltzschig, H.K.; Yuan, X. Purinergic Signaling in Pulmonary Inflammation. Front. Immunol. 2019, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, B.L.; Sammani, S.; Bermudez, T.; Dudek, S.M.; Camp, S.M.; Garcia, J.G. Genetic and epigenetic regulation of the non-muscle myosin light chain kinase isoform by lung inflammatory factors and mechanical stress. Clin. Sci. 2021, 135, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Liu, Y.; Kim, J.; Kim, Y.; Klouda, T.; Fisch, S.; Baek, S.H.; Liu, T.; Dahlberg, S.; Hu, C.-J.; et al. Pericytes contribute to pulmonary vascular remodeling via HIF2α signaling. EMBO Rep. 2024, 25, 616–645. [Google Scholar] [CrossRef]
- Huang, Y.; Kempen, M.B.-v.; Munck, A.B.-d.; Swagemakers, S.; Driegen, S.; Mahavadi, P.; Meijer, D.; van Ijcken, W.; van der Spek, P.; Grosveld, F.; et al. Hypoxia-Inducible Factor 2α Plays a Critical Role in the Formation of Alveoli and Surfactant. Am. J. Respir. Cell Mol. Biol. 2012, 46, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Zhu, H.; Xu, N.; Zhang, D.; Ou, J.; Wang, G.; Fang, X.; Zhou, J.; Song, Y.; Bai, C. Increased Lung Ischemia–Reperfusion Injury in Aquaporin 1–Null Mice Is Mediated via Decreased Hypoxia-Inducible Factor 2α Stability. Am. J. Respir. Cell Mol. Biol. 2016, 54, 882–891. [Google Scholar] [CrossRef]
- Wu, D.; Liao, X.; Gao, J.; Gao, Y.; Li, Q.; Gao, W. Potential pharmaceuticals targeting neuroimmune interactions in treating acute lung injury. Clin. Transl. Med. 2024, 14, e1808. [Google Scholar] [CrossRef]
- Lin, C.-K.; Lin, Y.-H.; Huang, T.-C.; Shi, C.-S.; Yang, C.-T.; Yang, Y.-L. VEGF mediates fat embolism-induced acute lung injury via VEGF receptor 2 and the MAPK cascade. Sci. Rep. 2019, 9, 11713. [Google Scholar] [CrossRef]
- Shi, H.; Lu, R.; Wang, S.; Chen, H.; Wang, F.; Liu, K. Effects of SDF-1/CXCR4 on Acute Lung Injury Induced by Cardiopulmonary Bypass. Inflammation 2017, 40, 937–945. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Yang, J.; Chen, X.; Guo, X.; Xu, K.; Wang, N.; Zhao, W.; Xia, C.; Lian, H.; et al. Endothelial cell-derived MMP19 promotes pulmonary fibrosis by inducing E(nd)MT and monocyte infiltration. Cell Commun. Signal. 2023, 21, 56. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, X.; He, Y.; Zhang, L.; Huang, X.; Xu, X.; Chen, M.; Chen, X.; Wang, L. Activation of A2aR attenuates bleomycin-induced pulmonary fibrosis via the SDF-1/CXCR4 axis-related pathway. Am. J. Transl. Res. 2017, 9, 4125–4136. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, J.; Zhang, Y.; Lu, Q.; Tian, S.; Zhao, Y.; Fan, H. Dual Roles of Hypoxia-Inducible Factor 1 in Acute Lung Injury: Tissue-Specific Mechanisms and Therapeutic Modulation. Cells 2025, 14, 1089. https://doi.org/10.3390/cells14141089
Jia J, Zhang Y, Lu Q, Tian S, Zhao Y, Fan H. Dual Roles of Hypoxia-Inducible Factor 1 in Acute Lung Injury: Tissue-Specific Mechanisms and Therapeutic Modulation. Cells. 2025; 14(14):1089. https://doi.org/10.3390/cells14141089
Chicago/Turabian StyleJia, Junjing, Yingyi Zhang, Qianying Lu, Sijia Tian, Yanmei Zhao, and Haojun Fan. 2025. "Dual Roles of Hypoxia-Inducible Factor 1 in Acute Lung Injury: Tissue-Specific Mechanisms and Therapeutic Modulation" Cells 14, no. 14: 1089. https://doi.org/10.3390/cells14141089
APA StyleJia, J., Zhang, Y., Lu, Q., Tian, S., Zhao, Y., & Fan, H. (2025). Dual Roles of Hypoxia-Inducible Factor 1 in Acute Lung Injury: Tissue-Specific Mechanisms and Therapeutic Modulation. Cells, 14(14), 1089. https://doi.org/10.3390/cells14141089