
Pseudohypoxia-Stabilized HIF2α Transcriptionally Inhibits MNRR1, a Druggable Target in MELAS

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Lines
2.2. Chemicals
2.3. Plasmids
2.4. Stable Cell Line Generation
2.5. MicroSource Spectrum Collection
2.6. Transient Transfection of MELAS Cells
2.7. Real-Time Polymerase Chain Reaction (RT-PCR)
2.8. Luciferase Reporter Assay
2.9. Immunoblotting
2.10. Immunofluorescence
2.11. Mitochondrial DNA Levels
2.12. Restriction Enzyme Digestion
2.13. ROS Measurements
2.14. Intact Cellular Oxygen Consumption and Measurement of ATP
2.15. Chromatin Immunoprecipitation-qPCR (ChIP-qPCR)
2.16. Statistical Analysis
3. Results
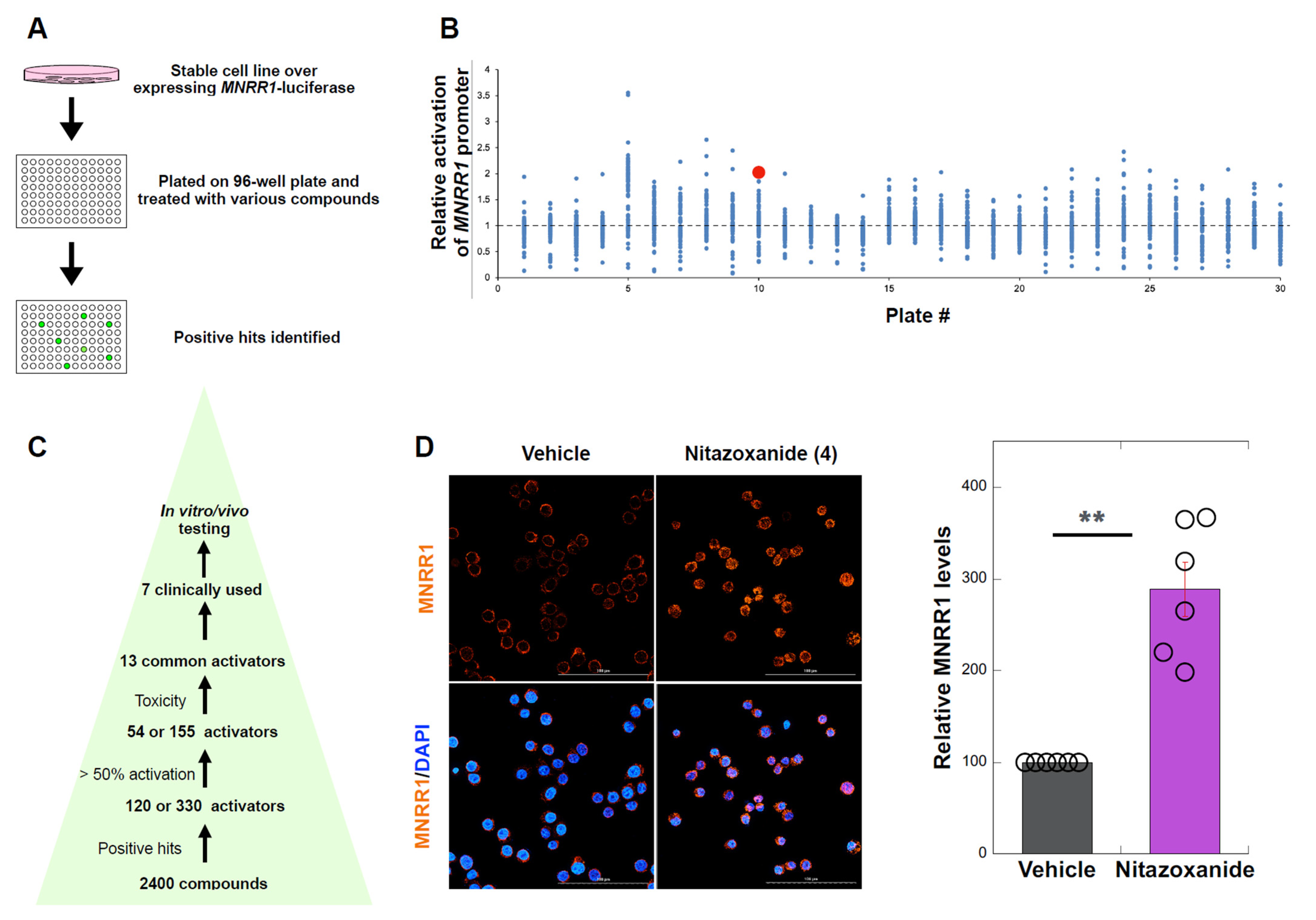
3.1. High Throughput Screen to Identify MNRR1 Activators
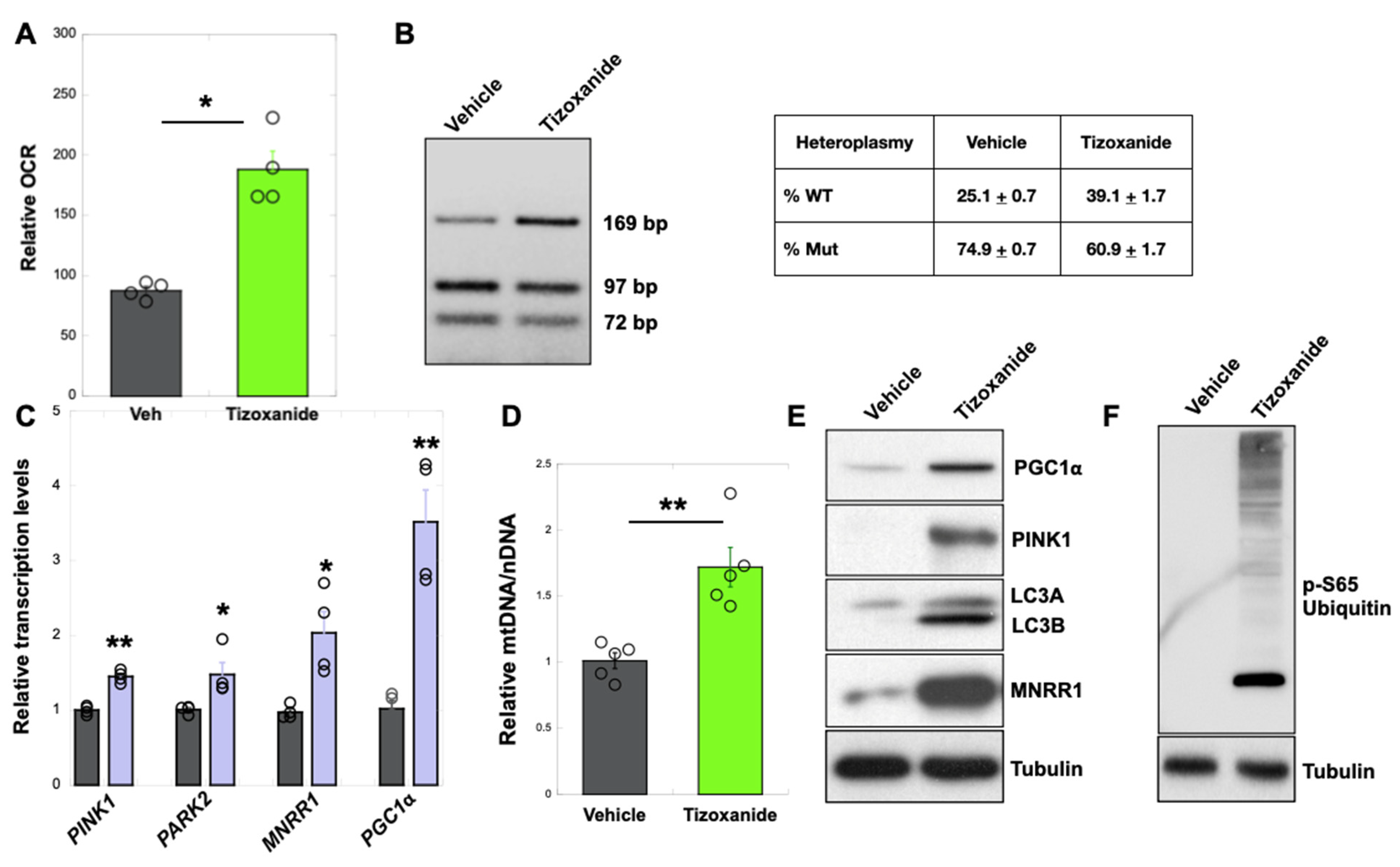
3.2. MNRR1 Activation Using Tizoxanide Enhances Mitochondrial Biogenesis and Mitophagy to Shift Heteroplasmy in MELAS Cybrid Cells
3.3. MNRR1 Activation Using Tizoxanide in MELAS Patient Fibroblasts Enhances Mitochondrial Function and Mitophagy and Protects from LPS-Induced Inflammation
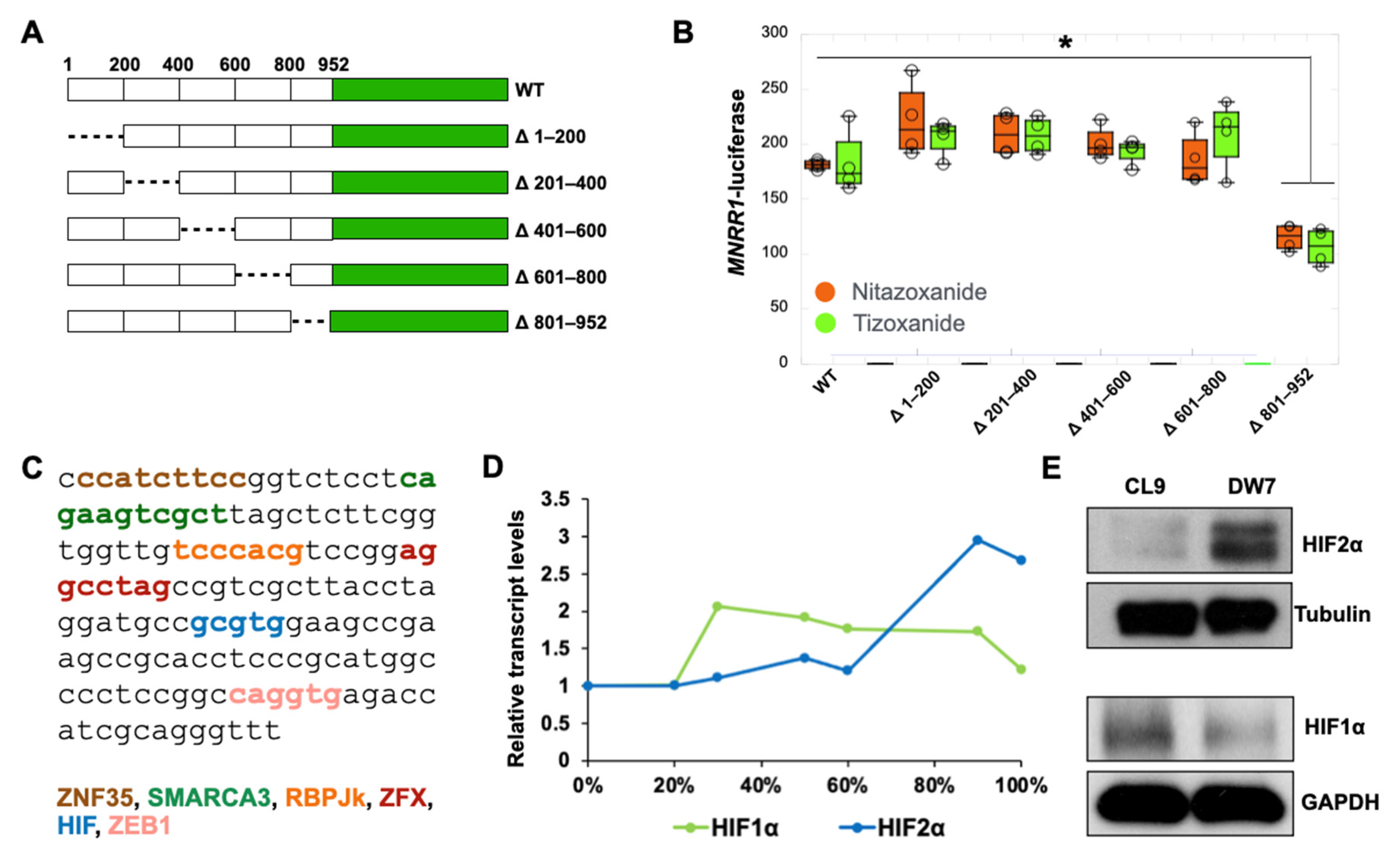
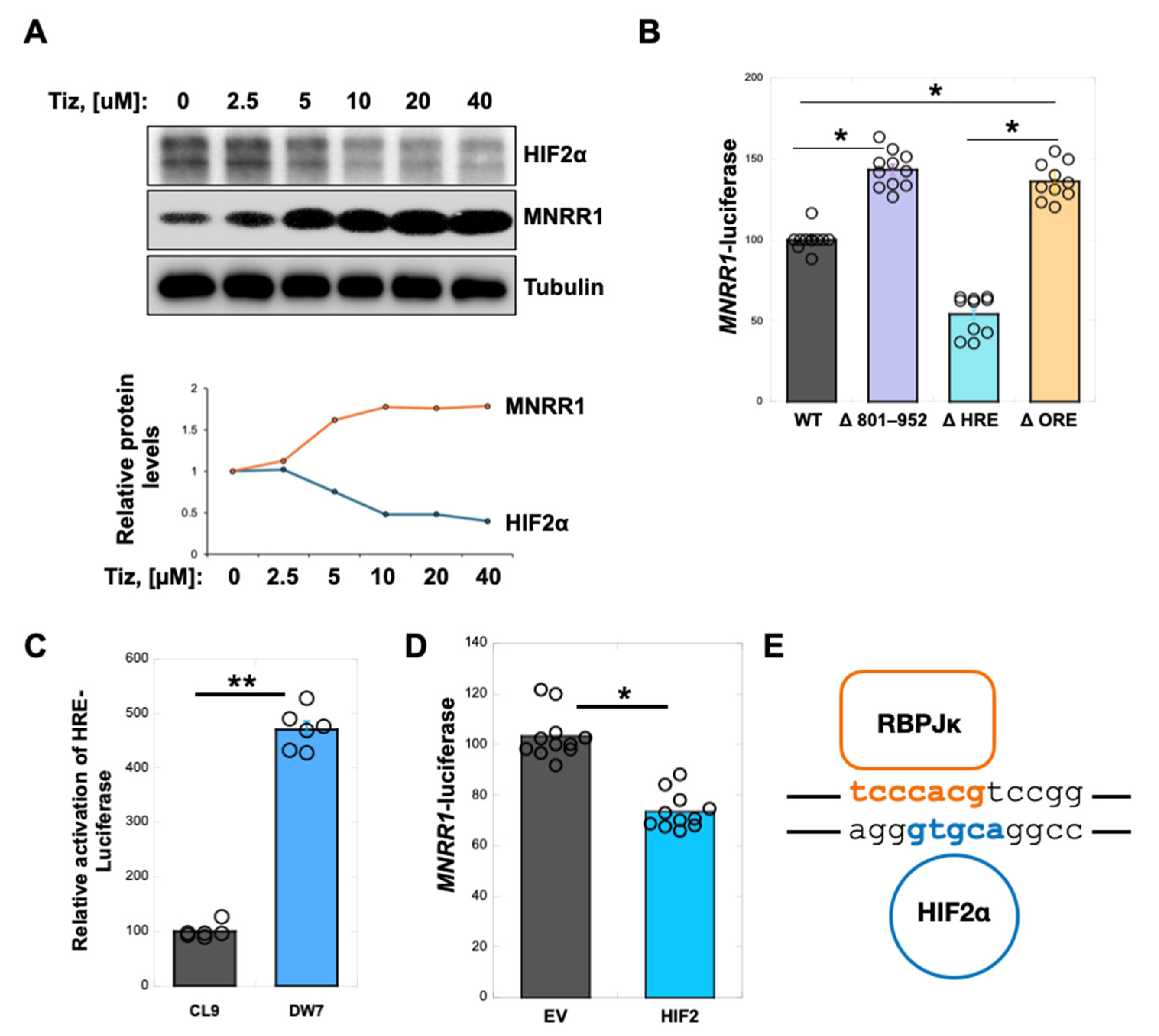
3.4. Nitazoxanide Acts by Reducing HIF2α Protein Levels in MELAS Cybrid Cells
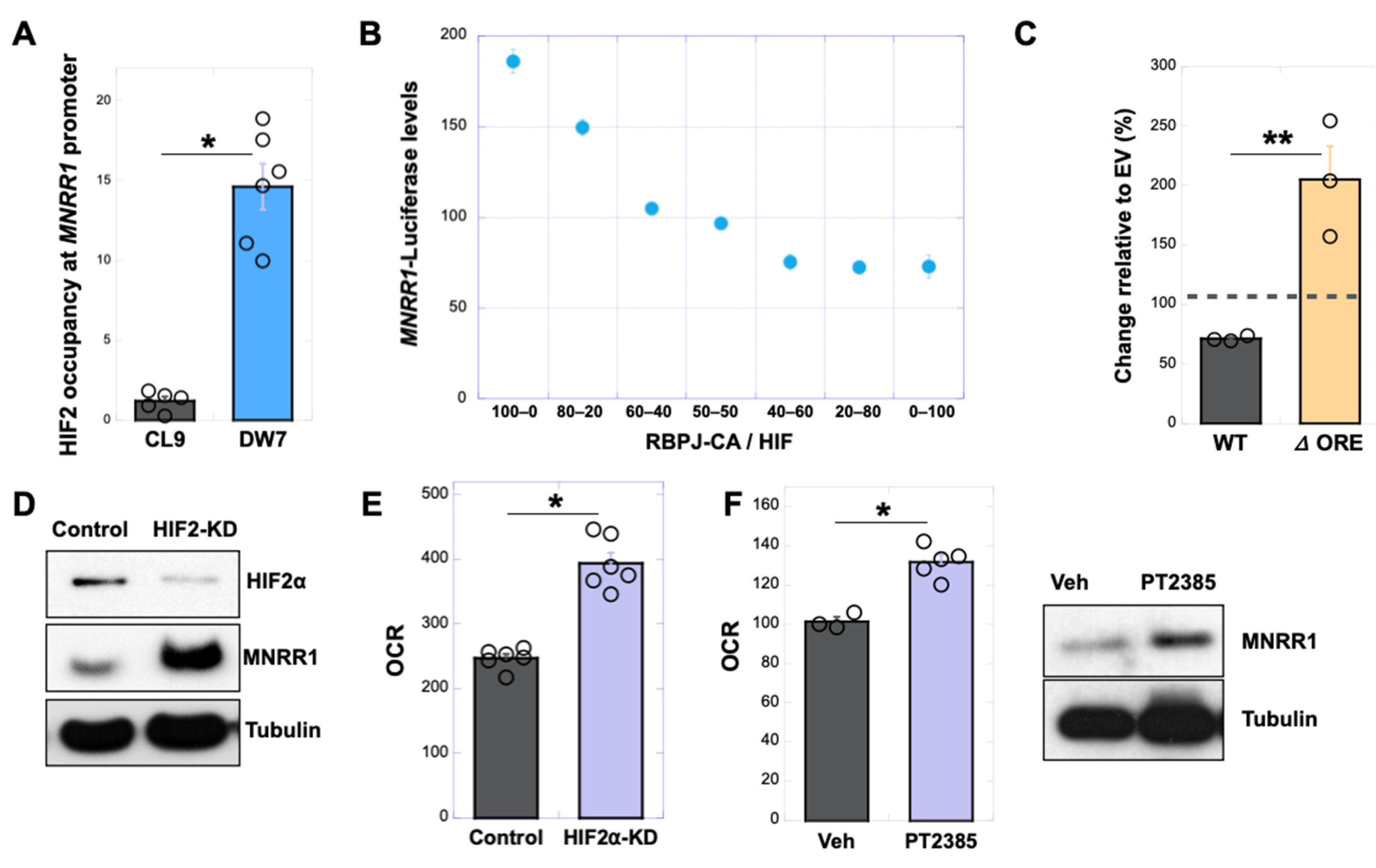
3.5. RBPJk and HIF2α Compete for Binding at the ORE in the MNRR1 Promoter to Regulate Transcription
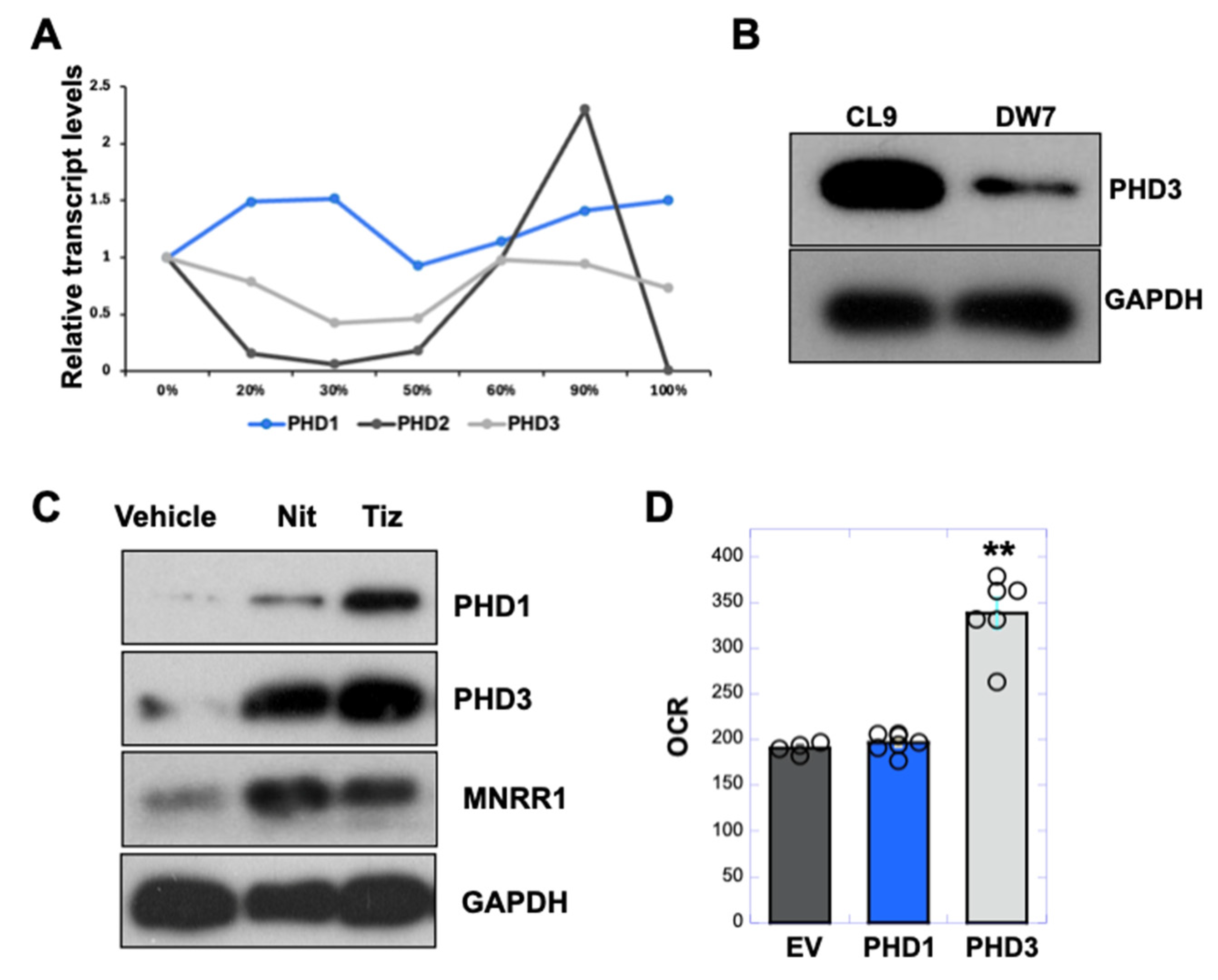
3.6. PHD3 Levels Are Reduced in MELAS Cybrid Cells and Enhanced by Nitazoxanide to Increase MNRR1 Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nass, S.; Nass, M.M. Intramitochondrial Fibers with DNA Characteristics. Ii. Enzymatic and Other Hydrolytic Treatments. J. Cell Biol. 1963, 19, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Nass, M.M.; Nass, S. Intramitochondrial Fibers with DNA Characteristics. I. Fixation and Electron Staining Reactions. J. Cell Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, R.M.; Schapira, A.H. Clinical, biochemical and molecular genetic features of Leber’s hereditary optic neuropathy. Biochim. Biophys. Acta 1999, 1410, 147–158. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; McFarland, R. Mitochondrial myopathies in adults and children: Management and therapy development. Curr. Opin. Neurol. 2014, 27, 576–582. [Google Scholar] [CrossRef]
- Tatuch, Y.; Christodoulou, J.; Feigenbaum, A.; Clarke, J.T.; Wherret, J.; Smith, C.; Rudd, N.; Petrova-Benedict, R.; Robinson, B.H. Heteroplasmic mtDNA mutation (T----G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am. J. Hum. Genet. 1992, 50, 852–858. [Google Scholar]
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders--past, present and future. Biochim. Biophys. Acta 2004, 1659, 115–120. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef]
- Zhao, M.; Zuo, C.; Hao, H.; Xing, X.; Zhao, L.; Li, N. A patient with MELAS syndrome combined with autoimmune abnormalities: A case report. Front. Neurol. 2023, 14, 1239664. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Almannai, M.; El-Hattab, A.W. Nitric Oxide Deficiency in Mitochondrial Disorders: The Utility of Arginine and Citrulline. Front. Mol. Neurosci. 2021, 14, 682780. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.C.; Pearson, J.Y.; Mandrekar, J.; Gavrilova, R.H. The clinical spectrum of MELAS and associated disorders across ages: A retrospective cohort study. Front. Neurol. 2023, 14, 1298569. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Nilsson, R.; Gohil, V.M.; Arlow, D.H.; Gauhar, Z.; Mootha, V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009, 5, e1000590. [Google Scholar] [CrossRef]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Huttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Aras, S.; Arrabi, H.; Purandare, N.; Huttemann, M.; Kamholz, J.; Zuchner, S.; Grossman, L.I. Abl2 kinase phosphorylates Bi-organellar regulator MNRR1 in mitochondria, stimulating respiration. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Clegg, H.V.; Leslie, P.L.; Di, J.; Tollini, L.A.; He, Y.; Kim, T.H.; Jin, A.; Graves, L.M.; Zheng, J.; et al. CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ. 2015, 22, 1035–1046. [Google Scholar] [CrossRef]
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Huttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic. Acids Res. 2013, 41, 2255–2266. [Google Scholar] [CrossRef]
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 32056–32065. [Google Scholar] [CrossRef]
- Emerling, B.M.; Weinberg, F.; Liu, J.L.; Mak, T.W.; Chandel, N.S. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc. Natl. Acad. Sci. USA 2008, 105, 2622–2627. [Google Scholar] [CrossRef]
- Kondo, K.; Klco, J.; Nakamura, E.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002, 1, 237–246. [Google Scholar] [CrossRef]
- Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M.S.; Sajan, M.P.; Farese, R.V.; Freeman, R.S.; Carter, B.D.; Kaelin, W.G., Jr.; et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005, 8, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, A.; Selfors, L.M.; Forster, T.; Wrobel, D.; Kennedy, C.J.; Shanks, E.; Santoyo-Lopez, J.; Dunican, D.J.; Long, A.; Kelleher, D.; et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat. Methods 2009, 6, 569–575. [Google Scholar] [CrossRef]
- Purandare, N.; Somayajulu, M.; Huttemann, M.; Grossman, L.I.; Aras, S. The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): Partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem. 2018, 293, 6517–6529. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, S.; Dai, X.; Gan, W.; Nie, X.; Wei, W.; Hu, G.; Guo, J. Tumor suppressor SPOP ubiquitinates and degrades EglN2 to compromise growth of prostate cancer cells. Cancer Lett. 2017, 390, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Liu, W.; Li, B.; Chen, D.; Hu, F.; Wang, L.; Liu, X.M.; Cui, R.; Liu, R. MicroRNA-1205, encoded on chromosome 8q24, targets EGLN3 to induce cell growth and contributes to risk of castration-resistant prostate cancer. Oncogene 2019, 38, 4820–4834. [Google Scholar] [CrossRef]
- Purandare, N.; Gomez-Lopez, N.; Arenas-Hernandez, M.; Galaz, J.; Romero, R.; Xi, Y.; Fribley, A.M.; Grossman, L.I.; Aras, S. The MNRR1 activator nitazoxanide abrogates lipopolysaccharide-induced preterm birth in mice. Placenta 2023, 140, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Stockis, A.; Deroubaix, X.; Lins, R.; Jeanbaptiste, B.; Calderon, P.; Rossignol, J.F. Pharmacokinetics of nitazoxanide after single oral dose administration in 6 healthy volunteers. Int. J. Clin. Pharmacol. Ther. 1996, 34, 349–351. [Google Scholar]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Purandare, N.; Kunji, Y.; Xi, Y.; Romero, R.; Gomez-Lopez, N.; Fribley, A.; Grossman, L.I.; Aras, S. Lipopolysaccharide induces placental mitochondrial dysfunction in murine and human systems by reducing MNRR1 levels via a TLR4-independent pathway. iScience 2022, 25, 105342. [Google Scholar] [CrossRef]
- Walker, M.A.; Slate, N.; Alejos, A.; Volpi, S.; Iyengar, R.S.; Sweetser, D.; Sims, K.B.; Walter, J.E. Predisposition to infection and SIRS in mitochondrial disorders: 8 years’ experience in an academic center. J. Allergy Clin. Immunol. Pract. 2014, 2, 465–468.e1. [Google Scholar] [CrossRef]
- Warren, E.B.; Gordon-Lipkin, E.M.; Cheung, F.; Chen, J.; Mukherjee, A.; Apps, R.; Tsang, J.S.; Jetmore, J.; Schlein, M.L.; Kruk, S.; et al. Inflammatory and interferon gene expression signatures in patients with mitochondrial disease. J. Transl. Med. 2023, 21, 331. [Google Scholar] [CrossRef] [PubMed]
- Hanaford, A.; Johnson, S.C. The immune system as a driver of mitochondrial disease pathogenesis: A review of evidence. Orphanet. J. Rare Dis. 2022, 17, 335. [Google Scholar] [CrossRef]
- van Gisbergen, M.W.; Offermans, K.; Voets, A.M.; Lieuwes, N.G.; Biemans, R.; Hoffmann, R.F.; Dubois, L.J.; Lambin, P. Mitochondrial Dysfunction Inhibits Hypoxia-Induced HIF-1alpha Stabilization and Expression of Its Downstream Targets. Front. Oncol. 2020, 10, 770. [Google Scholar] [CrossRef]
- Diaz-Trelles, R.; Scimia, M.C.; Bushway, P.; Tran, D.; Monosov, A.; Monosov, E.; Peterson, K.; Rentschler, S.; Cabrales, P.; Ruiz-Lozano, P.; et al. Notch-independent RBPJ controls angiogenesis in the adult heart. Nat. Commun. 2016, 7, 12088. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Gao, X.; Sun, D.; Zhang, Y.; Krausz, K.W.; Qin, X.; Gonzalez, F.J. Metabolic Profiling of the Novel Hypoxia-Inducible Factor 2alpha Inhibitor PT2385 In Vivo and In Vitro. Drug Metab. Dispos. 2018, 46, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Markova, D.; Anderson, D.G.; Chiba, K.; Toyama, Y.; Shapiro, I.M.; Risbud, M.V. Expression of prolyl hydroxylases (PHDs) is selectively controlled by HIF-1 and HIF-2 proteins in nucleus pulposus cells of the intervertebral disc: Distinct roles of PHD2 and PHD3 proteins in controlling HIF-1alpha activity in hypoxia. J. Biol. Chem. 2012, 287, 16975–16986. [Google Scholar] [CrossRef]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef]
- Miikkulainen, P.; Hogel, H.; Seyednasrollah, F.; Rantanen, K.; Elo, L.L.; Jaakkola, P.M. Hypoxia-inducible factor (HIF)-prolyl hydroxylase 3 (PHD3) maintains high HIF2A mRNA levels in clear cell renal cell carcinoma. J. Biol. Chem. 2019, 294, 3760–3771. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Finger, E.C.; Krieg, A.J.; Wu, C.; Diep, A.N.; LaGory, E.L.; Wei, K.; McGinnis, L.M.; Yuan, J.; Kuo, C.J.; et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat. Med. 2013, 19, 1325–1330. [Google Scholar] [CrossRef]
- Rossignol, J.F. Nitazoxanide: A first-in-class broad-spectrum antiviral agent. Antiviral. Res. 2014, 110, 94–103. [Google Scholar] [CrossRef]
- Singh, N.; Narayan, S. Nitazoxanide: A Broad Spectrum Antimicrobial. Med. J. Armed. Forces. India 2011, 67, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; He, R.Z.; Li, Y.; Ruan, Z.F. Tizoxanide mitigates inflammatory response in LPS-induced neuroinflammation in microglia via restraining p38/MAPK pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6446–6454. [Google Scholar] [CrossRef] [PubMed]
- Lokhande, A.S.; Devarajan, P.V. A review on possible mechanistic insights of Nitazoxanide for repurposing in COVID-19. Eur. J. Pharmacol. 2021, 891, 173748. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Hu, W.; Meng, F.; Li, X. Design, Synthesis, and Pharmacokinetic Evaluation of O-Carbamoyl Tizoxanide Prodrugs. Med. Chem. 2022, 18, 140–150. [Google Scholar] [CrossRef]
- Neary, M.; Arshad, U.; Tatham, L.; Pertinez, H.; Box, H.; Rajoli, R.K.R.; Valentijn, A.; Sharp, J.; Rannard, S.P.; Biagini, G.A.; et al. Quantitation of tizoxanide in multiple matrices to support cell culture, animal and human research. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2023, 1228, 123823. [Google Scholar] [CrossRef]
- Guo, S.; Li, F.; Wang, B.; Zhao, Y.; Wang, X.; Wei, H.; Yu, K.; Hai, X. Analysis of tizoxanide, active metabolite of nitazoxanide, in rat brain tissue and plasma by UHPLC-MS/MS. Biomed. Chromatogr. 2020, 34, e4716. [Google Scholar] [CrossRef]
- Koga, Y.; Povalko, N.; Inoue, E.; Nakamura, H.; Ishii, A.; Suzuki, Y.; Yoneda, M.; Kanda, F.; Kubota, M.; Okada, H.; et al. Therapeutic regimen of L-arginine for MELAS: 9-year, prospective, multicenter, clinical research. J. Neurol. 2018, 265, 2861–2874. [Google Scholar] [CrossRef]
- Barros, C.D.S.; Coutinho, A.; Tengan, C.H. Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms? Int. J. Mol. Sci. 2024, 25, 3629. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purandare, N.; Pasupathi, V.; Xi, Y.; Rajan, V.; Vegh, C.; Firestine, S.; Kozicz, T.; Fribley, A.M.; Grossman, L.I.; Aras, S. Pseudohypoxia-Stabilized HIF2α Transcriptionally Inhibits MNRR1, a Druggable Target in MELAS. Cells 2025, 14, 1078. https://doi.org/10.3390/cells14141078
Purandare N, Pasupathi V, Xi Y, Rajan V, Vegh C, Firestine S, Kozicz T, Fribley AM, Grossman LI, Aras S. Pseudohypoxia-Stabilized HIF2α Transcriptionally Inhibits MNRR1, a Druggable Target in MELAS. Cells. 2025; 14(14):1078. https://doi.org/10.3390/cells14141078
Chicago/Turabian StylePurandare, Neeraja, Vignesh Pasupathi, Yue Xi, Vikram Rajan, Caleb Vegh, Steven Firestine, Tamas Kozicz, Andrew M. Fribley, Lawrence I. Grossman, and Siddhesh Aras. 2025. "Pseudohypoxia-Stabilized HIF2α Transcriptionally Inhibits MNRR1, a Druggable Target in MELAS" Cells 14, no. 14: 1078. https://doi.org/10.3390/cells14141078
APA StylePurandare, N., Pasupathi, V., Xi, Y., Rajan, V., Vegh, C., Firestine, S., Kozicz, T., Fribley, A. M., Grossman, L. I., & Aras, S. (2025). Pseudohypoxia-Stabilized HIF2α Transcriptionally Inhibits MNRR1, a Druggable Target in MELAS. Cells, 14(14), 1078. https://doi.org/10.3390/cells14141078