

Resistance Mechanisms to BCMA Targeting Bispecific Antibodies and CAR T-Cell Therapies in Multiple Myeloma

Abstract
1. Introduction
2. BCMA as a Target for Immunotherapy in MM
3. Currently Approved BCMA Therapies in RRMM
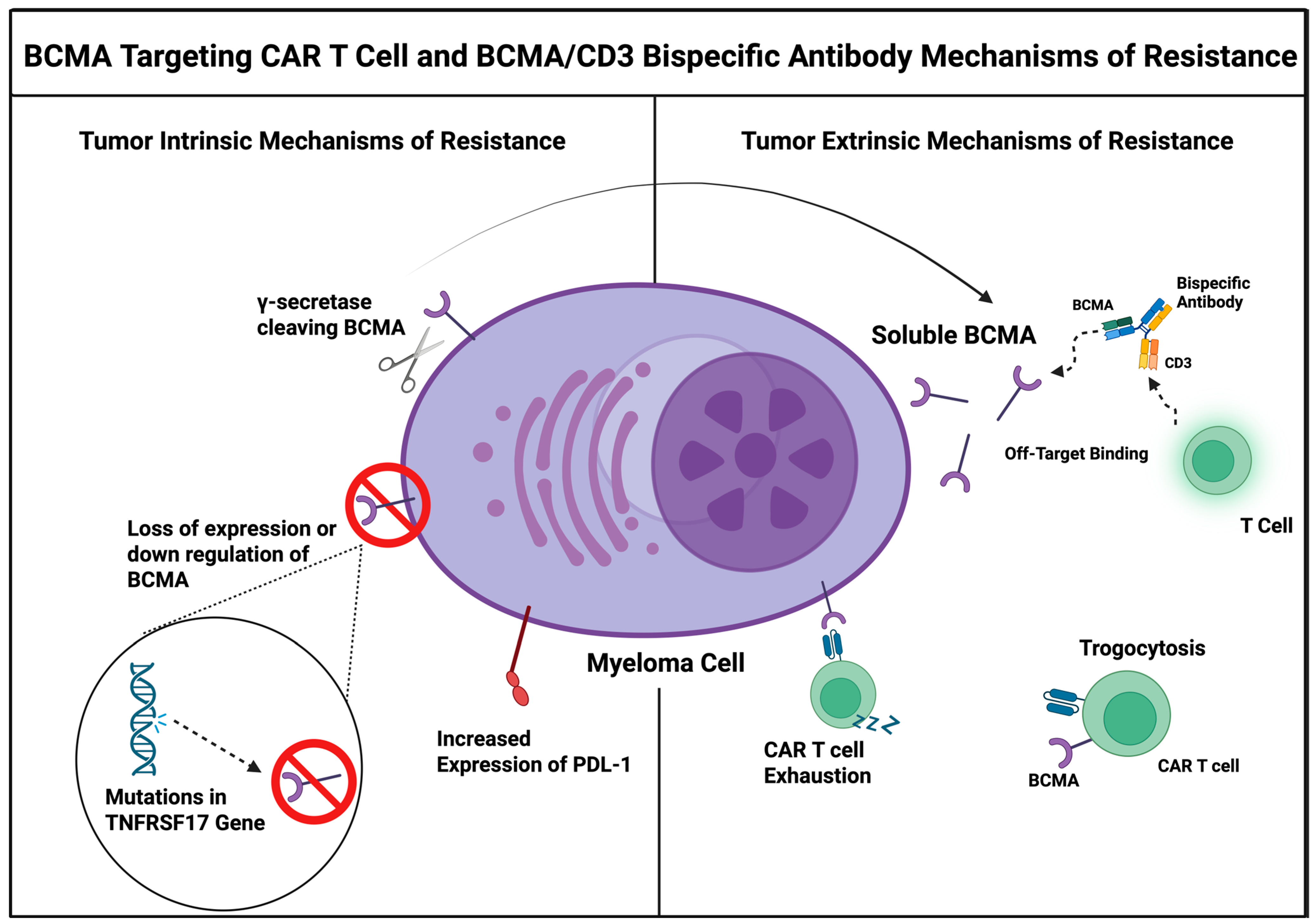
4. Key Mechanisms of Resistance to BCMA-Targeted Therapies
4.1. Tumor-Intrinsic Mechanisms of Resistance
4.1.1. Baseline BCMA Expression
4.1.2. Dynamic Changes in BCMA Expression with Treatment
4.1.3. Trogocytosis-Mediated Antigen Loss
4.1.4. Altered Intracellular Signaling Pathways and Antigen Presentation
4.2. Host Related Mechanisms of Resistance
4.2.1. T-Cell Fitness at Baseline
4.2.2. T-Cell Exhaustion During Treatment
4.2.3. CAR T-Cell Associated Resistance
5. Tumor Microenvironment Mediated Resistance
6. Key Strategies to Overcome Resistance to BCMA-Targeted Therapies
6.1. Preventing Antigen Escape
6.2. Overcoming T-Cell Exhaustion
6.3. Targeting TME
6.4. Sequencing BCMA-Targeted Therapies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laabi, Y.; Gras, M.P.; Brouet, J.C.; Berger, R.; Larsen, C.J.; Tsapis, A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res. 1994, 22, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef]
- Xu, S.; Lam, K.P. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol. Cell Biol. 2001, 21, 4067–4074. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Otero, P.; Ailawadhi, S.; Arnulf, B.; Patel, K.; Cavo, M.; Nooka, A.K.; Manier, S.; Callander, N.; Costa, L.J.; Vij, R.; et al. Ide-cel or Standard Regimens in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2023, 388, 1002–1014. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- San-Miguel, J.; Dhakal, B.; Yong, K.; Spencer, A.; Anguille, S.; Mateos, M.V.; Fernandez de Larrea, C.; Martinez-Lopez, J.; Moreau, P.; Touzeau, C.; et al. Cilta-cel or Standard Care in Lenalidomide-Refractory Multiple Myeloma. N. Engl. J. Med. 2023, 389, 335–347. [Google Scholar] [CrossRef]
- Moreau, P.; Garfall, A.L.; van de Donk, N.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Costello, C.L.; Raje, N.S.; Levy, M.Y.; Dholaria, B.; Solh, M.; Tomasson, M.H.; Damore, M.A.; Jiang, S.; Basu, C.; et al. Elranatamab in relapsed or refractory multiple myeloma: The MagnetisMM-1 phase 1 trial. Nat. Med. 2023, 29, 2570–2576. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Tomasson, M.H.; Arnulf, B.; Bahlis, N.J.; Miles Prince, H.; Niesvizky, R.; Rodriotaguez-Otero, P.; Martinez-Lopez, J.; Koehne, G.; Touzeau, C.; et al. Elranatamab in relapsed or refractory multiple myeloma: Phase 2 MagnetisMM-3 trial results. Nat. Med. 2023, 29, 2259–2267. [Google Scholar] [CrossRef]
- Laabi, Y.; Gras, M.P.; Carbonnel, F.; Brouet, J.C.; Berger, R.; Larsen, C.J.; Tsapis, A. A new gene, BCM, on chromosome 16 is fused to the interleukin 2 gene by a t(4;16)(q26;p13) translocation in a malignant T cell lymphoma. EMBO J. 1992, 11, 3897–3904. [Google Scholar] [CrossRef]
- Kozlow, E.J.; Wilson, G.L.; Fox, C.H.; Kehrl, J.H. Subtractive cDNA cloning of a novel member of the Ig gene superfamily expressed at high levels in activated B lymphocytes. Blood 1993, 81, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Day, E.S.; Cachero, T.G.; Qian, F.; Sun, Y.; Wen, D.; Pelletier, M.; Hsu, Y.M.; Whitty, A. Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry 2005, 44, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-kappa B, elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Guo, Y.; Qi, J.; Shi, W.; Wu, X.; Ju, S. Binding of B-cell maturation antigen to B-cell activating factor induces survival of multiple myeloma cells by activating Akt and JNK signaling pathways. Cell Biochem. Funct. 2016, 34, 104–110. [Google Scholar] [CrossRef]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Reme, T.; Lugagne, C.; Moine, P.; Rossi, J.F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef]
- Pan, J.; Sun, Y.; Zhang, N.; Li, J.; Ta, F.; Wei, W.; Yu, S.; Ai, L. Characteristics of BAFF and APRIL factor expression in multiple myeloma and clinical significance. Oncol. Lett. 2017, 14, 2657–2662. [Google Scholar] [CrossRef]
- Moreaux, J.; Sprynski, A.C.; Dillon, S.R.; Mahtouk, K.; Jourdan, M.; Ythier, A.; Moine, P.; Robert, N.; Jourdan, E.; Rossi, J.F.; et al. APRIL and TACI interact with syndecan-1 on the surface of multiple myeloma cells to form an essential survival loop. Eur. J. Haematol. 2009, 83, 119–129. [Google Scholar] [CrossRef]
- Bolkun, L.; Lemancewicz, D.; Jablonska, E.; Kulczynska, A.; Bolkun-Skornicka, U.; Kloczko, J.; Dzieciol, J. BAFF and APRIL as TNF superfamily molecules and angiogenesis parallel progression of human multiple myeloma. Ann. Hematol. 2014, 93, 635–644. [Google Scholar] [CrossRef]
- Marsters, S.A.; Yan, M.; Pitti, R.M.; Haas, P.E.; Dixit, V.M.; Ashkenazi, A. Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr. Biol. 2000, 10, 785–788. [Google Scholar] [CrossRef]
- Moreaux, J.; Cremer, F.W.; Reme, T.; Raab, M.; Mahtouk, K.; Kaukel, P.; Pantesco, V.; De Vos, J.; Jourdan, E.; Jauch, A.; et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood 2005, 106, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Lin, L.; Xing, L.; Cho, S.F.; Yu, T.; Acharya, C.; Wen, K.; Hsieh, P.A.; Dulos, J.; van Elsas, A.; et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: Therapeutic implications. Leukemia 2019, 33, 426–438. [Google Scholar] [CrossRef]
- Tedder, B. 2025. Available online: https://BioRender.com/a2ezh17 (accessed on 29 June 2025).
- Cortes-Selva, D.; Casneuf, T.; Vishwamitra, D.; Stein, S.; Perova, T.; Skerget, S.; Ramos, E.; van Steenbergen, L.; De Maeyer, D.; Boominathan, R.; et al. Teclistamab, a B-Cell Maturation Antigen (BCMA) x CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM): Correlative Analyses from MajesTEC-1. Blood 2022, 140, 241–243. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Salem, D.A.; Maric, I.; Yuan, C.M.; Liewehr, D.J.; Venzon, D.J.; Kochenderfer, J.; Stetler-Stevenson, M. Quantification of B-cell maturation antigen, a target for novel chimeric antigen receptor T-cell therapy in Myeloma. Leuk. Res. 2018, 71, 106–111. [Google Scholar] [CrossRef]
- Lee, H.; Durante, M.; Skerget, S.; Vishwamitra, D.; Benaoudia, S.; Ahn, S.; Poorebrahim, M.; Barakat, E.; Jung, D.; Leblay, N.; et al. Impact of soluble BCMA and non-T-cell factors on refractoriness to BCMA-targeting T-cell engagers in multiple myeloma. Blood 2024, 144, 2637–2651. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef]
- Lee, H.; Ahn, S.; Maity, R.; Leblay, N.; Ziccheddu, B.; Truger, M.; Chojnacka, M.; Cirrincione, A.; Durante, M.; Tilmont, R.; et al. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat. Med. 2023, 29, 2295–2306. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef]
- Samur, M.K.; Aktas Samur, A.; Corre, J.; Lannes, R.; Shah, P.; Anderson, K.; Avet-Loiseau, H.; Munshi, N. Monoallelic deletion of BCMA is a frequent feature in multiple myeloma. Blood Adv. 2023, 7, 6599–6603. [Google Scholar] [CrossRef]
- Li, W.; Zhang, B.; Cao, W.; Zhang, W.; Li, T.; Liu, L.; Xu, L.; Gao, F.; Wang, Y.; Wang, F.; et al. Identification of potential resistance mechanisms and therapeutic targets for the relapse of BCMA CAR-T therapy in relapsed/refractory multiple myeloma through single-cell sequencing. Exp. Hematol. Oncol. 2023, 12, 44. [Google Scholar] [CrossRef]
- Chen, Y.; Xin, Q.; Zhu, M.; Qiu, J.; Qiu, J.; Li, R.; Tu, J. Trogocytosis in CAR immune cell therapy: A key mechanism of tumor immune escape. Cell Commun. Signal 2024, 22, 521. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Zhai, Y.; Du, Y.; Li, G.; Yu, M.; Hu, H.; Pan, C.; Wang, D.; Shi, Z.; Yan, X.; Li, X.; et al. Trogocytosis of CAR molecule regulates CAR-T cell dysfunction and tumor antigen escape. Signal Transduct. Target. Ther. 2023, 8, 457. [Google Scholar] [CrossRef]
- Zhang, Z.; Su, M.; Jiang, P.; Wang, X.; Tong, X.; Wu, G. Unlocking Apoptotic Pathways: Overcoming Tumor Resistance in CAR-T-Cell Therapy. Cancer Med. 2024, 13, e70283. [Google Scholar] [CrossRef]
- Friedrich, M.J.; Neri, P.; Kehl, N.; Michel, J.; Steiger, S.; Kilian, M.; Leblay, N.; Maity, R.; Sankowski, R.; Lee, H.; et al. The pre-existing T cell landscape determines the response to bispecific T cell engagers in multiple myeloma patients. Cancer Cell 2023, 41, 711–725.e716. [Google Scholar] [CrossRef]
- Letouze, E.; Moreau, P.; Munshi, N.; Samur, M.; Minvielle, S.; Touzeau, C. Mechanisms of resistance to bispecific T-cell engagers in multiple myeloma and their clinical implications. Blood Adv. 2024, 8, 2952–2959. [Google Scholar] [CrossRef] [PubMed]
- Philipp, N.; Kazerani, M.; Nicholls, A.; Vick, B.; Wulf, J.; Straub, T.; Scheurer, M.; Muth, A.; Hanel, G.; Nixdorf, D.; et al. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood 2022, 140, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Finney, O.C.; Yeri, A.; Mao, P.; Pandya, C.; Alonzo, E.; Hopkins, G.; Hymson, S.; Hu, T.; Foos, M.; Bhadoriya, S.; et al. Molecular and Phenotypic Profiling of Drug Product and Post-Infusion Samples from CRB-402, an Ongoing: Phase I Clinical Study of bb21217 a BCMA-Directed CAR T Cell Therapy. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Leblay, N.; Maity, R.; Barakat, E.; McCulloch, S.; Duggan, P.; Jimenez-Zepeda, V.; Bahlis, N.J.; Neri, P. Cite-Seq Profiling of T Cells in Multiple Myeloma Patients Undergoing BCMA Targeting CAR-T or Bites Immunotherapy. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Mehta, P.H.; Fiorenza, S.; Koldej, R.M.; Jaworowski, A.; Ritchie, D.S.; Quinn, K.M. T Cell Fitness and Autologous CAR T Cell Therapy in Haematologic Malignancy. Front. Immunol. 2021, 12, 780442. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef]
- Vishwamitra, D.; Skerget, S.; Cortes, D.; Perova, T.; Lau, O.; Davis, C.; Guo, Y.; Miao, X.; Stephenson, T.; Hodin, C.; et al. Longitudinal Correlative Profiles of Responders, Nonresponders, and Those with Relapse on Treatment with Teclistamab in the Phase 1/2 MajesTEC-1 Study of Patients with Relapsed/Refractory Multiple Myeloma. Blood 2023, 142, 455. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; O’Neill, C.A.; Broekmans, M.E.C.; Frerichs, K.A.; Bruins, W.S.C.; Duetz, C.; Kruyswijk, S.; Baglio, S.R.; Skerget, S.; Montes de Oca, R.; et al. T-Cell Characteristics Impact Response and Resistance to T-Cell-Redirecting Bispecific Antibodies in Multiple Myeloma. Clin. Cancer Res. 2024, 30, 3006–3022. [Google Scholar] [CrossRef]
- Neri, P.; Ahn, S.; Lee, H.; Leblay, N.; Friedrich, M.; Maity, R.; Tilmont, R.; Barakat, E.; Raab, M.S.; Bahlis, N.J. Dysfunctional Hyper-Expanded Clonotypes and Lack of TCR Clonal Replacement Predict Resistance to T Cell Engagers in Multiple Myeloma. Blood 2022, 140, 2093–2094. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Scott, C.D.; Leonardi, A.J.; Yamamoto, T.N.; Cruz, A.C.; Ouyang, C.; Ramaswamy, M.; Roychoudhuri, R.; Ji, Y.; Eil, R.L.; et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig. 2016, 126, 318–334. [Google Scholar] [CrossRef]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef]
- Yue, T.; Sun, Y.; Dai, Y.; Jin, F. Mechanisms for resistance to BCMA-targeted immunotherapies in multiple myeloma. Blood Rev. 2025, 70, 101256. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Botta, C.; Zabaleta, A.; Puig, N.; Cedena, M.T.; Goicoechea, I.; Alameda, D.; San Jose-Eneriz, E.; Merino, J.; Rodriguez-Otero, P.; et al. Immunogenomic identification and characterization of granulocytic myeloid-derived suppressor cells in multiple myeloma. Blood 2020, 136, 199–209. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Cohen, A.D.; Kaushal, A.; Garfall, A.L.; Manalo, R.J.; Carr, A.R.; McCachren, S.S.; Stadtmauer, E.A.; Lacey, S.F.; Melenhorst, J.J.; et al. Changes in Bone Marrow Tumor and Immune Cells Correlate with Durability of Remissions Following BCMA CAR T Therapy in Myeloma. Blood Cancer Discov. 2022, 3, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.C.; Magen, H.; Gatt, M.; Sebag, M.; Kim, K.; Min, C.K.; Ocio, E.M.; Yoon, S.S.; Chu, M.P.; Rodriguez-Otero, P.; et al. Talquetamab plus Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2025, 392, 138–149. [Google Scholar] [CrossRef]
- Pillarisetti, R.; Yang, D.; Yao, J.; Smith, M.; Luistro, L.; Vulfson, P.; Testa, J., Jr.; Packman, K.; Brodeur, S.; Attar, R.M.; et al. Characterization of JNJ-79635322, a Novel BCMAxGPRC5DxCD3 T-Cell Redirecting Trispecific Antibody, for the Treatment of Multiple Myeloma. Blood 2023, 142, 456. [Google Scholar] [CrossRef]
- Pihlgren, M.; Hall, O.; Carretero, L.; Estoppey, C.; Drake, A.; Pais, D.; Loyau, J.; Berret, J.; Gruber, I.; Suere, P.; et al. ISB 2001, a First-in-Class Trispecific BCMA and CD38 T Cell Engager Designed to Overcome Mechanisms of Escape from Treatments for Multiple Myeloma By Targeting Two Antigens. Blood 2022, 140, 858–859. [Google Scholar] [CrossRef]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef]
- Garfall, A.L.; Cohen, A.D.; Susanibar-Adaniya, S.P.; Hwang, W.T.; Vogl, D.T.; Waxman, A.J.; Lacey, S.F.; Gonzalez, V.E.; Fraietta, J.A.; Gupta, M.; et al. Anti-BCMA/CD19 CAR T Cells with Early Immunomodulatory Maintenance for Multiple Myeloma Responding to Initial or Later-Line Therapy. Blood Cancer Discov. 2023, 4, 118–133. [Google Scholar] [CrossRef]
- Shi, M.; Wang, J.; Huang, H.; Liu, D.; Cheng, H.; Wang, X.; Chen, W.; Yan, Z.; Sang, W.; Qi, K.; et al. Bispecific CAR T cell therapy targeting BCMA and CD19 in relapsed/refractory multiple myeloma: A phase I/II trial. Nat. Commun. 2024, 15, 3371. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yin, H.; Zhao, X.; Jin, D.; Liang, Y.; Xiong, T.; Li, L.; Tang, W.; Zhang, J.; Liu, M.; et al. High efficacy and safety of CD38 and BCMA bispecific CAR-T in relapsed or refractory multiple myeloma. J. Exp. Clin. Cancer Res. 2022, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Huang, H.; Qin, H.; Tang, K.; Shi, X.; Zhu, T.; Gao, Y.; Zhang, Y.; Tian, X.; Fu, J.; et al. Chimeric antigen receptor T cells targeting FcRH5 provide robust tumour-specific responses in murine xenograft models of multiple myeloma. Nat. Commun. 2023, 14, 3642. [Google Scholar] [CrossRef] [PubMed]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Lee, L.; Draper, B.; Chaplin, N.; Philip, B.; Chin, M.; Galas-Filipowicz, D.; Onuoha, S.; Thomas, S.; Baldan, V.; Bughda, R.; et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood 2018, 131, 746–758. [Google Scholar] [CrossRef]
- Yang, Y.; McCloskey, J.E.; Yang, H.; Puc, J.; Alcaina, Y.; Vedvyas, Y.; Gomez Gallegos, A.A.; Ortiz-Sanchez, E.; de Stanchina, E.; Min, I.M.; et al. Bispecific CAR T Cells against EpCAM and Inducible ICAM-1 Overcome Antigen Heterogeneity and Generate Superior Antitumor Responses. Cancer Immunol. Res. 2021, 9, 1158–1174. [Google Scholar] [CrossRef]
- Chen, H.; Yu, T.; Lin, L.; Xing, L.; Cho, S.F.; Wen, K.; Aardalen, K.; Oka, A.; Lam, J.; Daley, M.; et al. gamma-secretase inhibitors augment efficacy of BCMA-targeting bispecific antibodies against multiple myeloma cells without impairing T-cell activation and differentiation. Blood Cancer J. 2022, 12, 118. [Google Scholar] [CrossRef]
- Offner, F.; Decaux, O.; Hulin, C.; Anguille, S.; Sophie Michallet, A.; Costa, L.; Touzeau, C.; Boyd, K.; Vishwamitra, D.; Guo, Y.; et al. S194: Teclistamab (Tec) + Nirogacestat (Niro) in Relapsed/Refractory Multiple Myeloma (Rrmm): The Phase 1b Majestec-2 Study. HemaSphere 2023, 7 (Suppl. S3). [Google Scholar] [CrossRef]
- Cowan, A.J.; Pont, M.J.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Libby, E.N., 3rd; Coffey, D.G.; Tuazon, S.A.; Wood, B.; Gooley, T.; et al. gamma-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: A phase 1, first-in-human trial. Lancet Oncol. 2023, 24, 811–822. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Gaffney, B.; Burnett, K.; Castiglioni, P.; Angelo, M.; Pierce, D.W.; Boss, I.W. Synergistic Antitumor Activity of Alnuctamab (ALNUC.; BMS-986349; CC-93269), a BCMA 2+1 T Cell Engager (TCE), and Celmod Agents in Multiple Myeloma (MM) Preclinical Models. Blood 2022, 140, 7054–7055. [Google Scholar] [CrossRef]
- Vrohlings, M.; Müller, J.; Jungmichel, S.; Senn, D.; Howald, A.B.; Schleier, T.; Scheifele, F.; Wendelspiess, S.; Richle, P.; Merten, H.; et al. Preclinical Assessment of CDR101—A BCMAxCD3xPD-L1 Trispecific Antibody with Superior Anti-Tumor Efficacy. Blood 2021, 138, 1583. [Google Scholar] [CrossRef]
- Hutchings, M.; Carlo-Stella, C.; Gritti, G.; Bosch, F.; Morschhauser, F.; Townsend, W.; Offner, F.; Walter, H.S.; Ghesquieres, H.; Houot, R.; et al. CD19 4-1BBL (RO7227166) a Novel Costimulatory Bispecific Antibody Can be Safely Combined with the T-Cell-Engaging Bispecific Antibody Glofitamab in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma. Blood 2022, 140, 9461–9463. [Google Scholar] [CrossRef]
- Waldschmidt, J.M.; Sotudeh, N.; Arora, S.; Vijaykumar, T.; Anand, P.; Stuart, H.; Frede, J.; Campbell, T.; Kaiser, S.M.; Zheng, X.; et al. Nivolumab to restore T-cell fitness in CAR-T refractory multiple myeloma. Blood Adv. 2025, 9, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Alsina, M.; Shah, N.; Raje, N.S.; Jagannath, S.; Madduri, D.; Kaufman, J.L.; Siegel, D.S.; Munshi, N.C.; Rosenblatt, J.; Lin, Y.; et al. Updated Results from the Phase I CRB-402 Study of Anti-Bcma CAR-T Cell Therapy bb21217 in Patients with Relapsed and Refractory Multiple Myeloma: Correlation of Expansion and Duration of Response with T Cell Phenotypes. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Alabanza, L.M.; Xiong, Y.; Vu, B.; Webster, B.; Wu, D.; Hu, P.; Zhu, Z.; Dropulic, B.; Dash, P.; Schneider, D. Armored BCMA CAR T Cells Eliminate Multiple Myeloma and Are Resistant to the Suppressive Effects of TGF-beta. Front. Immunol. 2022, 13, 832645. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e476. [Google Scholar] [CrossRef] [PubMed]
- Battram, A.M.; Bachiller, M.; Lopez, V.; Fernandez de Larrea, C.; Urbano-Ispizua, A.; Martin-Antonio, B. IL-15 Enhances the Persistence and Function of BCMA-Targeting CAR-T Cells Compared to IL-2 or IL-15/IL-7 by Limiting CAR-T Cell Dysfunction and Differentiation. Cancers 2021, 13, 3534. [Google Scholar] [CrossRef]
- Joedicke, J.J.; Grosskinsky, U.; Gerlach, K.; Kunkele, A.; Hopken, U.E.; Rehm, A. Accelerating clinical-scale production of BCMA CAR T cells with defined maturation stages. Mol. Ther. Methods Clin. Dev. 2022, 24, 181–198. [Google Scholar] [CrossRef]
- Frigault, M.J.; Bishop, M.R.; Rosenblatt, J.; O’Donnell, E.K.; Raje, N.; Cook, D.; Yee, A.J.; Logan, E.; Avigan, D.E.; Jakubowiak, A.; et al. Phase 1 study of CART-ddBCMA for the treatment of subjects with relapsed and refractory multiple myeloma. Blood Adv. 2023, 7, 768–777. [Google Scholar] [CrossRef]
- Tan, J.; Jia, Y.; Zhou, M.; Fu, C.; Tuhin, I.J.; Ye, J.; Monty, M.A.; Xu, N.; Kang, L.; Li, M.; et al. Chimeric antigen receptors containing the OX40 signalling domain enhance the persistence of T cells even under repeated stimulation with multiple myeloma target cells. J. Hematol. Oncol. 2022, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2023, 41, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, H.; Hansen, D.K.; Peres, L.C.; Puglianini, O.C.; Freeman, C.; De Avila, G.; Sidana, S.; Shune, L.; Sborov, D.W.; Davis, J.; et al. Factors associated with refractoriness or early progression after idecabtagene vicleucel in patients with relapsed/refractory multiple myeloma: US Myeloma Immunotherapy Consortium real world experience. Haematologica 2024, 109, 1514–1524. [Google Scholar] [CrossRef]
- Chakraborty, R.; Cheruvalath, H.; Patwari, A.; Szabo, A.; Schinke, C.; Dhakal, B.; Lentzsch, S.; D’Souza, A.; Mohyuddin, G.R.; Julian, K.; et al. Sustained remission following finite duration bispecific antibody therapy in patients with relapsed/refractory myeloma. Blood Cancer J. 2024, 14, 137. [Google Scholar] [CrossRef]
- Van Oekelen, O.; Nath, K.; Mouhieddine, T.H.; Farzana, T.; Aleman, A.; Melnekoff, D.T.; Ghodke-Puranik, Y.; Shah, G.L.; Lesokhin, A.; Giralt, S.; et al. Interventions and outcomes of patients with multiple myeloma receiving salvage therapy after BCMA-directed CAR T therapy. Blood 2023, 141, 756–765. [Google Scholar] [CrossRef]
- Cohen, A.D.; Mateos, M.V.; Cohen, Y.C.; Rodriguez-Otero, P.; Paiva, B.; van de Donk, N.; Martin, T.; Suvannasankha, A.; De Braganca, K.C.; Corsale, C.; et al. Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood 2023, 141, 219–230. [Google Scholar] [CrossRef]
- Ferreri, C.J.; Hildebrandt, M.A.T.; Hashmi, H.; Shune, L.O.; McGuirk, J.P.; Sborov, D.W.; Wagner, C.B.; Kocoglu, M.H.; Rapoport, A.; Atrash, S.; et al. Real-world experience of patients with multiple myeloma receiving ide-cel after a prior BCMA-targeted therapy. Blood Cancer J. 2023, 13, 117. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| BCMA Targeting Mechanisms of Resistance Table Summary | ||
|---|---|---|
| Tumor Intrinsic | Tumor Extrinsic/Host-Related | Tumor Microenvironment |
| Loss of BCMA expression through mutations in TNFRSF17 Gene | T Cell Fitness (CAR-T Therapy) | Presence of MDSCs |
| Downregulation of BCMA | T Cell Exhaustion (BsAb and CAR-T Therapy) | Activation of T-regulator cells |
| BCMA shedding through Secretase via Secretase Enzyme | Age | Increased Inhibitory Cytokine release such as IL-10, TGF-B |
| Trogocytosis | Genetic Factors that compromise immune fitness | Physical barriers causing poor therapeutic infiltration |
| Upregulation of Anti-Apoptotic Proteins and MHC Class I Loss | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tedder, B.; Bhutani, M. Resistance Mechanisms to BCMA Targeting Bispecific Antibodies and CAR T-Cell Therapies in Multiple Myeloma. Cells 2025, 14, 1077. https://doi.org/10.3390/cells14141077
Tedder B, Bhutani M. Resistance Mechanisms to BCMA Targeting Bispecific Antibodies and CAR T-Cell Therapies in Multiple Myeloma. Cells. 2025; 14(14):1077. https://doi.org/10.3390/cells14141077
Chicago/Turabian StyleTedder, Brandon, and Manisha Bhutani. 2025. "Resistance Mechanisms to BCMA Targeting Bispecific Antibodies and CAR T-Cell Therapies in Multiple Myeloma" Cells 14, no. 14: 1077. https://doi.org/10.3390/cells14141077
APA StyleTedder, B., & Bhutani, M. (2025). Resistance Mechanisms to BCMA Targeting Bispecific Antibodies and CAR T-Cell Therapies in Multiple Myeloma. Cells, 14(14), 1077. https://doi.org/10.3390/cells14141077