

Integrative Review of Molecular, Metabolic, and Environmental Factors in Spina Bifida and Congenital Diaphragmatic Hernia: Insights into Mechanisms and Emerging Therapeutics

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Revised Material and Methods
- Original human or animal studies presenting molecular, genetic, epigenetic, or metabolic data related to SB or CDH;
- Experimental in vitro or in vivo studies focusing on developmental pathways, gene expression, microRNAs, or chromosomal alterations;
- Systematic reviews or meta-analyses with primary molecular data or gene pathway integration.
- Clinical case reports or reviews without molecular relevance;
- Duplicates, abstracts only, non-English publications, or studies focusing solely on surgical outcomes or late clinical course.
- 41 human molecular studies,
- 29 animal or in vitro experimental studies,
- 26 systematic or mechanistic reviews.
2.2. Inclusion and Exclusion Criteria
2.3. Study Selection Process
2.4. Quality Assessment and Scoring
2.5. Data Extraction and Synthesis
3. Epidemiology
3.1. Spinal Bifida
3.2. Diaphragmatic Hernia
4. Spinal Bifida
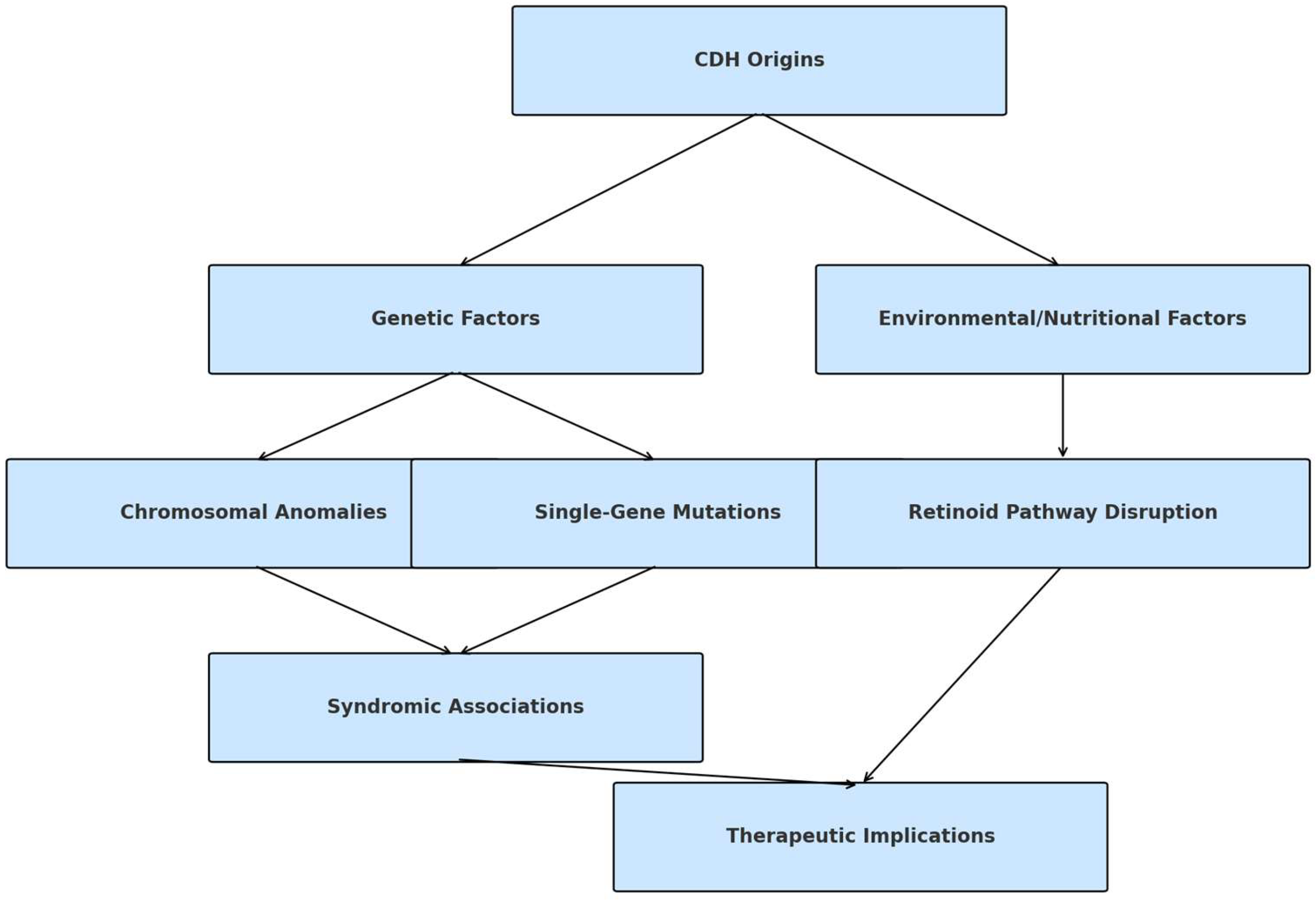
4.1. Etiology
4.2. Genetic-Based Studies
4.3. Folate Metabolism Associated Genes Analysis
4.4. Telomere Dynamics and SB: Unveiling the Genetic Link
4.5. Protein Profiling
5. Diaphragmatic Hernia
5.1. Etiology
5.2. Molecular Profiling
5.3. Protein Profiling
6. Discussion and Future Perspectives
6.1. In Vivo Models of SB
6.2. In Vivo Models of CDH: Targeting Novel Medical Approaches
6.3. Targeting Novel Medical Approaches
7. Conclusions
8. Key Take-Home Messages
- Shared mechanisms, distinct outcomes. While SB and CDH rarely co-occur, both are influenced by folate metabolism, oxidative stress, and miRNA-mediated regulation.
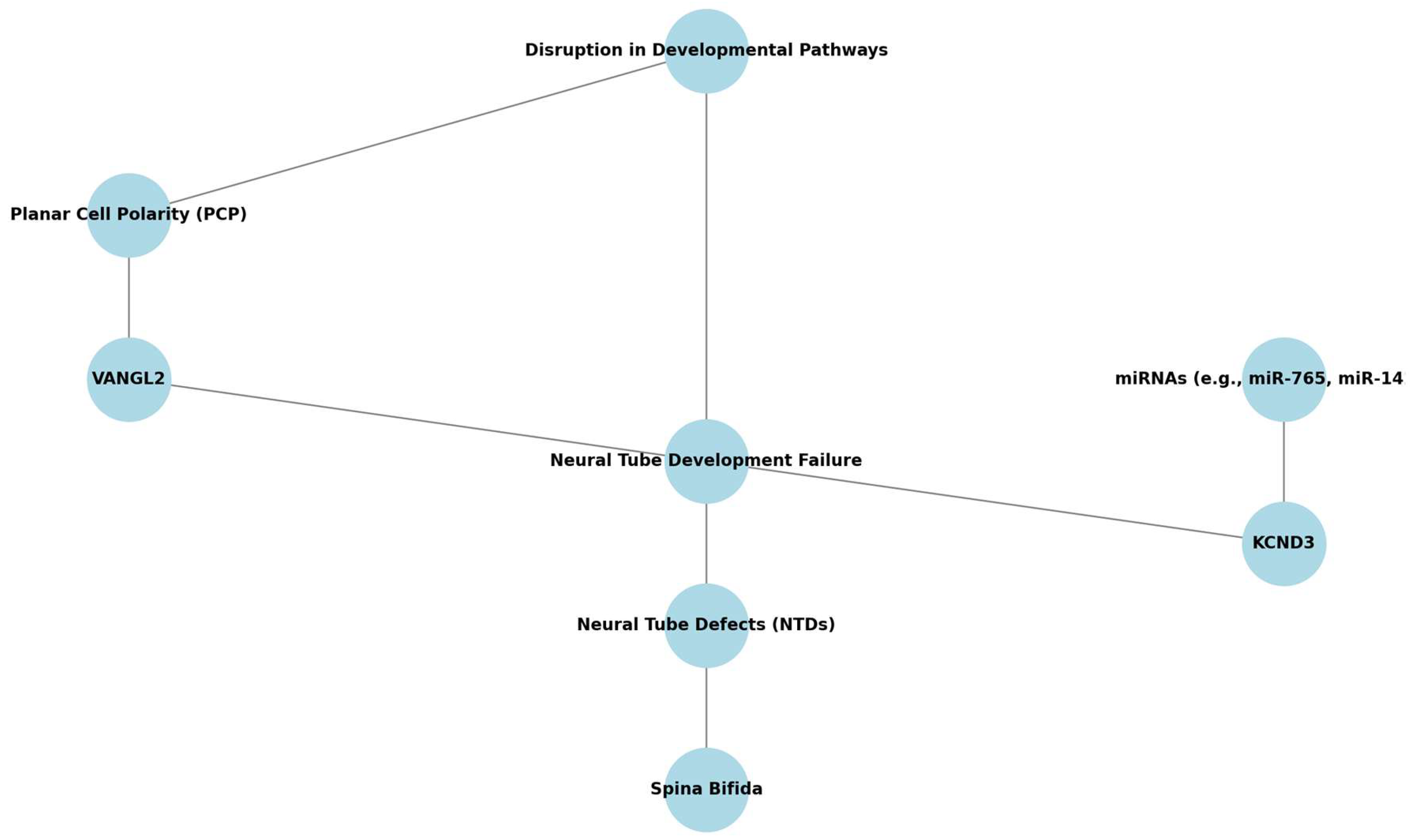
- Top gene–pathway candidates. VANGL2, KCND3 (SB), and GATA4, COUP-TFII (CDH) emerge as high-priority molecular targets for future functional studies.
- miRNAs as non-invasive biomarkers. Dysregulated circulating miRNAs (e.g., miR-320e in SB; miR-379-5p in CDH) show promise for early screening.
- Therapeutic horizons. Novel prenatal interventions—fetoscopic repair (SB), degradable tracheal plugs, and growth-factor modulation (CDH)—illustrate how mechanistic insights can drive clinical innovation.
- Research gaps. Systems-level studies that integrate multi-omics data with in vivo models are needed to unravel why dual SB + CDH presentations are exceptionally rare.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitchell, L.E.; Adzick, N.S.; Melchionne, J.; Pasquariello, P.S.; Sutton, L.N.; Whitehead, A.S. Spina bifida. Lancet 2004, 364, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, P.K.; Rawat, M.; Madappa, R.; Rothstein, D.H.; Lakshminrusimha, S. Congenital Diaphragmatic hernia—A review. Matern. Health Neonatol. Perinatol. 2017, 3, 532–540. [Google Scholar] [CrossRef]
- Rivas, J.F.G.; Clugston, R.D. The etiology of congenital diaphragmatic hernia: The retinoid hypothesis 20 years later. Pediatr. Res. 2023, 95, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, M.; Raffler, G.; Gaffi, M.S.; Antounians, L.; Lauriti, G.; Zani, A. Prevalence and risk factors for congenital diaphragmatic hernia: A global view. J. Pediatr. Surg. 2020, 55, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Shookhoff, J.; Gallicano, G.I. A new perspective on neural tube defects: Folic acid and microRNA misexpression. Genesis 2010, 48, 282–294. [Google Scholar] [CrossRef]
- Seidahmed, M.Z.; Abdelbasit, O.B.; Shaheed, M.M.; Alhussein, K.A.; Miqdad, A.M.; Samadi, A.S.; Khalil, M.I.; Al-Mardawi, E.; Salih, M.A. Genetic, chromosomal, and syndromic causes of neural tube defects. Saudi Med. J. 2014, 35, S49–S56. [Google Scholar]
- Mohd-Zin, S.W.; Marwan, A.I.; Chaar, M.K.A.; Ahmad-Annuar, A.; Abdul-Aziz, N.M. Spina Bifida: Pathogenesis, Mechanisms, and Genes in Mice and Humans. Scientifica 2017, 1, 5364827. [Google Scholar] [CrossRef]
- Perveen, S.; Frigeni, M.; Benveniste, H.; Kurepa, D. Cellular, molecular, and metabolic aspects of developing lungs in congenital diaphragmatic hernia. Front. Pediatr. 2022, 10, 932463. [Google Scholar] [CrossRef]
- Mukherjee, S.; Pasulka, J. Care for Adults with Spina Bifida: Current State and Future Directions. Top. Spinal Cord Inj. Rehabil. 2017, 23, 155–167. [Google Scholar] [CrossRef]
- Thompson, L.P.; Al-Hasan, Y. Impact of Oxidative Stress in Fetal Programming. J. Pregnancy 2012, 1, 582748. [Google Scholar] [CrossRef]
- Prasad, R.; Prasad, R. Congenital Diaphragmatic Hernia: A Major Challenge for Neonatologists. In Congenital Anomalies in Newborn Infants—Clinical and Etiopathological Perspectives; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Joyeux, L.; Chalouhi, G.; Ville, Y.; Sapin, E. Maternal-fetal surgery for spina bifida: Future perspectives. J. Gynecol. Obstet. Biol. Reprod. 2014, 43, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Renik-Jankowska, W.; Buczyńska, A.; Sidorkiewicz, I.; Kosiński, P.; Zbucka-Krętowska, M. Exploring new perspectives on congenital diaphragmatic hernia: A comprehensive review. Biochim. Biophys. Acta Mol. Basis. Dis. 2024, 1870, 167105. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Qi, Y.P.; Yeung, L.F.; Mai, C.T.; Zauche, L.H.; Wang, A.; Daniels, K.; Williams, J.L. Folic Acid and the Prevention of Birth Defects: 30 Years of Opportunity and Controversies. Annu. Rev. Nutr. 2022, 42, 423–452. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Feng, J.; Yuan, Z. Key Modules and Hub Genes Identified by Coexpression Network Analysis for Revealing Novel Biomarkers for Spina Bifida. Front. Genet. 2020, 11, 583316. [Google Scholar] [CrossRef]
- Fares, D.A.; Schalekamp-Timmermans, S.; Nawrot, T.S.; Steegers-Theunissen, R.P.M. Preconception telomere length as a novel maternal biomarker to assess the risk of spina bifida in the offspring. Birth Defects Res. 2020, 112, 645–651. [Google Scholar] [CrossRef]
- Buczyńska, A.; Sidorkiewicz, I.; Niemira, M.; Krętowski, A.J.; Węgrzyn, P.; Kosiński, P.; Zbucka-Krętowska, M. Identification of MicroRNA Profiles in Fetal Spina Bifida: The Role in Pathomechanism and Diagnostic Significance. Int. J. Mol. Sci. 2024, 25, 2896. [Google Scholar] [CrossRef]
- Wolujewicz, P.; Aguiar-Pulido, V.; AbdelAleem, A.; Nair, V.; Thareja, G.; Suhre, K.; Shaw, G.M.; Finnell, R.H.; Elemento, O.; Ross, M.E. Genome-wide investigation identifies a rare copy-number variant burden associated with human spina bifida. Genet. Med. 2021, 23, 1211–1218. [Google Scholar] [CrossRef]
- Scott, D.A.; Gofin, Y.; Berry, A.M.; Adams, A.D. Underlying Genetic Etiologies of Congenital Diaphragmatic Hernia. Prenat. Diagn. 2022, 42, 373–386. [Google Scholar] [CrossRef]
- Yu, L.; Wynn, J.; Ma, L.; Guha, S.; Mychaliska, G.B.; Crombleholme, T.M.; Azarow, K.S.; Lim, F.Y.; Chung, D.H.; Potoka, D.; et al. De novo copy number variants are associated with congenital diaphragmatic hernia. J. Med. Genet. 2012, 49, 650–659. [Google Scholar] [CrossRef]
- Zhu, Q.; High, F.A.; Zhang, C.; Cerveira, E.; Russell, M.K.; Longoni, M.; Joy, M.P.; Ryan, M.; Mil-Homens, A.; Bellfy, L.; et al. Systematic analysis of copy number variation associated with congenital diaphragmatic hernia. Proc. Natl. Acad. Sci. USA 2018, 115, 5247–5252. [Google Scholar] [CrossRef]
- Khawale, R.; Kanetkar, S.R.; Patil, M. Impact of Hypothyroidism in Pregnancy on Feto-Maternal Outcomes: A Prospective Observational Study. Cureus 2024, 16, e74494. [Google Scholar] [CrossRef]
- Miller, J.L.; Groves, M.L.; Baschat, A.A. Fetoscopic spina bifida repair. Minerva Obstet. Gynecol. 2019, 71, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Doné, E.; Gratacos, E.; Nicolaides, K.H.; Allegaert, K.; Valencia, C.; Castañon, M.; Martinez, J.; Jani, J.; Van Mieghem, T.; Greenough, A.; et al. Predictors of neonatal morbidity in fetuses with severe isolated congenital diaphragmatic hernia undergoing fetoscopic tracheal occlusion. Ultrasound Obstet. Gynecol. 2013, 42, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Altman, D.; Antes, G.; Atkins, D.; Barbour, V.; Barrowman, N.; Berlin, J.A.; et al. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Hutton, B.; Salanti, G.; Caldwell, D.M.; Chaimani, A.; Schmid, C.H.; Cameron, C.; Ioannidis, J.P.A.; Straus, S.; Thorlund, K.; Jansen, J.P.; et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: Checklist and explanations. Ann. Intern. Med. 2015, 162, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Stang, A.; Jonas, S.; Poole, C. Case study in major quotation errors: A critical commentary on the Newcastle–Ottawa scale. Eur. J. Epidemiol. 2018, 33, 1025–1031. [Google Scholar] [CrossRef]
- Atta, C.A.M.; Fiest, K.M.; Frolkis, A.D.; Jette, N.; Pringsheim, T.; Germaine-Smith, C.S.; Rajapakse, T.; Kaplan, G.G.; Metcalfe, A. Global birth prevalence of spina bifida by folic acid fortification status: A systematic review and meta-analysis. Am. J. Public Health 2016, 106, e24–e34. [Google Scholar] [CrossRef]
- Au, K.S.; Ashley-Koch, A.; Northrup, H. Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Dev. Disabil. Res. Rev. 2010, 16, 6–15. [Google Scholar] [CrossRef]
- Mai, C.T.; Isenburg, J.L.; Canfield, M.A.; Meyer, R.E.; Correa, A.; Alverson, C.J.; Lupo, P.J.; Riehle-Colarusso, T.; Cho, S.J.; Aggarwal, D.; et al. National population-based estimates for major birth defects, 2010–2014. Birth Defects Res. 2019, 111, 1420–1435. [Google Scholar] [CrossRef]
- Sahmat, A.; Gunasekaran, R.; Mohd-Zin, S.W.; Balachandran, L.; Thong, M.-K.; Engkasan, J.P.; Ganesan, D.; Omar, Z.; Azizi, A.B.; Ahmad-Annuar, A.; et al. The prevalence and distribution of spina bifida in a single major referral center in Malaysia. Front. Pediatr. 2017, 5, 301795. [Google Scholar] [CrossRef]
- Dumpa, V.; Chandrasekharan, P. Congenital Diaphragmatic Hernia. In StatPearls; StatPearls Publishing: Tampa/St. Petersburg, FL, USA, 2023. [Google Scholar]
- Kraemer, U.S.; Leeuwen, L.; Krasemann, T.B.; Wijnen, R.M.H.; Tibboel, D.; Ijsselstijn, H. Characteristics of Infants With Congenital Diaphragmatic Hernia Who Need Follow-Up of Pulmonary Hypertension. Pediatr. Crit. Care Med. 2018, 19, e219–e226. [Google Scholar] [CrossRef] [PubMed]
- Keijzer, R.; Puri, P. Congenital diaphragmatic hernia. Semin. Pediatr. Surg. 2010, 19, 180–185. [Google Scholar] [CrossRef]
- Copp, A.J.; Adzick, N.S.; Chitty, L.S.; Fletcher, J.M.; Holmbeck, G.N.; Shaw, G.M. Spina bifida. Nat. Rev. Dis. Prim. 2015, 1, 15007. [Google Scholar] [CrossRef] [PubMed]
- Brea, C.M.; Munakomi, S. Spina Bifida. In StatPearls; StatPearls Publishing: Tampa/St. Petersburg, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK559265/ (accessed on 19 October 2023).
- Harting, M.T. Congenital diaphragmatic hernia-associated pulmonary hypertension. Semin. Pediatr. Surg. 2017, 26, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Adzick, N.S. Prenatal diagnosis and treatment of spina bifida. Fetal Diagn. Ther. 2015, 37, 165. [Google Scholar] [CrossRef]
- Tinker, S.C.; Devine, O.; Mai, C.; Hamner, H.C.; Reefhuis, J.; Gilboa, S.M.; Dowling, N.F.; Honein, M.A. Estimate of the potential impact of folic acid fortification of corn masa flour on the prevention of neural tube defects. Birth Defects Res. Part A: Clin. Mol. Teratol. 2013, 97, 649–657. [Google Scholar] [CrossRef]
- Buczyńska, A.; Sidorkiewicz, I.; Hameed, A.; Krętowski, A.J.; Zbucka-Krętowska, M. Future Perspectives in Oxidative Stress in Trisomy 13 and 18 Evaluation. J. Clin. Med. 2022, 11, 1787. [Google Scholar] [CrossRef]
- Smets, K.; Duarri, A.; Deconinck, T.; Ceulemans, B.; van de Warrenburg, B.P.; Züchner, S.; Gonzalez, M.A.; Schüle, R.; Synofzik, M.; Van der Aa, N.; et al. First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Med. Genet. 2015, 16, 51. [Google Scholar] [CrossRef]
- Marini, N.J.; Hoffmann, T.J.; Lammer, E.J.; Hardin, J.; Lazaruk, K.; Stein, J.B.; Gilbert, D.A.; Wright, C.; Lipzen, A.; Pennacchio, L.A.; et al. A Genetic Signature of Spina Bifida Risk from Pathway-Informed Comprehensive Gene-Variant Analysis. PLoS ONE 2011, 6, e28408. [Google Scholar] [CrossRef]
- Price, E.M.; Peñaherrera, M.S.; Portales-Casamar, E.; Pavlidis, P.; Van Allen, M.I.; McFadden, D.E.; Robinson, W.P. Profiling placental and fetal DNA methylation in human neural tube defects. Epigenetics Chromatin 2016, 9, 6. [Google Scholar] [CrossRef]
- Scholten, R.H.; Møller, P.; Andersen, Z.J.; Dehlendorff, C.; Khan, J.; Brandt, J.; Ketzel, M.; Knudsen, L.E.; Mathiesen, L. Telomere length in newborns is associated with exposure to low levels of air pollution during pregnancy. Environ. Int. 2021, 146, 106202. [Google Scholar] [CrossRef]
- Zhao, X.-X.; Le Bai, L. Correlation between telomere shortening in maternal peripheral blood and fetal aneuploidy. BMC Pregnancy Childbirth 2024, 24, 2. [Google Scholar] [CrossRef]
- Chen, L.; Tan, K.M.L.; Gong, M.; Chong, M.F.F.; Tan, K.H.; Chong, Y.S.; Meaney, M.J.; Gluckman, P.D.; Eriksson, J.G.; Karnani, N. Variability in newborn telomere length is explained by inheritance and intrauterine environment. BMC Med. 2022, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Factor-Litvak, P.; Susser, E.; Aviv, A. Environmental Exposures, Telomere Length at Birth, and Disease Susceptibility in Later Life. JAMA Pediatr. 2017, 171, 1143–1144. [Google Scholar] [CrossRef]
- Hemann, M.T.; Strong, M.A.; Hao, L.-Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Alder, J.K.; Hanumanthu, V.S.; Strong, M.A.; DeZern, A.E.; Stanley, S.E.; Takemoto, C.M.; Danilova, L.; Applegate, C.D.; Bolton, S.G.; Mohr, D.W.; et al. Diagnostic utility of telomere length testing in a hospital-based setting. Proc. Natl. Acad. Sci. USA 2018, 115, E2358–E2365. [Google Scholar] [CrossRef] [PubMed]
- Bhala, S.; Savage, S.A. What is the future of telomere length testing in telomere biology disorders? Expert Rev. Hematol. 2023, 16, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5, 89. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef]
- Thielen, E.; Oria, M.; Watanabe-Chailland, M.; Lampe, K.; Romick-Rosendale, L.; Peiro, J.L. Non-Targeted Metabolic Profiling of Cerebellum in Spina Bifida Fetal Rats. Metabolites 2023, 13, 670. [Google Scholar] [CrossRef]
- Punda, H.; Mardesic, S.; Filipovic, N.; Kosovic, I.; Benzon, B.; Ogorevc, M.; Bocina, I.; Kolic, K.; Vukojevic, K.; Saraga-Babic, M. Expression pattern of 5-ht (Serotonin) receptors during normal development of the human spinal cord and ganglia and in fetus with cervical spina bifida. Int. J. Mol. Sci. 2021, 22, 7320. [Google Scholar] [CrossRef] [PubMed]
- Clugston, R.D.; Zhang, W.; Álvarez, S.; de Lera, A.R.; Greer, J.J. Understanding abnormal retinoid signaling as a causative mechanism in congenital diaphragmatic hernia. Am. J. Respir. Cell Mol. Biol. 2010, 42, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Coste, K.; Beurskens, L.W.J.E.; Blanc, P.; Gallot, D.; Delabaere, A.; Blanchon, L.; Tibboel, D.; Labbé, A.; Rottier, R.J.; Sapin, V. Metabolic disturbances of the vitamin A pathway in human diaphragmatic hernia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L147–L157. [Google Scholar] [CrossRef]
- Friedmacher, F.; Puri, P. Disruptions in retinoic acid signaling pathway contribute to abnormal lung development in congenital diaphragmatic hernia: A therapeutic potential for retinoids to attenuate pulmonary hypoplasia. Pediatr. Res. 2024, 95, 1415–1417. [Google Scholar] [CrossRef]
- Ackerman, K.G.; Herron, B.J.; O Vargas, S.; Huang, H.; Tevosian, S.G.; Kochilas, L.; Rao, C.; Pober, B.R.; Babiuk, R.P.; A Epstein, J.; et al. Fog2 Is Required for Normal Diaphragm and Lung Development in Mice and Humans. PLoS Genet. 2005, 1, e10–e65. [Google Scholar] [CrossRef]
- Yu, L.; Wynn, J.; Cheung, Y.H.; Shen, Y.; Mychaliska, G.B.; Crombleholme, T.M.; Azarow, K.S.; Lim, F.Y.; Chung, D.H.; Potoka, D.; et al. Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Hum. Genet. 2013, 132, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.M.; Mathieu, Y.; Luzón-Toro, B.; Núñez-Torres, R.; González-Meneses, A.; Antiñolo, G.; Amiel, J.; Borrego, S.; Veitia, R.A. Contributions of PHOX2B in the Pathogenesis of Hirschsprung Disease. PLoS ONE 2013, 8, e54043. [Google Scholar] [CrossRef]
- Kantarci, S.; Ragge, N.K.; Thomas, N.S.; Robinson, D.O.; Noonan, K.M.; Russell, M.K.; Donnai, D.; Raymond, F.L.; Walsh, C.A.; Donahoe, P.K.; et al. Donnai–Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am. J. Med. Genet. Part A 2008, 146, 1842–1847. [Google Scholar] [CrossRef]
- Pober, B. Genetic aspects of human congenital diaphragmatic hernia. Clin. Genet. 2008, 74, 1. [Google Scholar] [CrossRef]
- Zaiss, I.; Kehl, S.; Link, K.; Neff, W.; Schaible, T.; Sütterlin, M.; Siemer, J. Associated malformations in congenital diaphragmatic hernia. Am. J. Perinatol. 2011, 28, 211–218. [Google Scholar] [CrossRef]
- Scott, D.A.; Klaassens, M.; Holder, A.M.; Lally, K.P.; Fernandes, C.J.; Galjaard, R.-J.; Tibboel, D.; de Klein, A.; Lee, B. Genome-wide oligonucleotide-based array comparative genome hybridization analysis of non-isolated congenital diaphragmatic hernia. Hum. Mol. Genet. 2007, 16, 424–430. [Google Scholar] [CrossRef]
- Wynn, J.; Yu, L.; Chung, W.K. Genetic causes of congenital diaphragmatic hernia. Semin. Fetal Neonatal Med. 2014, 19, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.O.; Bazeley, P.; Erzurum, S.C.; Asosingh, K. Single-cell transcriptomic profiling of microvascular endothelial cell heterogeneity in congenital diaphragmatic hernia. Sci. Rep. 2023, 13, 9581. [Google Scholar] [CrossRef]
- Dalmer, T.R.A.; Clugston, R.D. Gene ontology enrichment analysis of congenital diaphragmatic hernia-associated genes. Pediatr. Res. 2018, 85, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Fabietti, I.; Nardi, T.; Favero, C.; Dioni, L.; Cantone, L.; Pergoli, L.; Hoxha, M.; Pinatel, E.; Mosca, F.; Bollati, V.; et al. Extracellular vesicles and their mirna content in amniotic and tracheal fluids of fetuses with severe congenital diaphragmatic hernia undergoing fetal intervention. Cells 2021, 10, 1493. [Google Scholar] [CrossRef]
- Tian, L.; Zhang, L.; Liu, J.; Guo, T.; Gao, C.; Ni, J. Effects of TSH on the function of human umbilical vein endothelial cells. J. Mol. Endocrinol. 2014, 52, 215–222. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhao, L.; Yue, L.; Zhang, W.; Wang, W.; Fu, Y.; Feng, Y.; Fu, F. The relationship between IGF1 and the expression spectrum of miRNA in the placenta of preeclampsia patients. Ginekol. Pol. 2019, 90, 596–603. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, Y.; Li, L.; Guo, M.; Zou, C.; Xu, Y.; Yang, Z. MicroRNA-889-3p restrains the proliferation and epithelial—Mesenchymal transformation of lung cancer cells via down-regulation of Homeodomain-interacting protein kinase 1. Bioengineered 2021, 12, 10945–10958. [Google Scholar] [CrossRef]
- Srisupundit, K.; Brady, P.D.; Devriendt, K.; Fryns, J.; Cruz-Martinez, R.; Gratacos, E.; Deprest, J.A.; Vermeesch, J.R. Targeted array comparative genomic hybridisation (array CGH) identifies genomic imbalances associated with isolated congenital diaphragmatic hernia (CDH). Prenat. Diagn. 2010, 30, 1198–1206. [Google Scholar] [CrossRef]
- Bhutada, S.; Tran-Lundmark, K.; Kramer, B.; Conner, P.; Lowry, A.M.; Blackstone, E.; Frenckner, B.; Mesas-Burgos, C.; Apte, S.S. Identification of protein biomarkers associated with congenital diaphragmatic hernia in human amniotic fluid. Sci. Rep. 2023, 13, 15483. [Google Scholar] [CrossRef]
- Wagner, R.; Lieckfeldt, P.; Piyadasa, H.; Markel, M.; Riedel, J.; Stefanovici, C.; Peukert, N.; Patel, D.; Derraugh, G.; Min, S.A.L.; et al. Proteomic Profiling of Hypoplastic Lungs Suggests an Underlying Inflammatory Response in the Pathogenesis of Abnormal Lung Development in Congenital Diaphragmatic Hernia. Ann. Surg. 2023, 278, e411–e421. [Google Scholar] [CrossRef]
- Jayashankar, S.S.; Nasaruddin, M.L.; Hassan, M.F.; Dasrilsyah, R.A.; Shafiee, M.N.; Ismail, N.A.S.; Alias, E. Non-Invasive Prenatal Testing (NIPT): Reliability, Challenges, and Future Directions. Diagnostics 2023, 13, 2570. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Leonard, A.; Essawi, K.; Tisdale, J.F. CRISPR-Cas9 to induce fetal hemoglobin for the treatment of sickle cell disease. Mol. Ther. Methods Clin. Dev. 2021, 23, 276–285. [Google Scholar] [CrossRef]
- Papizan, J.B.; Porter, S.N.; Sharma, A.; Pruett-Miller, S.M. Therapeutic gene editing strategies using CRISPR-Cas9 for the β-hemoglobinopathies. J. Biomed. Res. 2020, 35, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Galea, G.L.; Nychyk, O.; Mole, M.A.; Moulding, D.; Savery, D.; Nikolopoulou, E.; Henderson, D.J.; Greene, N.D.E.; Copp, A.J. Vangl2 disruption alters the biomechanics of late spinal neurulation leading to spina bifida in mouse embryos. Dis. Model. Mech. 2018, 11, dmm032219. [Google Scholar] [CrossRef] [PubMed]
- Campiglio, C.E.; Villonio, M.; Dellacà, R.L.; Mosca, F.; Draghi, L. An injectable, degradable hydrogel plug for tracheal occlusion in congenital diaphragmatic hernia (CDH). Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 99, 430–439. [Google Scholar] [CrossRef]
- Jesudason, E.C.; Connell, M.; Fernig, D.G.; Lloyd, D.A.; Losty, P.D. In vitro effects of growth factors on lung hypoplasia in a model of congenital diaphragmatic hernia. J. Pediatr. Surg. 2000, 35, 914–922. [Google Scholar] [CrossRef]
- Peiro, J.L.; Oria, M.; Aydin, E.; Joshi, R.; Cabanas, N.; Schmidt, R.; Schroeder, C.; Marotta, M.; Varisco, B.M. Proteomic profiling of tracheal fluid in an ovine model of congenital diaphragmatic hernia and fetal tracheal occlusion. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L1028–L1041. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Condition | Prevalence | Classification | Key Characteristics | References |
|---|---|---|---|---|
| SB | Global incidence: 1.0–10.0 per 1000 newborns Approx. 1427 cases annually (1 in 2758 live births) | NTDs: Anencephaly: Incomplete closure in the cranial region SB: Incomplete closure below the cranial region SB Subtypes: Myelomeningocele: >90% of cases Meningocele - Lipomeningocele | Myelomeningocele: Protrusion of nervous tissue and protective coverings through a vertebral defect Most common type of SB | [1,35,36] |
| CDH | Global incidence: 1–4 per 10,000 live births | Types of Hernias: - Bochdalek Hernia: 70–75%; postero-lateral defect (mainly left side) - Morgagni Hernia: 20–25%; anteromedial defect - Central Hernia: 2–5%; rare and severe | - Bochdalek Hernia: Most prevalent, affecting diaphragm development - Central Hernia: Rare, associated with grave prognosis | [11,32,34,37] |
| Aspect/Source | [15] | [16] | [17,42] |
|---|---|---|---|
| Objective | Analyze transcription profiles from human amniocytes to identify genes related to SB and healthy controls. | Investigate how periconceptional factors affect NTD pathogenesis and prevention. | Examine the persistence of SB in a folate-replete population and explore potential epigenetic factors. |
| Data Source | Gene Expression Omnibus database | Experiments with embryonic mice and literature review | Fetal tissue samples collected from different NTD status groups in Canada |
| Initial Data Processing | Outlier data identified and removed using PCA and sample clustering. | N/A | Analysis of DNA methylation (DNAm) using the Illumina Infinium HumanMethylation450 array. |
| Analysis Method | WGCNA | Telomere length analysis and experimental observations | Assessment of DNAm in chorionic villi and kidney samples, and evaluation of Methylenetetrahydrofolate reductase genotypes. |
| Dataset Analyzed | GSE4182 dataset | Observations from embryonic mice deficient in the telomerase gene | Fetal tissue DNAm data and Methylenetetrahydrofolate reductase genotypes. |
| Genes Analyzed | 5407 genes categorized into 19 distinct modules | Telomere length associated with developmental processes | Differentially methylated sites in anencephaly and spina bifida compared to controls. |
| Findings | Identified 967 candidate genes associated with SB; miRNA–mRNA network with 4 miRNAs and 39 mRNAs. | Excessive TL shortening linked to neural tube closure failure in mice. | No significant DNAm differences in repetitive elements; specific DNAm changes observed in chorionic villi and kidneys. |
| Key Genes/MiRNAs Identified | KCND3 (upregulated in SB); associated miRNAs: miR-765 and miR-142-3p. | N/A | Notable DNAm changes in spina bifida kidneys and anencephaly. |
| Validation Method | Quantitative real-time PCR (qRT-PCR) | N/A | Analysis of DNAm and genotyping results. |
| Hypothesis/Conclusions | KCND3 and its associated miRNAs are promising for early detection and noninvasive screening in SB. | Long-term exposure to harmful factors accelerates maternal aging, increasing NTD risk. Alternatively, higher NTD risk may correlate with advanced biological age before pregnancy. | Persistent NTDs in folate-sufficient populations may involve alternative epigenetic mechanisms, with specific DNAm changes observed in spina bifida kidneys. |
| Aspect/Source | [64] | [66] | [67,72] | [68] |
|---|---|---|---|---|
| Objective | Detect and validate chromosomal abnormalities in CDH patients. | Identify transcriptomic signatures in microvascular endothelial cells (mvECs) related to CDH. | Investigate genes associated with CDH and their pathways. | Examine extracellular vesicle (EV) concentrations and miRNA expression in CDH fetuses. |
| Data Source | 26 patients with CDH | Single-cell RNA sequencing of mvECs from CDH and control samples | Literature review and gene ontology analysis | Amniotic and tracheal fluids of fetuses undergoing FETO |
| Analysis Method | Genome-wide oligonucleotide-based array comparative genomic hybridization (aCGH) and quantitative real-time PCR. | Unbiased clustering approach in single-cell RNA sequencing. | Gene ontology analysis and review of existing literature on CDH-associated genes. | Analysis of EV concentrations and miRNA expression levels. |
| Findings | Identified genes on chromosomes 2q37, 6p22-25, and 14q; minimal deleted region on 15q26 includes COUP-TFII. | Unique inflammatory transcriptomic signature in CDH-associated mvECs; reduced mvCa4 + ECs in CDH cases. | 218 genes associated with CDH identified; distinct pathways related to various CDH types. | Increased EV concentration in non-surviving CDH infants; upregulation of miR-379-5p and miR-889-3p in pre-FETO amniotic fluid. |
| Key Genes/MiRNAs Identified | COUP-TFII and genes on chromosomes 2q37, 6p22-25, 14q; 15q26 deletions. | miR-379-5p, miR-889-3p; inflammatory markers in mvECs. | 218 CDH-associated genes; pathways related to retinoic acid signaling, myogenesis, and angiogenesis. | miR-379-5p targets IGF1; miR-889-3p targets FGFR2. |
| Validation Method | Comparison of chromosomal abnormalities using aCGH and PCR. | Comparison of transcriptomic profiles between CDH and control endothelial cells. | Gene ontology analysis and comparison of gene functions. | Analysis of miRNA expression in pre-FETO amniotic fluid and association with survival outcomes. |
| Hypothesis/Conclusions | Oligonucleotide-based aCGH and PCR are effective for identifying and mapping CDH-related chromosomal changes. | Distinct mvEC clusters in CDH have unique inflammatory signatures and reduced mvCa4 + ECs. | Gene pathways are significantly associated with different forms of CDH, providing insights into disease mechanisms. | Elevated EV concentrations and specific miRNAs in CDH fetuses are linked to survival outcomes and disease mechanisms. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buczyńska, A.; Sidorkiewicz, I.; Kosiński, P.; Krętowski, A.J.; Zbucka-Krętowska, M. Integrative Review of Molecular, Metabolic, and Environmental Factors in Spina Bifida and Congenital Diaphragmatic Hernia: Insights into Mechanisms and Emerging Therapeutics. Cells 2025, 14, 1059. https://doi.org/10.3390/cells14141059
Buczyńska A, Sidorkiewicz I, Kosiński P, Krętowski AJ, Zbucka-Krętowska M. Integrative Review of Molecular, Metabolic, and Environmental Factors in Spina Bifida and Congenital Diaphragmatic Hernia: Insights into Mechanisms and Emerging Therapeutics. Cells. 2025; 14(14):1059. https://doi.org/10.3390/cells14141059
Chicago/Turabian StyleBuczyńska, Angelika, Iwona Sidorkiewicz, Przemysław Kosiński, Adam Jacek Krętowski, and Monika Zbucka-Krętowska. 2025. "Integrative Review of Molecular, Metabolic, and Environmental Factors in Spina Bifida and Congenital Diaphragmatic Hernia: Insights into Mechanisms and Emerging Therapeutics" Cells 14, no. 14: 1059. https://doi.org/10.3390/cells14141059
APA StyleBuczyńska, A., Sidorkiewicz, I., Kosiński, P., Krętowski, A. J., & Zbucka-Krętowska, M. (2025). Integrative Review of Molecular, Metabolic, and Environmental Factors in Spina Bifida and Congenital Diaphragmatic Hernia: Insights into Mechanisms and Emerging Therapeutics. Cells, 14(14), 1059. https://doi.org/10.3390/cells14141059