

From Brain to Blood: Uncovering Potential Therapeutical Targets and Biomarkers for Huntington’s Disease Using an Integrative RNA-Seq Analytical Platform (BDASeq®)

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
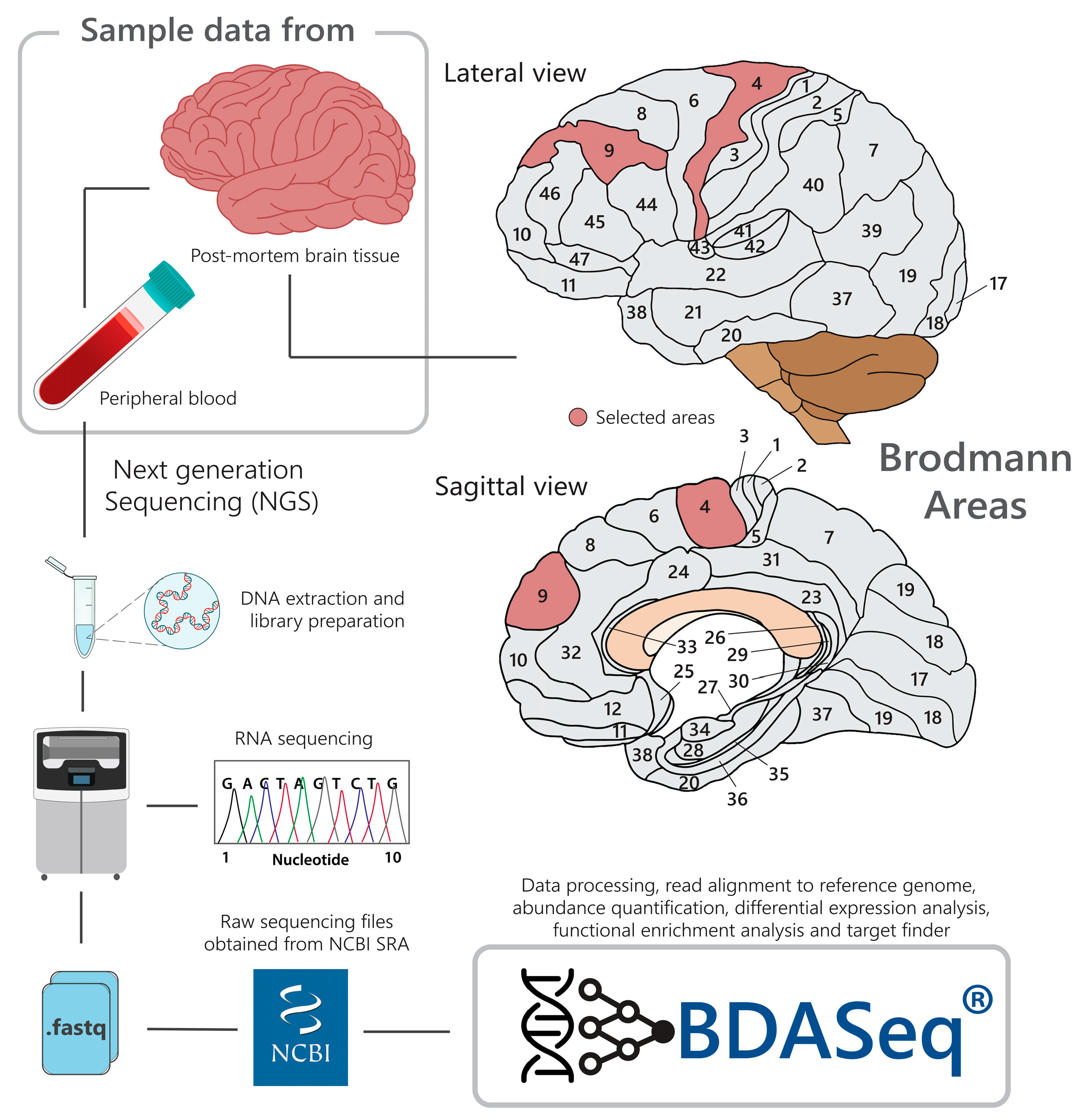
2.2. BioProject (Data) Description
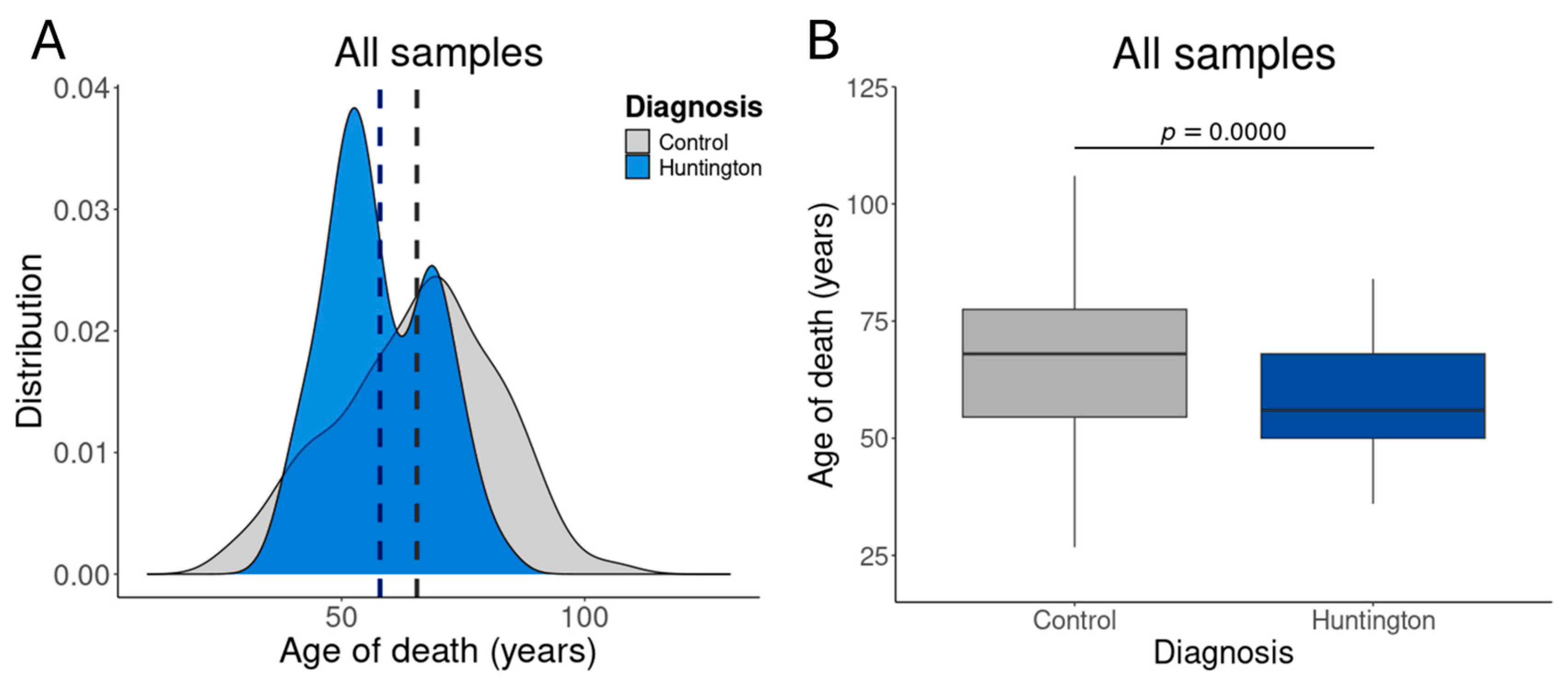
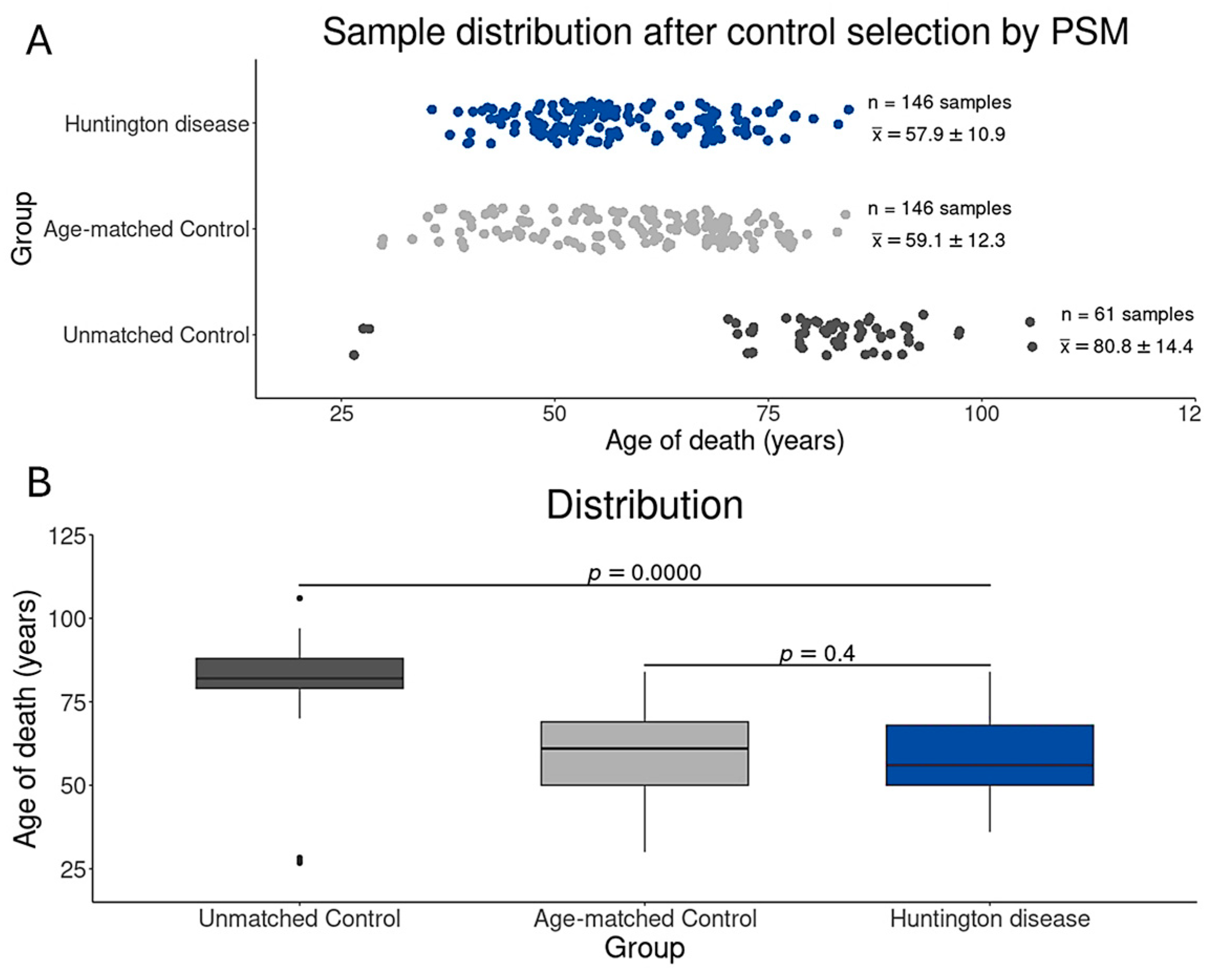
2.3. Control Sample Selection by Age Distribution
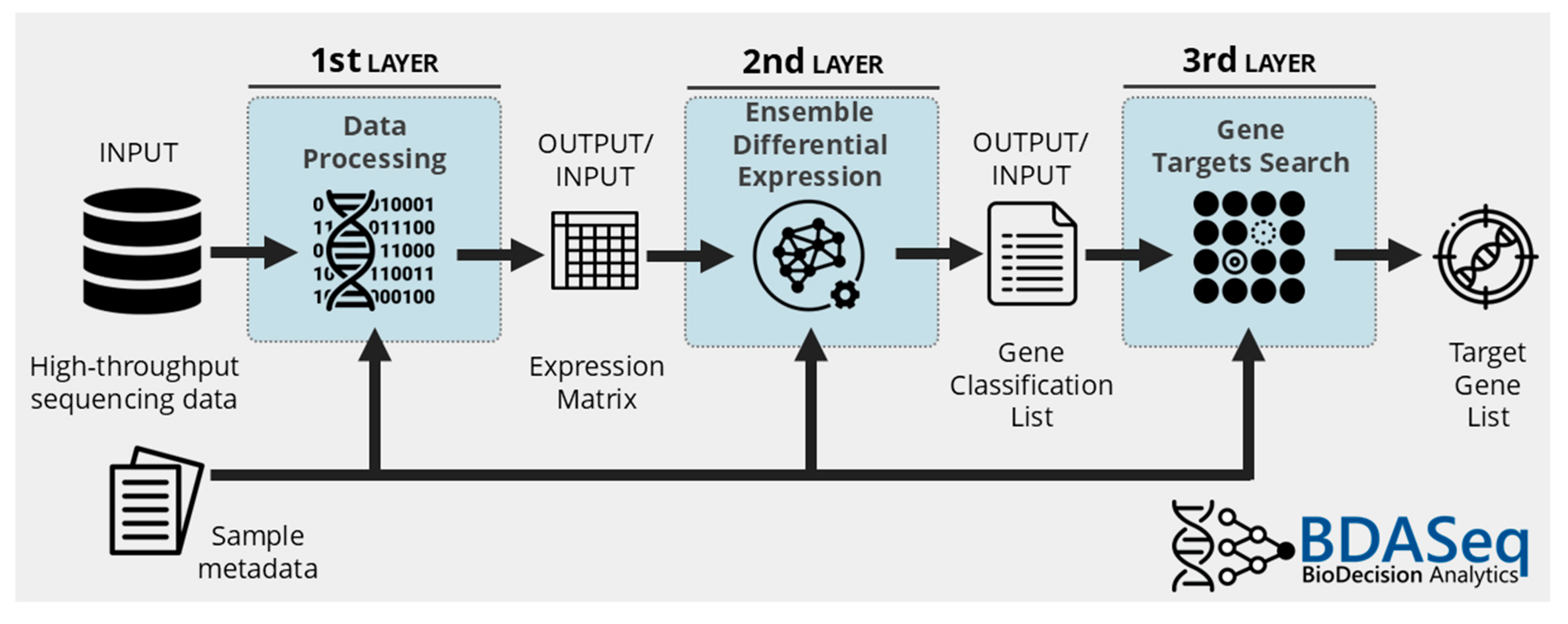
2.4. Data Processing, Differential Expression Analysis, and Target Discovery Using BDASeq®
3. Results
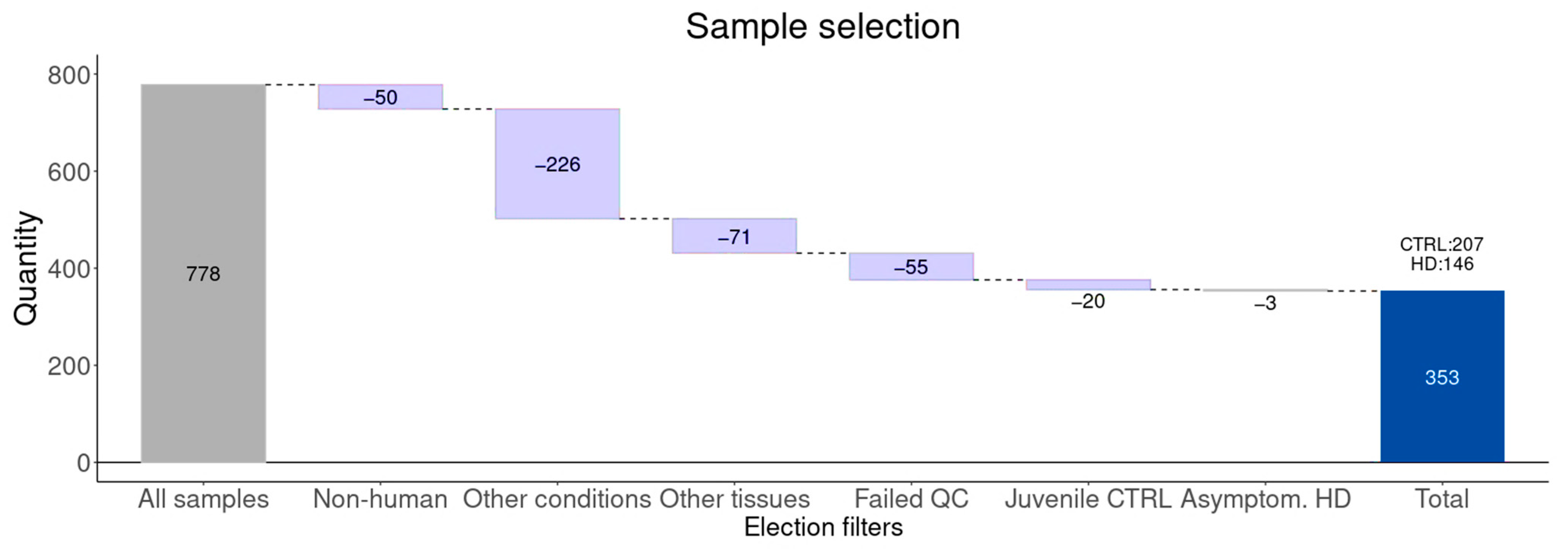
3.1. Study Design and Sample Selection
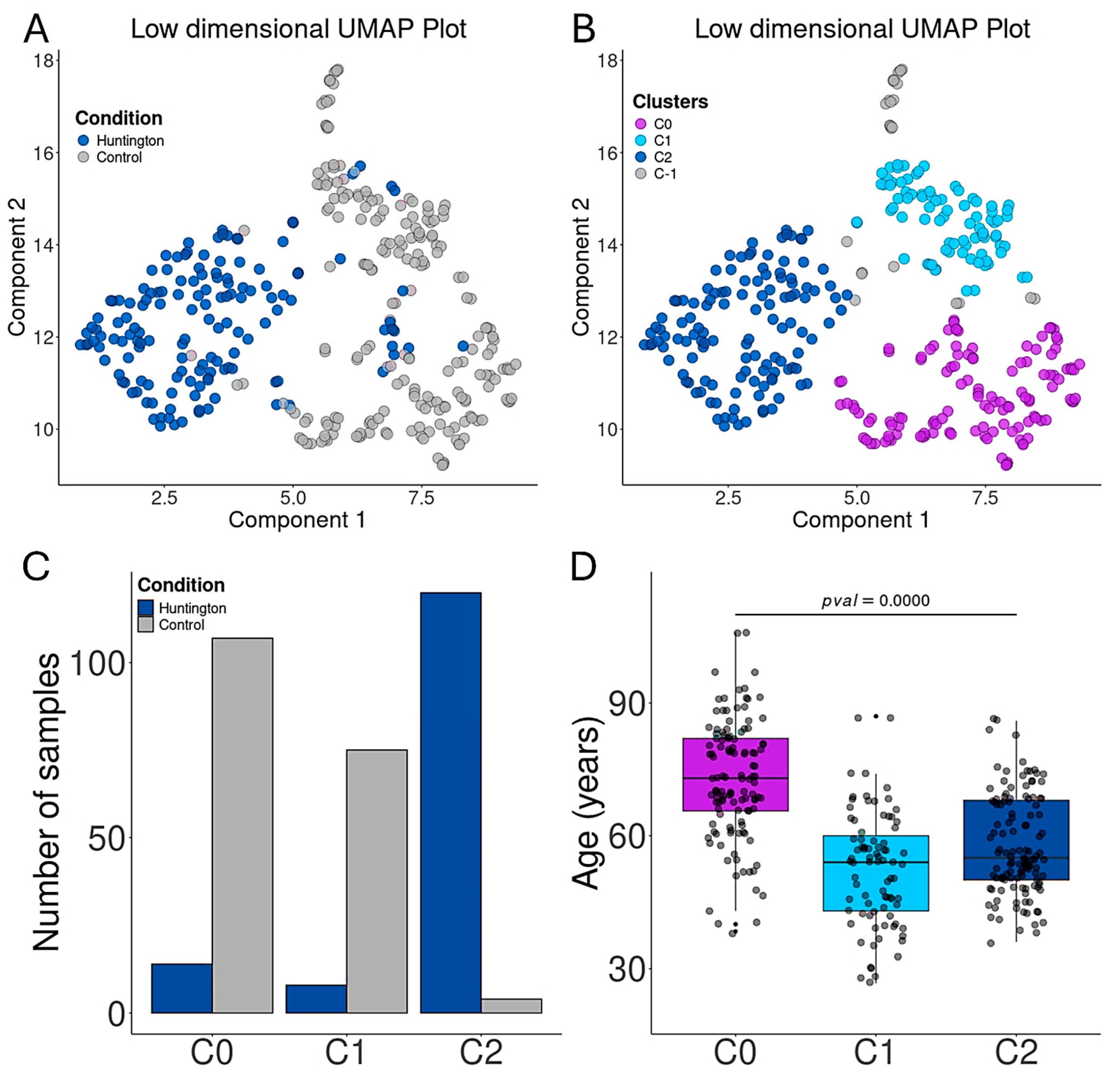
3.2. Aging Promotes Transcriptional Alterations in the Prefrontal Cortex
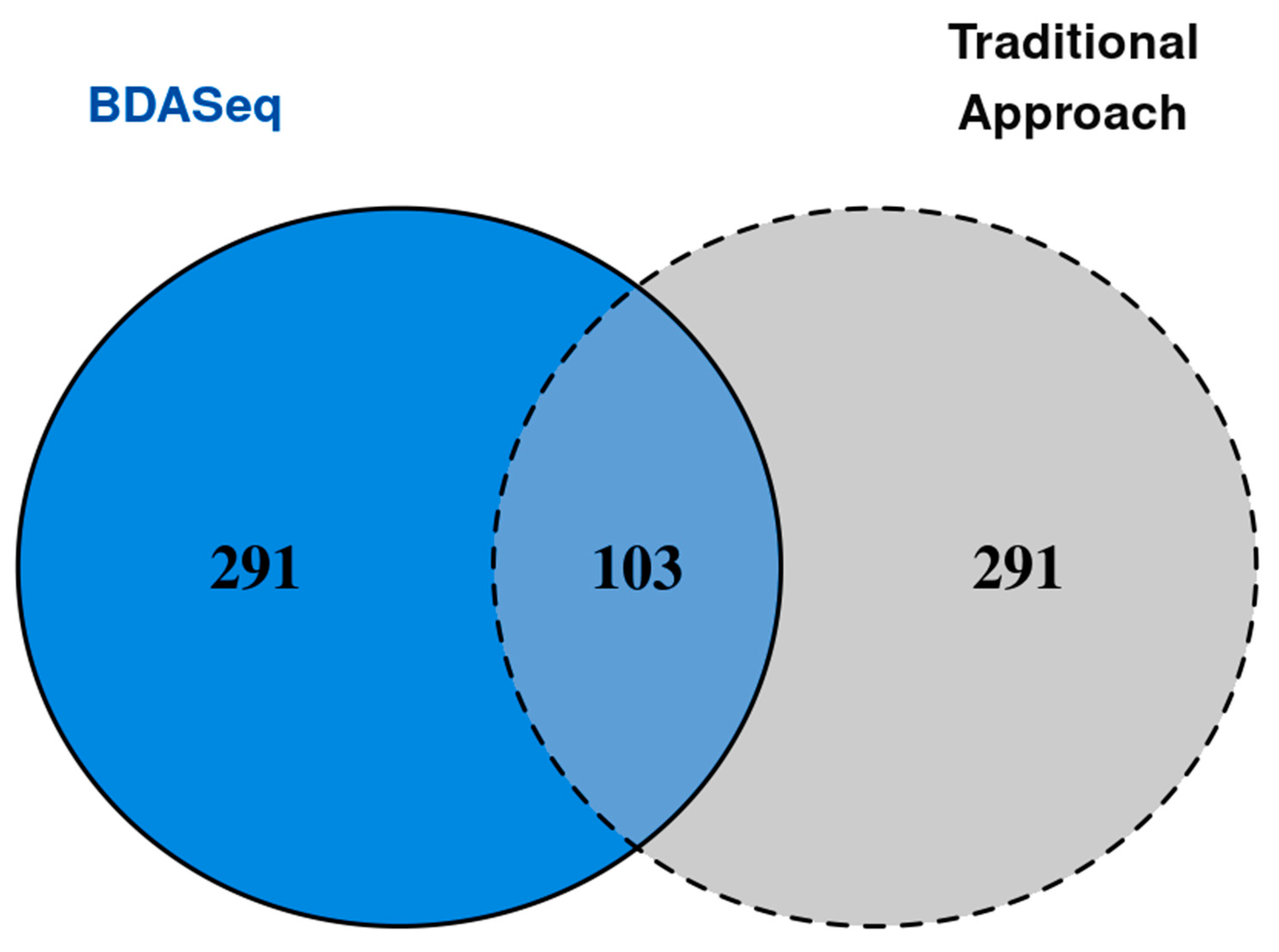
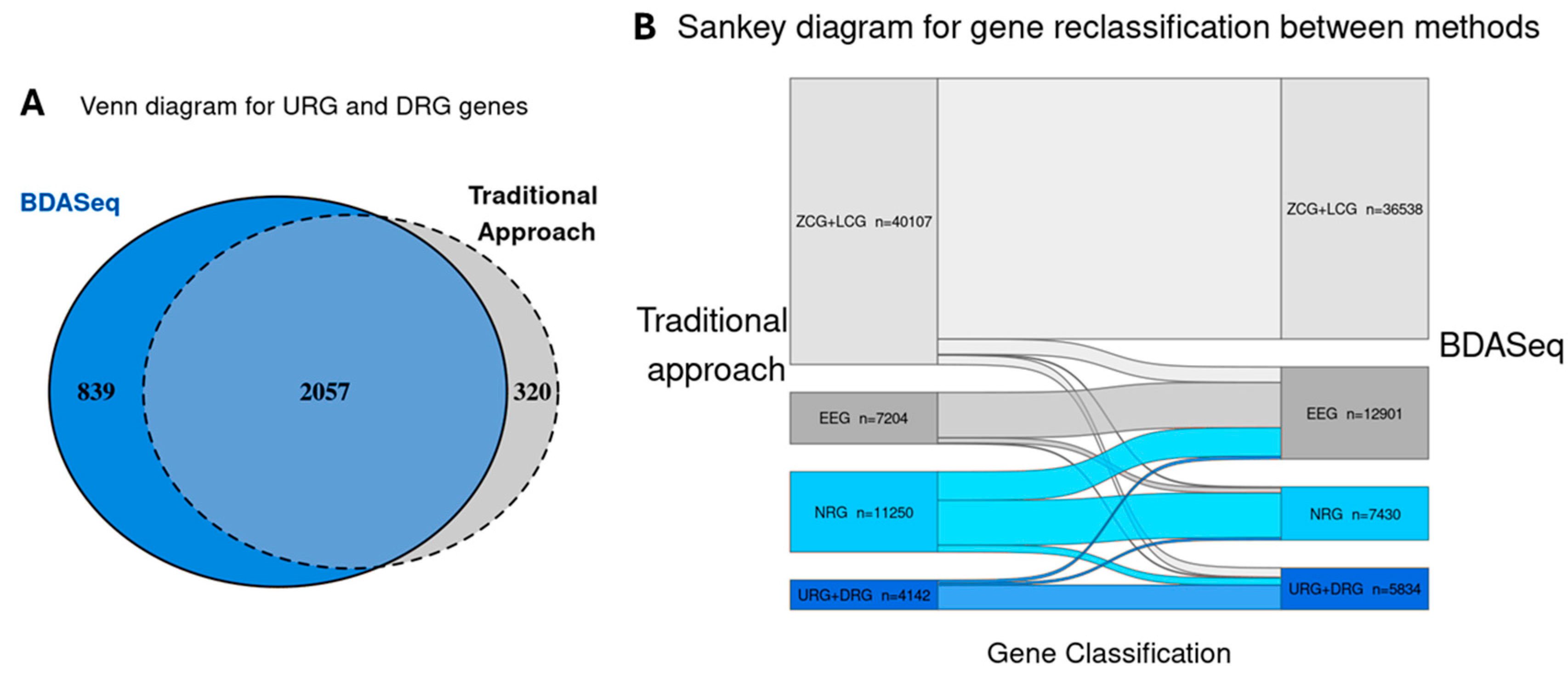
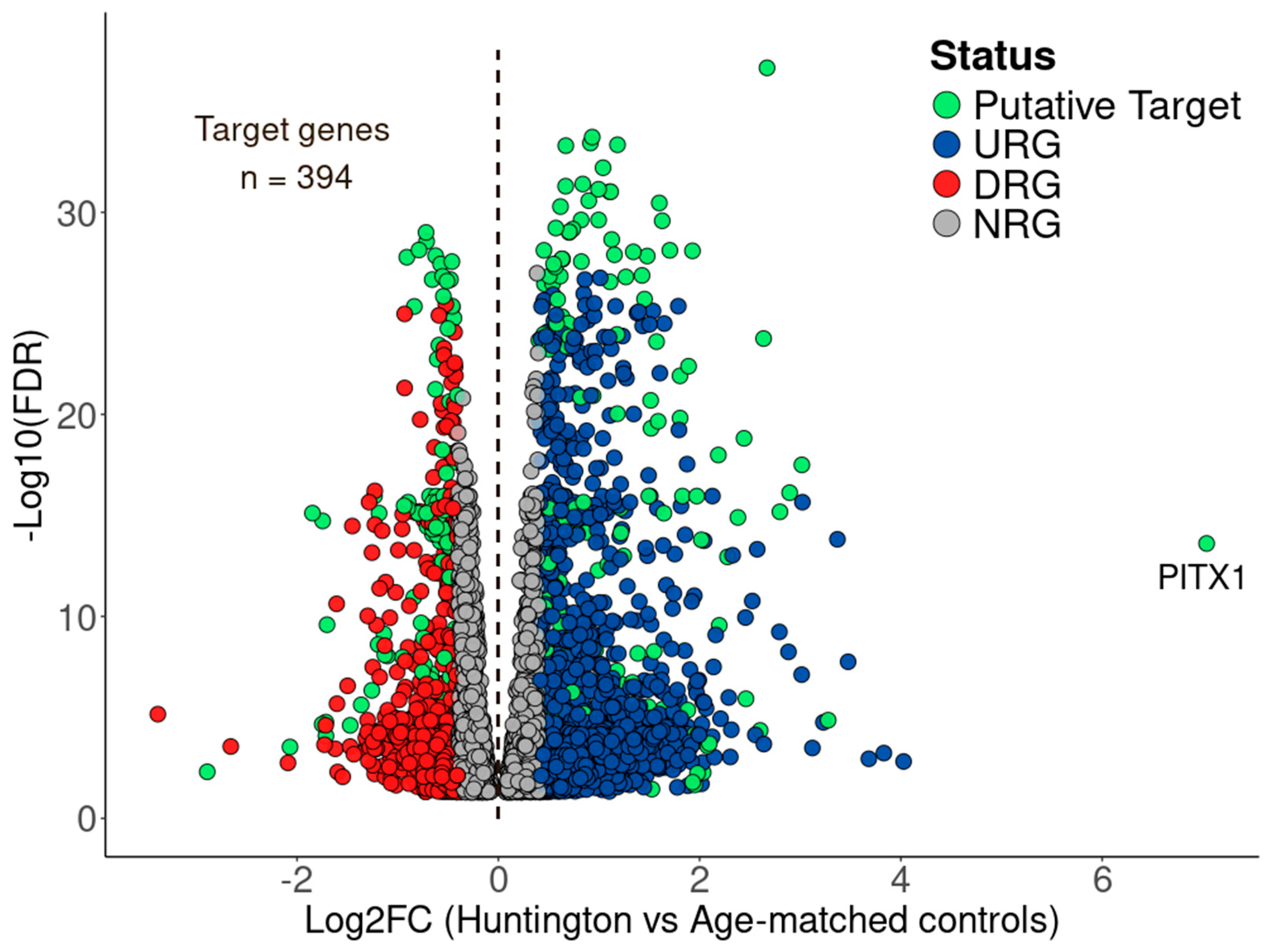
3.3. BDASeq® Identified 394 Potential Druggable Targets for HD
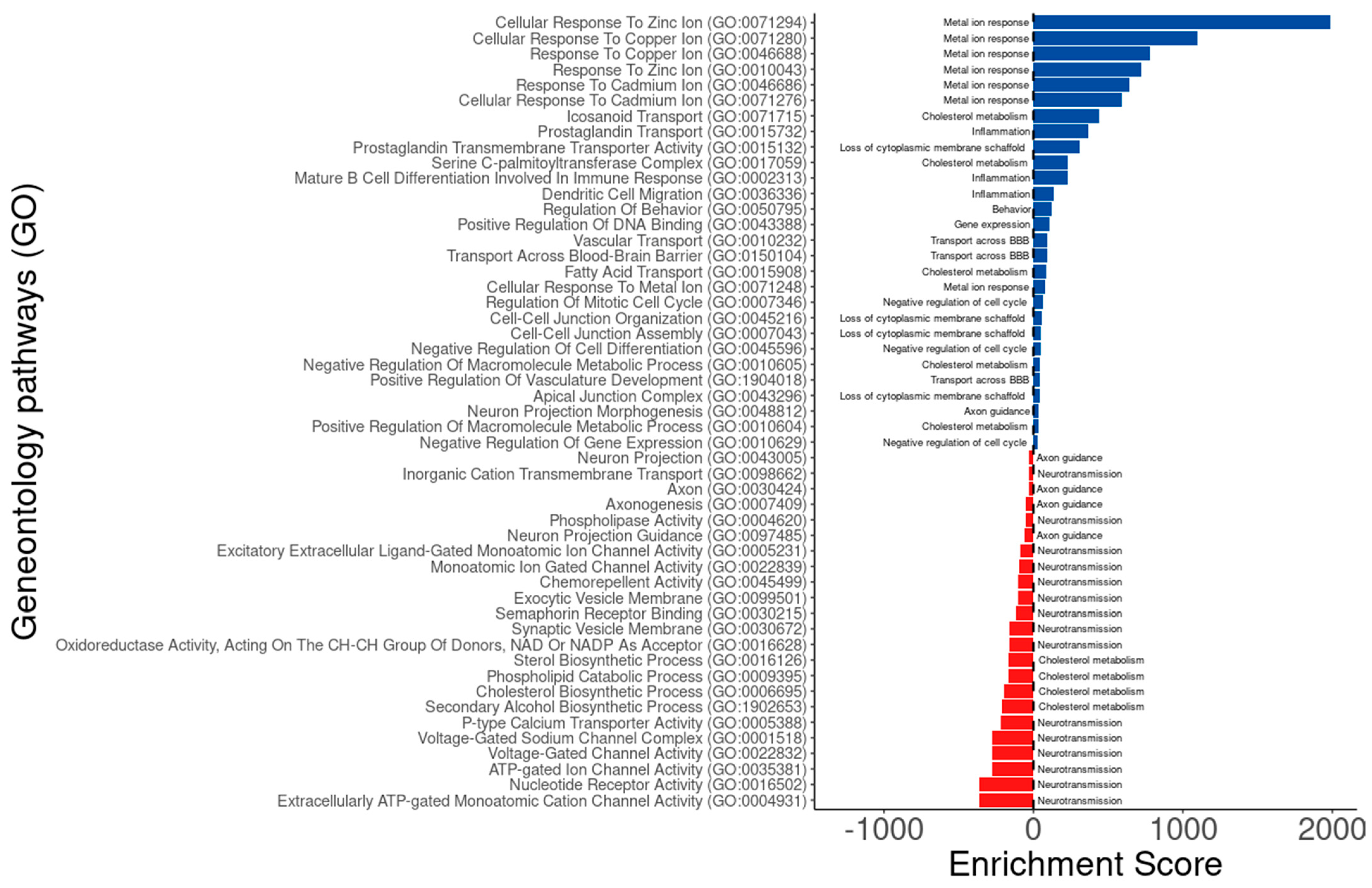
3.4. Putative Druggable Genes Identified by BDASeq® Are Associated with Dysregulated Biological Processes in HD
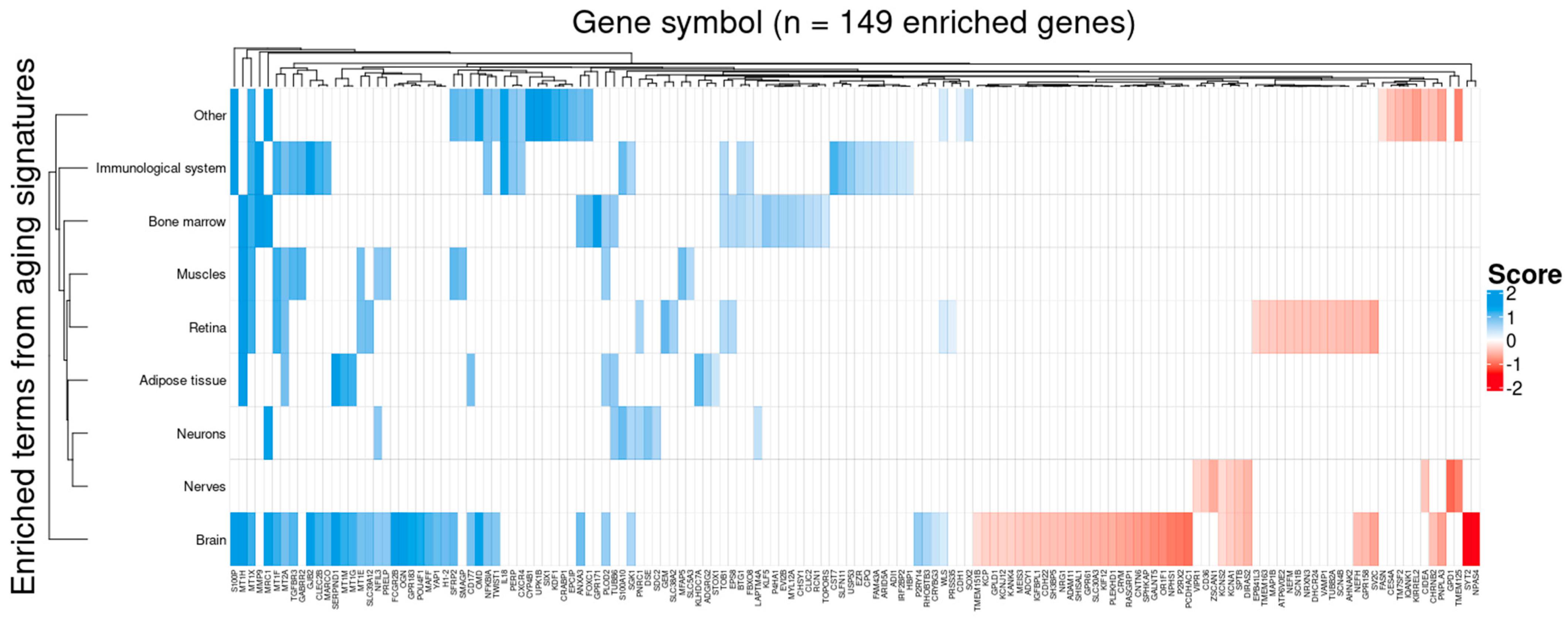
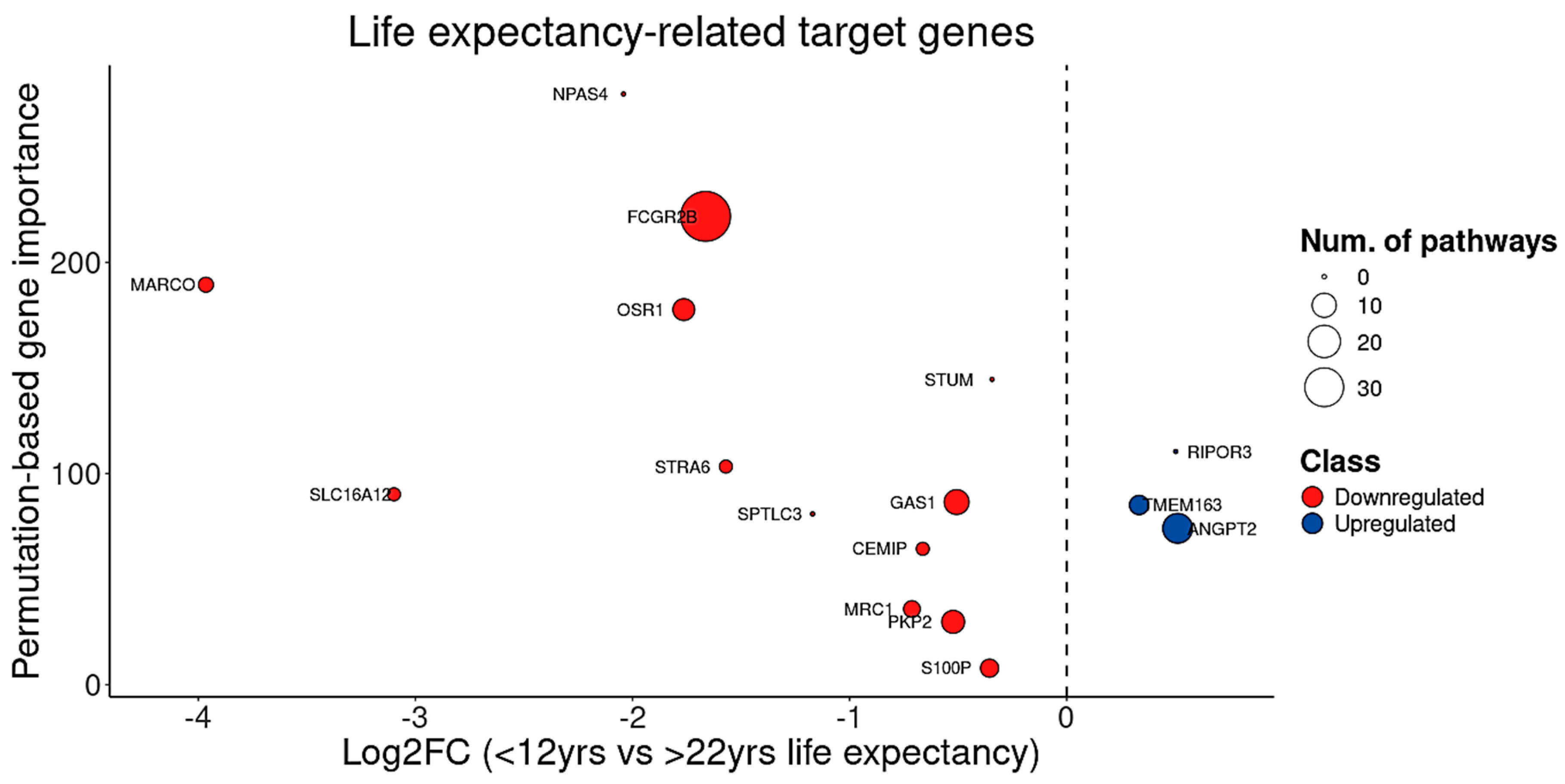
3.5. Target Genes Identified by BDASeq Are Involved in Aging-Related Processes Exacerbated in HD
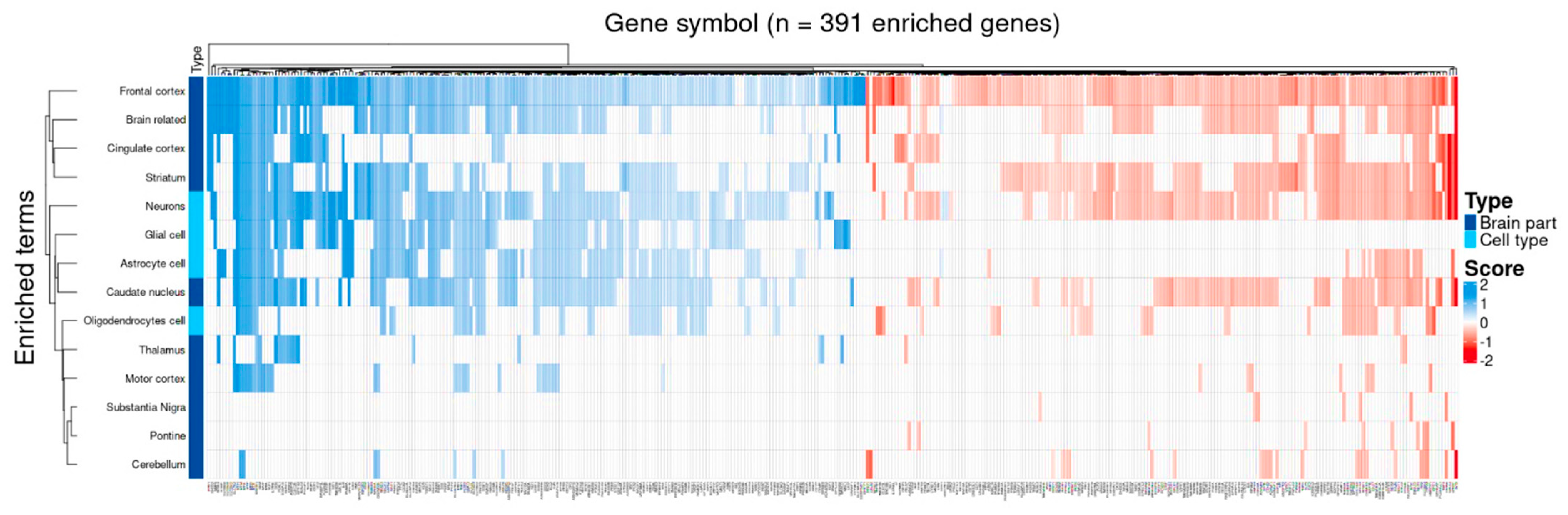
3.6. Target Genes Identified by BDASeq Are Deregulated in Brain Regions Affected by HD
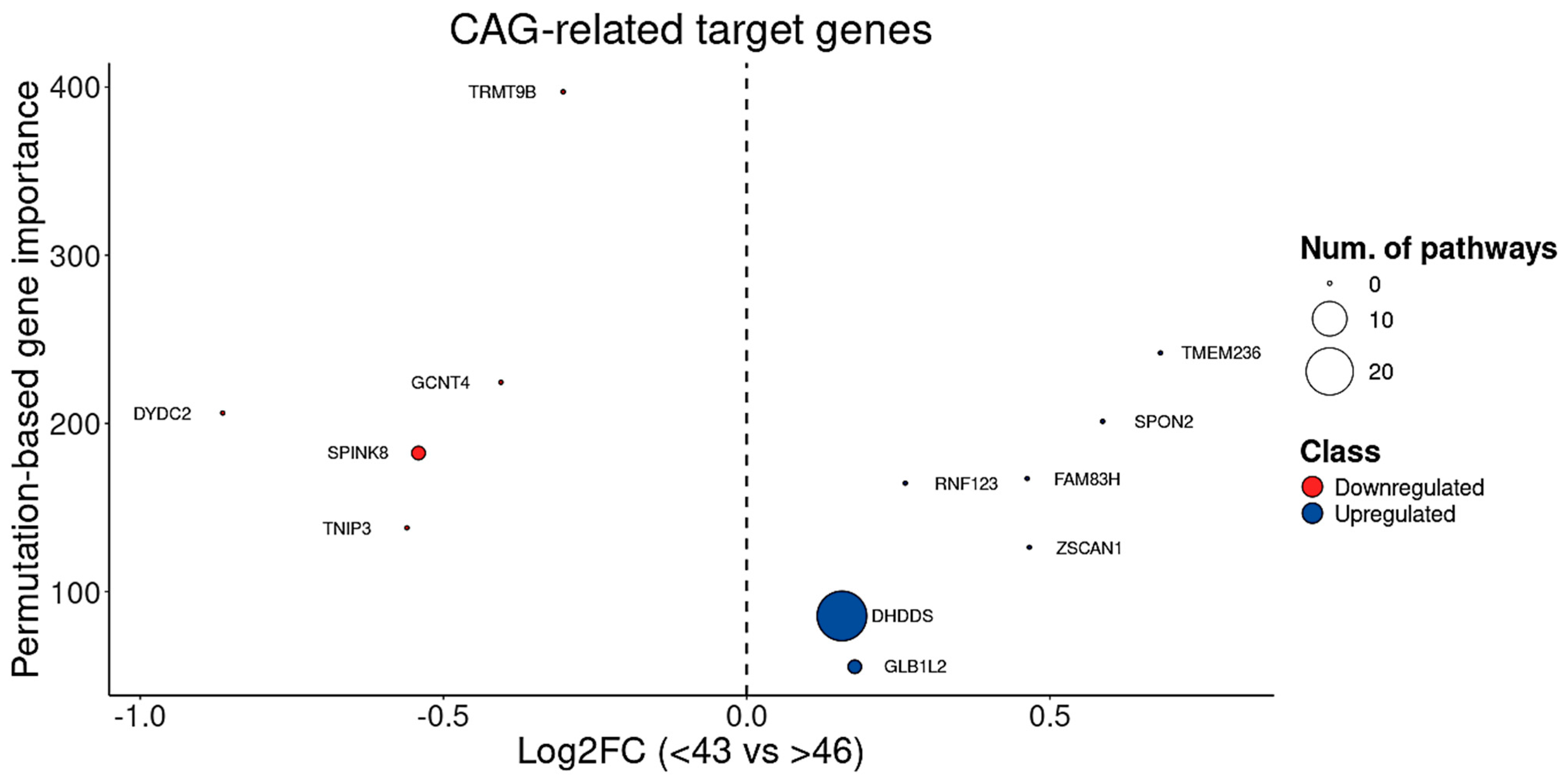
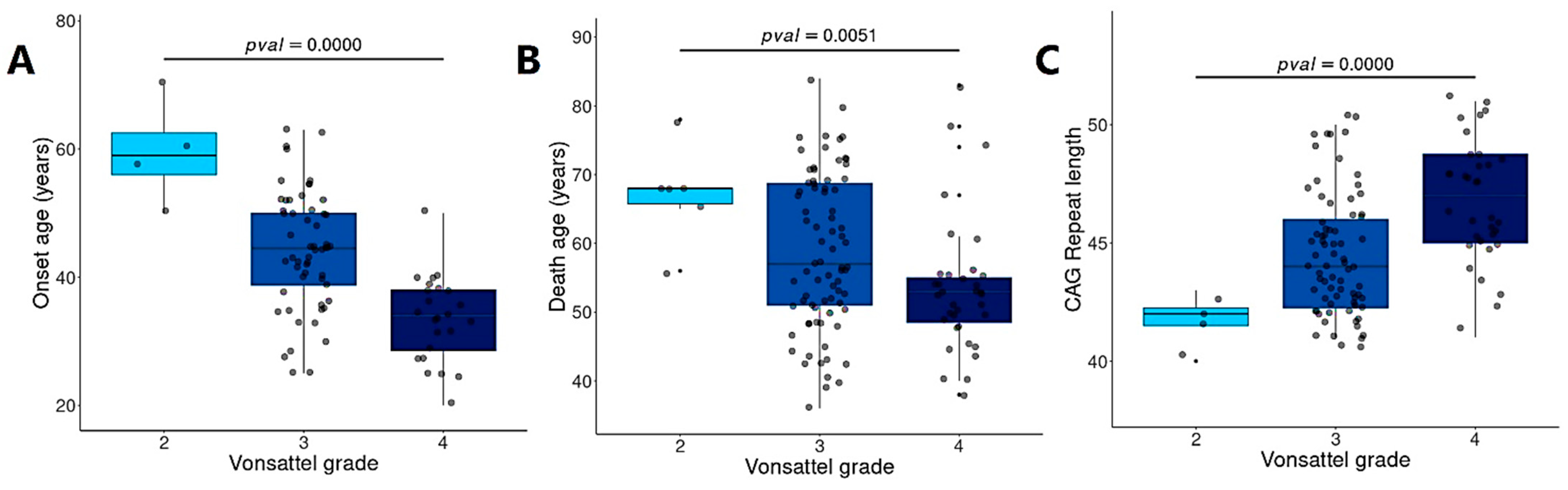
3.7. BDASeq® Identified CAG- and Onset-Age-Related Target Genes
3.8. BDASeq® Identified Target Genes Associated with Brain Degeneration
3.9. BDASeq® Identified for First-Time Blood Biomarkers for HD, Including FTH1
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABH | Brazil Huntington Association |
| AD | Alzheimer’s Disease |
| BDASeq® | Biomarker Discovery Algorithm |
| CADD | Computer-aided Drug Design |
| CAG | Cytosine–Adenine–Guanine |
| DEA | Differential Expression Analysis |
| DEG | Differentially Expressed Gene |
| FTH1 | Ferritin Have Chain 1 |
| HD | Huntington’s Disease |
| IHA | International Huntington Association |
| PD | Parkinson’s Disease |
| PSM | Propensity Score Matching |
| RMC | Recursive Method Combination |
| TFC | Total Function Capacity |
| TMS | Total Motor Score |
| UHDRS | Unified Huntington’s Disease Rate Scale |
Appendix
References
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Jurcau, C. Mitochondria in Huntington’s Disease: Implications in Pathogenesis and Mitochondrial-Targeted Therapeutic Strategies. Neural Regen. Res. 2023, 18, 1472. [Google Scholar] [CrossRef]
- Harper, P.S. The Epidemiology of Huntington’s Disease. Hum. Genet. 1992, 89, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, K. The Story of George Huntington and His Disease. Ann. Indian Acad. Neurol. 2016, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, M. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Claassen, D.O.; Ayyagari, R.; Garcia-Horton, V.; Zhang, S.; Alexander, J.; Leo, S. Real-World Adherence to Tetrabenazine or Deutetrabenazine Among Patients With Huntington’s Disease: A Retrospective Database Analysis. Neurol. Ther. 2022, 11, 435–448. [Google Scholar] [CrossRef]
- Furr Stimming, E.; Claassen, D.O.; Kayson, E.; Goldstein, J.; Mehanna, R.; Zhang, H.; Liang, G.S.; Haubenberger, D.; Adams, J.; Beck, C.; et al. Safety and Efficacy of Valbenazine for the Treatment of Chorea Associated with Huntington’s Disease (KINECT-HD): A Phase 3, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2023, 22, 494–504. [Google Scholar] [CrossRef]
- Heim, B.; Seppi, K. Valbenazine as Treatment for Huntington’s Disease Chorea. Lancet Neurol. 2023, 22, 459–460. [Google Scholar] [CrossRef]
- Kaur, N.; Kumar, P.; Jamwal, S.; Deshmukh, R.; Gauttam, V. Tetrabenazine: Spotlight on Drug Review. Ann. Neurosci. 2016, 23, 176–185. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Duarte, G.S.; Costa, J.; Ferreira, J.J.; Wild, E.J. Tetrabenazine Versus Deutetrabenazine for Huntington’s Disease: Twins or Distant Cousins? Mov. Disord. Clin. Pract. 2017, 4, 582–585. [Google Scholar] [CrossRef]
- Frank, S.; Testa, C.; Edmondson, M.C.; Goldstein, J.; Kayson, E.; Leavitt, B.R.; Oakes, D.; O’Neill, C.; Vaughan, C.; Whaley, J.; et al. The Safety of Deutetrabenazine for Chorea in Huntington Disease: An Open-Label Extension Study. CNS Drugs 2022, 36, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Login, I.S.; Cronin, M.J.; MacLeod, R.M. Tetrabenazine Has Properties of a Dopamine Receptor Antagonist. Ann. Neurol. 1982, 12, 257–262. [Google Scholar] [CrossRef]
- Claassen, D.O.; Carroll, B.; De Boer, L.M.; Wu, E.; Ayyagari, R.; Gandhi, S.; Stamler, D. Indirect Tolerability Comparison of Deutetrabenazine and Tetrabenazine for Huntington Disease. J. Clin. Mov. Disord. 2017, 4, 3. [Google Scholar] [CrossRef]
- Shahani, L. Tetrabenazine and Suicidal Ideation. J. Neuropsychiatry Clin. Neurosci. 2013, 25, E30. [Google Scholar] [CrossRef]
- Dalton, M.P.; Cheng, M.H.; Bahar, I.; Coleman, J. Structural Mechanisms for VMAT2 Inhibition by Tetrabenazine. Biophys. J. 2024, 123, 116a. [Google Scholar] [CrossRef]
- Niazi, S.K.; Mariam, Z. Computer-Aided Drug Design and Drug Discovery: A Prospective Analysis. Pharmaceuticals 2023, 17, 22. [Google Scholar] [CrossRef]
- Faustinoni-Neto, B.; Dias-Pinto, J.R.; Araldi, R.P. Pharma Data Analytics (Pharma Intelligence): Opportunities and Challenges in the Multi-Omics Era of Drug Discovery and Development. Pharmacologia 2023, 14, 29–39. [Google Scholar] [CrossRef]
- Dias-Pinto, J.R.; Faustinoni-Neto, B.; Sanches-Fernandes, J.M.; Kerkis, I.; Araldi, R.P. How Does the Age of Control Individuals Hinder the Identification of Target Genes for Huntington’s Disease? Front. Genet. 2024, 15, 1377237. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA Sequencing: The Teenage Years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Maza, E. In Papyro Comparison of TMM (EdgeR), RLE (DESeq2), and MRN Normalization Methods for a Simple Two-Conditions-Without-Replicates RNA-Seq Experimental Design. Front. Genet. 2016, 7, 164. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.-C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M. TPM, FPKM, or Normalized Counts? A Comparative Study of Quantification Measures for the Analysis of RNA-Seq Data from the NCI Patient-Derived Models Repository. J. Transl. Med. 2021, 19, 269. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Dias-Pinto, J.R.; Araldi, R.P.; Faustinoni-Neto, B. Biomarker Discovery Algorithm (BDASeq): An Integrative Analytics-Based Tool Designed to Identify Potential Diagnostic and Therapeutic Targets for Diseases. MENA Congr. Rare Dis. 2025, 1. [Google Scholar]
- Dias-Pinto, J.R.; Faustinoni-Neto, B.; Araldi, R.P. BDASeq: A Novel and Poweful Tool Able to Accurately Discover Theraeputic Targets for the Treatment of Rare Diseases. In Proceedings of the MENA Rare Disease Congress, Abu Dhabi, United Arab Emirates, 16–19 May 2024; p. 5. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 23 June 2025).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Tarazona, S.; García, F.; Ferrer, A.; Dopazo, J.; Conesa, A. NOIseq: A RNA-Seq Differential Expression Method Robust for Sequencing Depth Biases. EMBnet J. 2012, 17, 18. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An Empirical Bayes Hierarchical Model for Inference in RNA-Seq Experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Gauthier, M.; Agniel, D.; Thiébaut, R.; Hejblum, B.P. Dearseq: A Variance Component Score Test for RNA-Seq Differential Analysis That Effectively Controls the False Discovery Rate. NAR Genom. Bioinform. 2020, 2, lqaa093. [Google Scholar] [CrossRef] [PubMed]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. Biom. Bull. 1945, 1, 80. [Google Scholar] [CrossRef]
- Firth, D. Bias Reduction of Maximum Likelihood Estimates. Biometrika 1993, 80, 27–38. [Google Scholar] [CrossRef]
- Rosenbaum, P.R.; Rubin, D.B. The Central Role of the Propensity Score in Observational Studies for Causal Effects. Biometrika 1983, 70, 41–55. [Google Scholar] [CrossRef]
- Walsh, M.C.; Trentham-Dietz, A.; Newcomb, P.A.; Gangnon, R.; Palta, M. Using Propensity Scores to Reduce Case-Control Selection Bias. Epidemiology 2012, 23, 772–773. [Google Scholar] [CrossRef]
- Araldi, R.P.; Faustinoni-Neto, B.; Dias-Pinto, J.R. Novel Pharmaceutical Tragets and Prognostic Biomarkers for Huntington’s Disease: New Perspectives for the Drug Development. In Proceedings of the MENA Rare Disease Congress, Abu Dhabi, United Arab Emirates, 16–19 May 2024; p. 4. [Google Scholar]
- Ho, D.E.; Imai, K.; King, G.; Stuart, E.A. Matching as Nonparametric Preprocessing for Reducing Model Dependence in Parametric Causal Inference. Political Anal. 2007, 15, 199–236. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A Survey of Best Practices for RNA-Seq Data Analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Akond, Z.; Alam, M.; Mollah, M.N.H. Biomarker Identification from RNA-Seq Data Using a Robust Statistical Approach. Bioinformation 2018, 14, 153–163. [Google Scholar] [CrossRef]
- Amaratunga, D.; Göhlmann, H.; Peeters, P.J. Microarrays. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; pp. 87–106. [Google Scholar]
- Pfalzer, A.C.; Yan, Y.; Kang, H.; Totten, M.; Silverman, J.; Bowman, A.B.; Erikson, K.; Claassen, D.O. Alterations in Metal Homeostasis Occur Prior to Canonical Markers in Huntington Disease. Sci. Rep. 2022, 12, 10373. [Google Scholar] [CrossRef]
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of Copper Ion Mediated Huntington’s Disease Progression. PLoS ONE 2007, 2, e334. [Google Scholar] [CrossRef]
- Rosas, H.D.; Chen, Y.I.; Doros, G.; Salat, D.H.; Chen, N.; Kwong, K.K.; Bush, A.; Fox, J.; Hersch, S.M. Alterations in Brain Transition Metals in Huntington Disease. Arch. Neurol. 2012, 69, 887–893. [Google Scholar] [CrossRef]
- Cordeiro, L.M.; Soares, M.V.; da Silva, A.F.; dos Santos, L.V.; de Souza, L.I.; da Silveira, T.L.; Baptista, F.B.O.; de Oliveira, G.V.; Pappis, C.; Dressler, V.L.; et al. Toxicity of Copper and Zinc Alone and in Combination in Caenorhabditis Elegans Model of Huntington’s Disease and Protective Effects of Rutin. Neurotoxicology 2023, 97, 120–132. [Google Scholar] [CrossRef]
- Kacher, R.; Mounier, C.; Caboche, J.; Betuing, S. Altered Cholesterol Homeostasis in Huntington’s Disease. Front. Aging Neurosci. 2022, 14, 797220. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. The Impairment of Cholesterol Metabolism in Huntington Disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Karasinska, J.M.; Hayden, M.R. Cholesterol Metabolism in Huntington Disease. Nat. Rev. Neurol. 2011, 7, 561–572. [Google Scholar] [CrossRef]
- Jia, Q.; Li, S.; Li, X.-J.; Yin, P. Neuroinflammation in Huntington’s Disease: From Animal Models to Clinical Therapeutics. Front. Immunol. 2022, 13, 1088124. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A Novel Pathogenic Pathway of Immune Activation Detectable before Clinical Onset in Huntington’s Disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Silvestroni, A.; Faull, R.L.M.; Strand, A.D.; Möller, T. Distinct Neuroinflammatory Profile in Post-Mortem Human Huntington’s Disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef]
- Du, G.; Dong, W.; Yang, Q.; Yu, X.; Ma, J.; Gu, W.; Huang, Y. Altered Gut Microbiota Related to Inflammatory Responses in Patients With Huntington’s Disease. Front. Immunol. 2021, 11, 603594. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The Role of the Immune System in Huntington’s Disease. Clin. Dev. Immunol. 2013, 2013, 541259. [Google Scholar] [CrossRef]
- Katt, M.E.; Mayo, L.N.; Ellis, S.E.; Mahairaki, V.; Rothstein, J.D.; Cheng, L.; Searson, P.C. The Role of Mutations Associated with Familial Neurodegenerative Disorders on Blood–Brain Barrier Function in an IPSC Model. Fluids Barriers CNS 2019, 16, 20. [Google Scholar] [CrossRef]
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’Driscoll, C.M. The Blood-Brain Barrier in Aging and Neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673. [Google Scholar] [CrossRef]
- Hua, J.; Unschuld, P.G.; Margolis, R.L.; van Zijl, P.C.M.; Ross, C.A. Elevated Arteriolar Cerebral Blood Volume in Prodromal Huntington’s Disease. Mov. Disord. 2014, 29, 396–401. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Hsu, Y.-H.; Lin, M.-H.; Yang, T.-H.; Chen, H.-M.; Chen, Y.-C.; Hsiao, H.-Y.; Chen, C.-C.; Chern, Y.; Chang, C. Neurovascular Abnormalities in Humans and Mice with Huntington’s Disease. Exp. Neurol. 2013, 250, 20–30. [Google Scholar] [CrossRef]
- Vignone, D.; Gonzalez Paz, O.; Fini, I.; Cellucci, A.; Auciello, G.; Battista, M.R.; Gloaguen, I.; Fortuni, S.; Cariulo, C.; Khetarpal, V.; et al. Modelling the Human Blood–Brain Barrier in Huntington Disease. Int. J. Mol. Sci. 2022, 23, 7813. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Scalabrì, F.; Pepe, G.; Castaldo, S.; Elifani, F.; Capocci, L.; De Sanctis, C.; Comerci, L.; Pompeo, F.; et al. Impairment of Blood-Brain Barrier Is an Early Event in R6/2 Mouse Model of Huntington Disease. Sci. Rep. 2017, 7, 41316. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Sun, J.; Dong, Q.; Cui, M. Blood–Brain Barrier Endothelial Cells in Neurodegenerative Diseases: Signals from the “Barrier”. Front. Neurosci. 2023, 17, 1047778. [Google Scholar] [CrossRef]
- Capizzi, M.; Carpentier, R.; Denarier, E.; Adrait, A.; Kassem, R.; Mapelli, M.; Couté, Y.; Humbert, S. Developmental Defects in Huntington’s Disease Show That Axonal Growth and Microtubule Reorganization Require NUMA1. Neuron 2022, 110, e5. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, S.-H.; Yu, Z.-X.; Shelbourne, P.; Li, X.-J. Huntingtin Aggregate-Associated Axonal Degeneration Is an Early Pathological Event in Huntington’s Disease Mice. J. Neurosci. 2001, 21, 8473–8481. [Google Scholar] [CrossRef] [PubMed]
- Garret, M.; Du, Z.; Chazalon, M.; Cho, Y.H.; Baufreton, J. Alteration of GABAergic Neurotransmission in Huntington’s Disease. CNS Neurosci. Ther. 2018, 24, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Bruyn, G.W. Neurotransmitters in Huntington’s Chorea—A Clinician’s View. Prog. Brain Res. 1982, 55, 445–464. [Google Scholar]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.M.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s Disease Accelerates Epigenetic Aging of Human Brain and Disrupts DNA Methylation Levels. Aging 2016, 8, 1485–1512. [Google Scholar] [CrossRef] [PubMed]
- Machiela, E.; Southwell, A.L. Biological Aging and the Cellular Pathogenesis of Huntington’s Disease. J. Huntingt. Dis. 2020, 9, 115–128. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, X.; Ye, K.; Orth, M.; Gu, Z. Accelerated Expansion of Pathogenic Mitochondrial DNA Heteroplasmies in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2014610118. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, A.; Pineda, S.S.; Crittenden, J.R.; Lee, H.; Galani, K.; Mantero, J.; Tombaugh, G.; Kellis, M.; Heiman, M.; Graybiel, A.M. Transcriptional Vulnerabilities of Striatal Neurons in Human and Rodent Models of Huntington’s Disease. Nat. Commun. 2023, 14, 282. [Google Scholar] [CrossRef]
- Nopoulos, P.C. Huntington Disease: A Single-Gene Degenerative Disorder of the Striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar] [CrossRef]
- Hobbs, N.Z.; Pedrick, A.V.; Say, M.J.; Frost, C.; Dar Santos, R.; Coleman, A.; Sturrock, A.; Craufurd, D.; Stout, J.C.; Leavitt, B.R.; et al. The Structural Involvement of the Cingulate Cortex in Premanifest and Early Huntington’s Disease. Mov. Disord. 2011, 26, 1684–1690. [Google Scholar] [CrossRef]
- Lim, R.G.; Al-Dalahmah, O.; Wu, J.; Gold, M.P.; Reidling, J.C.; Tang, G.; Adam, M.; Dansu, D.K.; Park, H.-J.; Casaccia, P.; et al. Huntington Disease Oligodendrocyte Maturation Deficits Revealed by Single-Nucleus RNAseq Are Rescued by Thiamine-Biotin Supplementation. Nat. Commun. 2022, 13, 7791. [Google Scholar] [CrossRef]
- Weinberger, D.R.; Berman, K.F.; Iadarola, M.; Driesen, N.; Zec, R.F. Prefrontal Cortical Blood Flow and Cognitive Function in Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 94–104. [Google Scholar] [CrossRef]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.F.; Akbarian, S.; et al. RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS ONE 2015, 10, e0143563. [Google Scholar] [CrossRef] [PubMed]
- Agus, F.; Crespo, D.; Myers, R.H.; Labadorf, A. The Caudate Nucleus Undergoes Dramatic and Unique Transcriptional Changes in Human Prodromal Huntington’s Disease Brain. BMC Med. Genom. 2019, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Thiruvady, D.R.; Georgiou-Karistianis, N.; Egan, G.F.; Ray, S.; Sritharan, A.; Farrow, M.; Churchyard, A.; Chua, P.; Bradshaw, J.L.; Brawn, T.-L.; et al. Functional Connectivity of the Prefrontal Cortex in Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 127–133. [Google Scholar] [CrossRef]
- Blumenstock, S.; Dudanova, I. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Wake, C.; Labadorf, A.; Dumitriu, A.; Hoss, A.G.; Bregu, J.; Albrecht, K.H.; DeStefano, A.L.; Myers, R.H. Novel MicroRNA Discovery Using Small RNA Sequencing in Post-Mortem Human Brain. BMC Genom. 2016, 17, 776. [Google Scholar] [CrossRef]
- Lin, L.; Park, J.W.; Ramachandran, S.; Zhang, Y.; Tseng, Y.T.; Shen, S.; Waldvogel, H.J.; Curtis, M.A.; Richard, R.L.; Troncoso, J.C.; et al. Transcriptome Sequencing Reveals Aberrant Alternative Splicing in Huntington’s Disease. Hum. Mol. Genet. 2016, 25, 3454–3466. [Google Scholar] [CrossRef]
- Selemon, L.D.; Rajkowska, G.; Goldman-Rakic, P.S. Evidence for Progression in Frontal Cortical Pathology in Late-stage Huntington’s Disease. J. Comp. Neurol. 2004, 468, 190–204. [Google Scholar] [CrossRef]
- Delmaire, C.; Dumas, E.M.; Sharman, M.A.; van den Bogaard, S.J.A.; Valabregue, R.; Jauffret, C.; Justo, D.; Reilmann, R.; Stout, J.C.; Craufurd, D.; et al. The Structural Correlates of Functional Deficits in Early Huntington’s Disease. Hum. Brain Mapp. 2013, 34, 2141–2153. [Google Scholar] [CrossRef]
- Andrade-Navarro, M.A.; Mühlenberg, K.; Spruth, E.J.; Mah, N.; González-López, A.; Andreani, T.; Russ, J.; Huska, M.R.; Muro, E.M.; Fontaine, J.-F.; et al. RNA Sequencing of Human Peripheral Blood Cells Indicates Upregulation of Immune-Related Genes in Huntington’s Disease. Front. Neurol. 2020, 11, 573560. [Google Scholar] [CrossRef]
- Hensman Moss, D.J.; Flower, M.D.; Lo, K.K.; Miller, J.R.C.; van Ommen, G.-J.B.; ’t Hoen, P.A.C.; Stone, T.C.; Guinee, A.; Langbehn, D.R.; Jones, L.; et al. Huntington’s Disease Blood and Brain Show a Common Gene Expression Pattern and Share an Immune Signature with Alzheimer’s Disease. Sci. Rep. 2017, 7, 44849. [Google Scholar] [CrossRef]
- Mitchell, C.T.; Krier, I.; Arjomand, J.; Borowsky, B.; Tabrizi, S.J.; Leavitt, B.R.; Arran, N.; Axelson, E.; Bardinet, E.; Bechtel, N.; et al. Longitudinal Expression Changes Are Weak Correlates of Disease Progression in Huntington’s Disease. Brain Commun. 2020, 2, fcaa172. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.C.; Lo, K.K.; Andre, R.; Hensman Moss, D.J.; Träger, U.; Stone, T.C.; Jones, L.; Holmans, P.; Plagnol, V.; Tabrizi, S.J. RNA-Seq of Huntington’s Disease Patient Myeloid Cells Reveals Innate Transcriptional Dysregulation Associated with Proinflammatory Pathway Activation. Hum. Mol. Genet. 2016, 25, 2893–2904. [Google Scholar] [CrossRef]
- Ahmad, I.; Singh, R.; Pal, S.; Prajapati, S.; Sachan, N.; Laiq, Y.; Husain, H. Exploring the Role of Glycolytic Enzymes PFKFB3 and GAPDH in the Modulation of Aβ and Neurodegeneration and Their Potential of Therapeutic Targets in Alzheimer’s Disease. Appl. Biochem. Biotechnol. 2023, 195, 4673–4688. [Google Scholar] [CrossRef] [PubMed]
- Kee, T.; Khan, S.; Neidhart, M.; Masters, B.; Zhao, V.; Kim, Y.; Percy, K.; Woo, J. The Multifaceted Functions of β-Arrestins and Their Therapeutic Potential in Neurodegenerative Diseases. Exp. Mol. Med. 2024, 56, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, Y.; Peng, Q.; Li, F.; Chen, F. β-Arrestin2 Mediates the Hippocampal Dopaminergic System in Autistic Mouse through the ERK Signaling Pathway. Behav. Brain Res. 2022, 25, 113888. [Google Scholar] [CrossRef]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron Accumulates in Huntington’s Disease Neurons: Protection by Deferoxamine. PLoS ONE 2013, 8, e77023. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, M.; Liao, X.; Gao, S.; Hua, J.; Wu, X.; Guo, Q.; Xu, W.; Sun, J.; He, Y.; et al. Locally Organised and Activated Fth1hi Neutrophils Aggravate Inflammation of Acute Lung Injury in an IL-10-Dependent Manner. Nat. Commun. 2022, 13, 7703. [Google Scholar] [CrossRef]
- Mukherjee, C.; Kling, T.; Russo, B.; Miebach, K.; Kess, E.; Schifferer, M.; Pedro, L.D.; Weikert, U.; Fard, M.K.; Kannaiyan, N.; et al. Oligodendrocytes Provide Antioxidant Defense Function for Neurons by Secreting Ferritin Heavy Chain. Cell Metab. 2020, 32, 259–272.e10. [Google Scholar] [CrossRef]
- Palsa, K.; Baringer, S.L.; Shenoy, G.; Spiegelman, V.S.; Simpson, I.A.; Connor, J.R. Exosomes Are Involved in Iron Transport from Human Blood–Brain Barrier Endothelial Cells and Are Modified by Endothelial Cell Iron Status. J. Biol. Chem. 2023, 299, 102868. [Google Scholar] [CrossRef]
- Jamal, M.H.; Dhupar, M.; Aran, K.R. Exploring the Role of Ferroptosis Pathways in Huntington’s Disease: Insight of Pathophysiology to Emerging Treatment. Brain Disord. 2025, 18, 100207. [Google Scholar] [CrossRef]
- Dang, Y.; He, Q.; Yang, S.; Sun, H.; Liu, Y.; Li, W.; Tang, Y.; Zheng, Y.; Wu, T. FTH1- and SAT1-Induced Astrocytic Ferroptosis Is Involved in Alzheimer’s Disease: Evidence from Single-Cell Transcriptomic Analysis. Pharmaceuticals 2022, 15, 1177. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, K.; Ge, J.; Luo, X.; Wu, M.; Wang, N.; Zeng, J. Targeting Ferroptosis in Parkinson’s Disease: Mechanisms and Emerging Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 13042. [Google Scholar] [CrossRef]
- Li, X.; Si, W.; Li, Z.; Tian, Y.; Liu, X.; Ye, S.; Huang, Z.; Ji, Y.; Zhao, C.; Hao, X.; et al. MiR-335 Promotes Ferroptosis by Targeting Ferritin Heavy Chain 1 in in Vivo and in Vitro Models of Parkinson’s Disease. Int. J. Mol. Med. 2021, 47, 61. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Estévez, J.; Suárez, H.; Morales, L.M.; de Bonilla, L.C.; Villalobos, R.; Dávila, J.O. Serum Ferritin Deficiency in Huntington’s Disease Patients. Neurosci. Lett. 1991, 129, 22–24. [Google Scholar] [CrossRef]
- Morrison, P.J.; Nevin, N.C. Serum Iron, Total Iron Binding Capacity and Ferritin in Early Huntington Disease Patients. Ir. J. Med. Sci. 1994, 163, 236–237. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Criteria |
|---|---|
| Zero-count genes (ZCGs) | Genes with row counts = 0 |
| Low-count genes (LCGs) | Genes with normalized counts < 10 |
| Equally expressed genes (EEGs) | Genes with no statistical differences between cases and controls, adjusted p-value (FRD 1) > 0.05) |
| Non-relevant genes (NRGs) | Genes with adjusted p-value (FRD) < 0.05, but |log2FC| < 0.40 |
| Downregulated genes (DRGs) | Genes with adjusted p-value < 0.05 and log2FC < −0.40 |
| Upregulated genes (URGs) | Genes with adjusted p-value < 0.05 and log2FC < 0.40 |
| Process | Target Genes Identified | Pathophysiological Process | Reference |
|---|---|---|---|
| Metal ion response | CDH1, MMP9, MT1, MTIF, MT1G, MTIH, MT1M, MT1X, MT2A | - Copper, manganese, and zinc are increased in CSF in early HD - Copper and iron are increased in striatum of HD, causing pro-oxidant effects and neurodegeneration - Copper and zinc increased mHTT aggregation | Pfalzer et al. [44] Fox et al. [45] Rosas et al. [46] Cordeiro et al. [47] |
| Cholesterol metabolism | CD36, DHCR24, FASN, HMGCS1, MVD, TM7SF2 | - Cholesterol is an essential membrane component in the CNS - Cholesterol cannot cross the blood–brain barrier (BBB) due to its association with lipoproteins, so it is synthesized locally in the CNS - HTT protein is a key regulator of lipid metabolism - plasma 24S-hydroxycholesterol is reduced in HD patients proportionally to disease progression - mHTT decreases the maturation of SREBP and the upregulation of LXR and LXR-targeted genes (SREBP, ABCG1, and ABCG4, HMGCoA reductase, ApoE), resulting into a lower synthesis and transport of cholesterol from astrocytes to neurons via ApoE - HD attenuates brain sterol synthesis and accumulation of cholesterol in neuronal membranes, leading to neuronal loss | Kacher et al. [48] Leoni et al. [49] Karasinka et al. [50] |
| Neuronflammation | ADCY1, CAMKK2, CHRNB2, PB41L3, KCNA1, KCNC3, KCNC3 KCNJ12, NRG1, NRXN3, PPIA4, PRKKCG, SYT2 | - mHTT causes inflammatory responses in both CNS and peripheral tissues - increase in multiple cytokines in the plasma (IL-1β, IL-4, IL-6, IL-8, TNF-α, IL-10) and striatum (IL-6, IL-8 and TNF-α) of HD patients - Patients with HD have significantly lower plasma concentrations of IL-4, a marker of responses from T-helper-2 cells, than healthy control | Jia et al. [51] Björkqvist et al. [52] Silvestrini et al. [53] Du et al. [54] Ellrichmann et al. [55] Björkqvist et al. [52] |
| Transport across the brain-blood barrier | ABCC4, ANXA3, BTG1, ITGB8, SFRP2, SIRT1, SLC16A12, SLC22A2, SLC22A8, SLC38A2, SLC5A3, SCL7A2, TWIST1 | - mHTT expression alters the neurovasculature by increasing cerebral blood volume, small vessel density, and BBB permeability in rodent models of HD and patient tissue | Katt et al. [56] Knox et al. [57] Hua et al. [58] Lin et al. [59] Vignone et al. [60] Pardo et al. [61] Yaun et al. [62] |
| Axon guidance | AZIN2, CHRNB2, CNTN6, DCTN1, DPYSL5, EPHA10, HTR2C, KCNA1, KCNA3, MAP1B, NEFH, PARD3, PARD6B, POU4F1, PRKCG, RGS11, RIT2, ROCK1, SCN1B, SDC2, SEMA4D, SEMA5B, SGK1, SLC30A3, SLC39A12, SYT2 | - mHTT causes developmental defects affecting axon growth - mHTT causes axon growth dysfunctions | Capizzi et al. [63] Li et al. [64] |
| Neurotransmission | ATP2B2, ATP2C2, ATP6V0E2, ACNA1H, CALHM1, CHRNB2, DHCR24, GPLD1, KCNA1, KCNC3, KCNS2, MECR, NRG1, OTOF, P2RX2, P2RX6, PLA2G4C, PLBD2, PNPLA3, RIT2, SCN1B, SCN4B, SEMA4D, SEMA5B, SLC30A3, SMPD3, SV2C, SYT2, TM7SF2, TMEM163, TRPM1, VAMP1 | - neurotransmission is altered in HD, mainly GABAergic neurotransmission | Garret et al. [65] Bruyin [66] |
| Gene | Classification | p-Value | Log2FC | FDR | Pathways 1 |
|---|---|---|---|---|---|
| TRMT9B | DRG | 0.0388 | −0.5504 | 0.0001 | 0 |
| SPINK8 | DRG | 0.0480 | 1.1801 | 0.0000 | 1 |
| GCNT4 | DRG | 0.0388 | −0.4312 | 0.0004 | 0 |
| DYDC2 | DRG | 0.0321 | −0.8206 | 0.0003 | 0 |
| TNIP3 | DRG | 0.0264 | −1.7133 | 0.0000 | 0 |
| FAM83H | URG | 0.0189 | −0.5040 | 0.0001 | 0 |
| RNF123 | URG | 0.0216 | −0.4283 | 0.0000 | 0 |
| SPON2 | URG | 0.0099 | −0.8406 | 0.0000 | 0 |
| ZSCAN1 | URG | 0.0215 | −0.7667 | 0.0000 | 0 |
| GLB1L2 | URG | 0.0264 | −0.5157 | 0.0000 | 1 |
| TMEM236 | URG | 0.0123 | 1.5482 | 0.0000 | 0 |
| DHDDS | URG | 0.0263 | −0.4200 | 0.0000 | 23 |
| Gene | Classification | p-Value | Log2FC | FDR | Pathways 1 |
|---|---|---|---|---|---|
| OR3A2 | DRG | 0.0292 | −0.5151 | 0.0064 | 0 |
| PRCD | DRG | 0.0007 | −0.4182 | 0.0025 | 0 |
| CD177 | DRG | 0.0092 | 1.9313 | 0.0162 | 0 |
| VWA7 | DRG | 0.0216 | −0.7744 | 0.0001 | 0 |
| PDZD7 | DRG | 0.0052 | −0.5045 | 0.0000 | 1 |
| NRBP2 | DRG | 0.0415 | −0.4667 | 0.0000 | 0 |
| EPHA10 | DRG | 0.0178 | −0.6829 | 0.0000 | 2 |
| SCN4B | DRG | 0.0439 | −0.5889 | 0.0000 | 22 |
| CIDEA | DRG | 0.0086 | −0.6417 | 0.0004 | 1 |
| PLEKHD1 | DRG | 0.0006 | −0.7914 | 0.0001 | 0 |
| CES4A | DRG | 0.0292 | −0.6246 | 0.0000 | 1 |
| FASN | DRG | 0.0464 | −0.4322 | 0.0000 | 2 |
| FTH1 | DRG | 0.0392 | −0.5208 | 0.0000 | 0 |
| CORO6 | DRG | 0.0099 | −0.4891 | 0.0000 | 0 |
| MVD | DRG | 0.0027 | −0.5133 | 0.0004 | 5 |
| MAMDC2 | DRG | 0.0042 | 0.5831 | 0.0004 | 0 |
| MTHFD2L | DRG | 0.0121 | 0.4417 | 0.0002 | 0 |
| TRIM17 | DRG | 0.0292 | −0.4646 | 0.0000 | 0 |
| NRG1 | DRG | 0.0121 | −0.6523 | 0.0000 | 2 |
| CEP112 | DRG | 0.0403 | 0.5611 | 0.0000 | 0 |
| SYT2 | DRG | 0.0049 | −1.7452 | 0.0000 | 37 |
| STAC2 | DRG | 0.0002 | −0.5169 | 0.0000 | 6 |
| CBLN3 | DRG | 0.0049 | −1.7130 | 0.0001 | 0 |
| VAMP1 | DRG | 0.0002 | −0.6175 | 0.0000 | 25 |
| TP53TG5 | DRG | 0.0464 | −0.7439 | 0.0000 | 0 |
| WNT3 | DRG | 0.0086 | 0.5000 | 0.0001 | 21 |
| SCN1B | DRG | 0.0027 | −0.5948 | 0.0000 | 56 |
| PLA2G4C | DRG | 0.0025 | −0.4849 | 0.0000 | 3 |
| SEC14L5 | DRG | 0.0167 | −0.5010 | 0.0000 | 0 |
| P2RX6 | DRG | 0.0086 | −0.5360 | 0.0000 | 13 |
| MYO15A | DRG | 0.0001 | −0.5266 | 0.0000 | 6 |
| RGS11 | DRG | 0.0080 | −0.4597 | 0.0001 | 1 |
| TNIP3 | DRG | 0.0074 | −1.7133 | 0.0000 | 0 |
| MRC1 | URG | 0.0464 | 1.8902 | 0.0000 | 4 |
| STUM | URG | 0.0229 | −0.8045 | 0.0000 | 0 |
| KCNJ12 | URG | 0.0392 | −0.5354 | 0.0001 | 8 |
| TMEM151B | URG | 0.0244 | −0.4485 | 0.0000 | 0 |
| FAM91A1 | URG | 0.0167 | 0.4735 | 0.0000 | 2 |
| TCP11L1 | URG | 0.0147 | −0.4481 | 0.0000 | 1 |
| PDGFD | URG | 0.0427 | 1.0627 | 0.0007 | 9 |
| SHOX2 | URG | 0.0006 | 2.6702 | 0.0000 | 16 |
| ABCA1 | URG | 0.0370 | 0.9330 | 0.0000 | 16 |
| CA10 | URG | 0.0019 | −0.6081 | 0.0000 | 0 |
| SPHKAP | URG | 0.0275 | −0.9092 | 0.0000 | 0 |
| SRD5A1 | URG | 0.0329 | −0.4246 | 0.0000 | 1 |
| ARPP19 | URG | 0.0329 | −0.4803 | 0.0000 | 1 |
| FEZF1 | URG | 0.0002 | 1.6361 | 0.0000 | 6 |
| SIX1 | URG | 0.0310 | 1.9791 | 0.0058 | 5 |
| TWIST1 | URG | 0.0027 | 1.0096 | 0.0009 | 68 |
| SLC9A2 | URG | 0.0229 | 0.6608 | 0.0007 | 0 |
| GABRR2 | URG | 0.0339 | 1.3858 | 0.0000 | 1 |
| SMPD3 | URG | 0.0121 | −0.4803 | 0.0000 | 6 |
| TBX5 | URG | 0.0109 | 2.0175 | 0.0000 | 37 |
| PPEF1 | URG | 0.0001 | −1.1849 | 0.0000 | 0 |
| TMEM38A | URG | 0.0349 | −0.5346 | 0.0000 | 7 |
| ARHGAP10 | URG | 0.0010 | 0.6159 | 0.0000 | 4 |
| PITX1 | URG | 0.0025 | 7.0377 | 0.0000 | 19 |
| RCN1 | URG | 0.0023 | 0.6193 | 0.0000 | 4 |
| NRXN3 | URG | 0.0392 | −0.5461 | 0.0000 | 10 |
| Gene | Classification | p-Value | Log2FC | FDR | Pathways 1 |
|---|---|---|---|---|---|
| NUDT18 | DRG | 0.0060 | −0.7345 | 0.0000 | 0 |
| CLEC2L | DRG | 0.0008 | −0.7127 | 0.0000 | 0 |
| TIMM23B | DRG | 0.0067 | −0.4999 | 0.0013 | 2 |
| IQANK1 | DRG | 0.0406 | −0.7817 | 0.0000 | 0 |
| SMIM10L2B | DRG | 0.0309 | −0.6233 | 0.0000 | 0 |
| KAZN | DRG | 0.0084 | −0.4590 | 0.0000 | 1 |
| PDZD7 | DRG | 0.0141 | −0.5045 | 0.0000 | 1 |
| AHNAK2 | DRG | 0.0372 | −0.6515 | 0.0000 | 0 |
| NRBP2 | DRG | 0.0256 | −0.4667 | 0.0000 | 0 |
| EPHA10 | DRG | 0.0093 | −0.6829 | 0.0000 | 2 |
| NXPH4 | DRG | 0.0339 | −1.0172 | 0.0000 | 1 |
| SCN4B | DRG | 0.0339 | −0.5889 | 0.0000 | 22 |
| CIDEA | DRG | 0.0042 | −0.6417 | 0.0004 | 1 |
| PLEKHD1 | DRG | 0.0083 | −0.7914 | 0.0001 | 0 |
| PHOSPHO1 | DRG | 0.0406 | −0.5368 | 0.0008 | 2 |
| CES4A | DRG | 0.0444 | −0.6246 | 0.0000 | 1 |
| LDLRAD4 | DRG | 0.0075 | −0.4421 | 0.0000 | 0 |
| HR | DRG | 0.0030 | −0.7181 | 0.0000 | 0 |
| CORO6 | DRG | 0.0174 | −0.4891 | 0.0000 | 0 |
| MVD | DRG | 0.0192 | −0.5133 | 0.0004 | 5 |
| NRG1 | DRG | 0.0142 | −0.6523 | 0.0000 | 2 |
| ZSCAN1 | DRG | 0.0093 | −0.7667 | 0.0000 | 0 |
| RIT2 | DRG | 0.0406 | −0.7547 | 0.0000 | 3 |
| TMEM163 | DRG | 0.0075 | −0.5153 | 0.0001 | 6 |
| PLBD2 | DRG | 0.0157 | −0.4840 | 0.0000 | 0 |
| PCYOX1L | DRG | 0.0079 | −0.6316 | 0.0001 | 0 |
| SYT2 | DRG | 0.0309 | −1.7452 | 0.0000 | 37 |
| STAC2 | DRG | 0.0014 | −0.5169 | 0.0000 | 6 |
| VAMP1 | DRG | 0.0030 | −0.6175 | 0.0000 | 25 |
| KIF12 | DRG | 0.0484 | −0.8271 | 0.0000 | 4 |
| ANGPTL2 | DRG | 0.0233 | −0.8151 | 0.0004 | 1 |
| ACP2 | DRG | 0.0192 | −0.5411 | 0.0000 | 1 |
| KCNC3 | DRG | 0.0256 | −0.5356 | 0.0000 | 30 |
| ARHGEF2 | DRG | 0.0093 | −0.4720 | 0.0000 | 8 |
| MECR | DRG | 0.0038 | −0.4410 | 0.0000 | 12 |
| SLC30A3 | DRG | 0.0484 | −0.7125 | 0.0000 | 6 |
| VIPR1 | DRG | 0.0115 | −0.4111 | 0.0001 | 1 |
| SCN1B | DRG | 0.0002 | −0.5948 | 0.0000 | 56 |
| NAPA | DRG | 0.0030 | −0.4614 | 0.0000 | 11 |
| NEFM | DRG | 0.0128 | −0.6006 | 0.0000 | 2 |
| SEC14L5 | DRG | 0.0038 | −0.5010 | 0.0000 | 0 |
| NUP93 | DRG | 0.0128 | −0.4756 | 0.0000 | 1 |
| GSTZ1 | DRG | 0.0406 | −0.5657 | 0.0001 | 1 |
| P2RX6 | DRG | 0.0173 | −0.5360 | 0.0000 | 13 |
| MYO15A | DRG | 0.0406 | −0.5266 | 0.0000 | 6 |
| ADAM11 | DRG | 0.0354 | −0.5504 | 0.0000 | 4 |
| F7 | DRG | 0.0060 | −0.8341 | 0.0000 | 1 |
| SLC5A3 | URG | 0.0164 | 0.8459 | 0.0000 | 4 |
| HMGN5 | URG | 0.0256 | 0.5588 | 0.0002 | 2 |
| TOPORS | URG | 0.0067 | 0.4285 | 0.0001 | 13 |
| PTAR1 | URG | 0.0211 | 0.6316 | 0.0000 | 0 |
| PLEKHF2 | URG | 0.0115 | 0.6301 | 0.0000 | 0 |
| GPR171 | URG | 0.0067 | 1.8061 | 0.0000 | 2 |
| SLFN11 | URG | 0.0406 | 1.0408 | 0.0000 | 7 |
| BCL2 | URG | 0.0060 | 0.5718 | 0.0000 | 95 |
| ARHGAP42 | URG | 0.0142 | 0.7404 | 0.0000 | 7 |
| STOX1 | URG | 0.0211 | 0.5138 | 0.0000 | 14 |
| RHOBTB3 | URG | 0.0192 | 0.7695 | 0.0000 | 2 |
| RANBP3L | URG | 0.0157 | 1.2461 | 0.0000 | 5 |
| PPP4R2 | URG | 0.0309 | 0.4286 | 0.0000 | 4 |
| GRAMD2B | URG | 0.0060 | 0.8249 | 0.0000 | 0 |
| GABPA | URG | 0.0060 | 0.4045 | 0.0002 | 7 |
| BCL2L11 | URG | 0.0339 | 0.5653 | 0.0002 | 35 |
| GXYLT1 | URG | 0.0053 | 0.4231 | 0.0002 | 1 |
| PARD3 | URG | 0.0339 | 0.9958 | 0.0000 | 8 |
| SLC39A12 | URG | 0.0233 | 1.1799 | 0.0000 | 5 |
| G3BP1 | URG | 0.0034 | 0.4533 | 0.0000 | 8 |
| USP53 | URG | 0.0406 | 0.8390 | 0.0000 | 2 |
| YAP1 | URG | 0.0115 | 1.1163 | 0.0000 | 57 |
| SLC38A2 | URG | 0.0173 | 0.8775 | 0.0000 | 8 |
| MAP7D3 | URG | 0.0030 | 0.5214 | 0.0019 | 0 |
| FEZF1 | URG | 0.0309 | 1.6361 | 0.0000 | 6 |
| GNA13 | URG | 0.0104 | 0.7054 | 0.0000 | 15 |
| UPK1B | URG | 0.0173 | 2.6019 | 0.0000 | 1 |
| GMNN | URG | 0.0339 | 0.7335 | 0.0001 | 9 |
| GABRR2 | URG | 0.0174 | 1.3858 | 0.0000 | 1 |
| DSE | URG | 0.0084 | 0.9154 | 0.0000 | 14 |
| ITGB8 | URG | 0.0021 | 0.6760 | 0.0000 | 20 |
| ATP11C | URG | 0.0371 | 0.6688 | 0.0000 | 1 |
| EZR | URG | 0.0406 | 0.7495 | 0.0000 | 20 |
| TGFBR3 | URG | 0.0211 | 1.1844 | 0.0000 | 34 |
| LAPTM4A | URG | 0.0443 | 0.5393 | 0.0000 | 0 |
| IL17RB | URG | 0.0173 | 1.1067 | 0.0000 | 5 |
| RCN1 | URG | 0.0142 | 0.6193 | 0.0000 | 4 |
| Gene | Classification | p-Value | Log2FC | FDR | Pathways 1 |
|---|---|---|---|---|---|
| MRC1 | DRG | 0.0412 | 1.8902 | 0.0000 | 4 |
| STUM | DRG | 0.0090 | −0.8045 | 0.0000 | 0 |
| GAS1 | DRG | 0.0152 | 1.0313 | 0.0001 | 11 |
| NPAS4 | DRG | 0.0075 | −2.8925 | 0.0049 | 0 |
| SPTLC3 | DRG | 0.0075 | 1.9498 | 0.0001 | 0 |
| S100P | DRG | 0.0427 | 2.4416 | 0.0000 | 5 |
| SLC16A12 | DRG | 0.0112 | 3.0162 | 0.0000 | 2 |
| OSR1 | DRG | 0.0028 | 1.3507 | 0.0036 | 8 |
| STRA6 | DRG | 0.0026 | 1.3572 | 0.0138 | 2 |
| CEMIP | DRG | 0.0152 | 0.7861 | 0.0000 | 2 |
| FCGR2B | DRG | 0.0023 | 2.3809 | 0.0000 | 53 |
| PKP2 | DRG | 0.0230 | 1.0042 | 0.0001 | 9 |
| MARCO | DRG | 0.0128 | 2.1951 | 0.0000 | 3 |
| TMEM163 | URG | 0.0341 | −0.5153 | 0.0001 | 6 |
| ANGPT2 | URG | 0.0165 | 1.4528 | 0.0000 | 17 |
| RIPOR3 | URG | 0.0427 | 1.2127 | 0.0000 | 0 |
| Gene ID | Gene | Log2FC | FDR | Classification |
|---|---|---|---|---|
| ENSG00000250568 1 | PABPC1 | 1.2687 | 0.0267 | URG |
| ENSG00000260035 | Lnc-DUOXA1-2 | 2.3900 | 0.0003 | URG |
| ENSG00000149925 | ALDOA | 0.8515 | 0.0014 | URG |
| ENSG00000141480 | ARRB2 | 0.4655 | 0.0009 | URG |
| ENSG00000135636 | DYSF | 0.6159 | 0.0109 | URG |
| ENSG00000132589 | FLOT2 | 0.4394 | 0.0369 | URG |
| ENSG00000167996 | FTH1 | 1.1435 | 0.0024 | URG |
| ENSG00000174021 | GNG5 | 0.4621 | 0.0295 | URG |
| ENSG00000170837 | GPR27 | 0.6579 | 0.0127 | URG |
| ENSG00000030582 | GRN | 0.5643 | 0.0495 | URG |
| ENSG00000248099 | INSL3 | 1.4028 | 0.0014 | URG |
| ENSG00000251230 | MIR3945HG | 0.8796 | 0.0493 | URG |
| ENSG00000170909 | OSCAR | 0.5463 | 0.0403 | URG |
| ENSG00000170525 | PFKFB3 | 0.8153 | 0.0006 | URG |
| ENSG00000105287 | PRKD2 | 0.4599 | 0.0077 | URG |
| ENSG00000112053 | SLC26A8 | 0.9719 | 0.0495 | URG |
| ENSG00000266402 | SNHG25 | 1.6604 | 0.0156 | URG |
| ENSG00000184292 | TACSTD2 | 3.8049 | 0.0000 | URG |
| ENSG00000156414 | TDRD9 | 1.1963 | 0.0403 | URG |
| ENSG00000170873 | MTSS1 | −0.6198 | 0.0011 | DRG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias Pinto, J.R.; Faustinoni Neto, B.; Munhoz, L.; Kerkis, I.; Araldi, R.P. From Brain to Blood: Uncovering Potential Therapeutical Targets and Biomarkers for Huntington’s Disease Using an Integrative RNA-Seq Analytical Platform (BDASeq®). Cells 2025, 14, 976. https://doi.org/10.3390/cells14130976
Dias Pinto JR, Faustinoni Neto B, Munhoz L, Kerkis I, Araldi RP. From Brain to Blood: Uncovering Potential Therapeutical Targets and Biomarkers for Huntington’s Disease Using an Integrative RNA-Seq Analytical Platform (BDASeq®). Cells. 2025; 14(13):976. https://doi.org/10.3390/cells14130976
Chicago/Turabian StyleDias Pinto, João Rafael, Benedito Faustinoni Neto, Luciana Munhoz, Irina Kerkis, and Rodrigo Pinheiro Araldi. 2025. "From Brain to Blood: Uncovering Potential Therapeutical Targets and Biomarkers for Huntington’s Disease Using an Integrative RNA-Seq Analytical Platform (BDASeq®)" Cells 14, no. 13: 976. https://doi.org/10.3390/cells14130976
APA StyleDias Pinto, J. R., Faustinoni Neto, B., Munhoz, L., Kerkis, I., & Araldi, R. P. (2025). From Brain to Blood: Uncovering Potential Therapeutical Targets and Biomarkers for Huntington’s Disease Using an Integrative RNA-Seq Analytical Platform (BDASeq®). Cells, 14(13), 976. https://doi.org/10.3390/cells14130976