
A Cell Biologist’s View on APOL1: What We Know and What We Still Need to Address



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Evolution of the APOL Gene Family
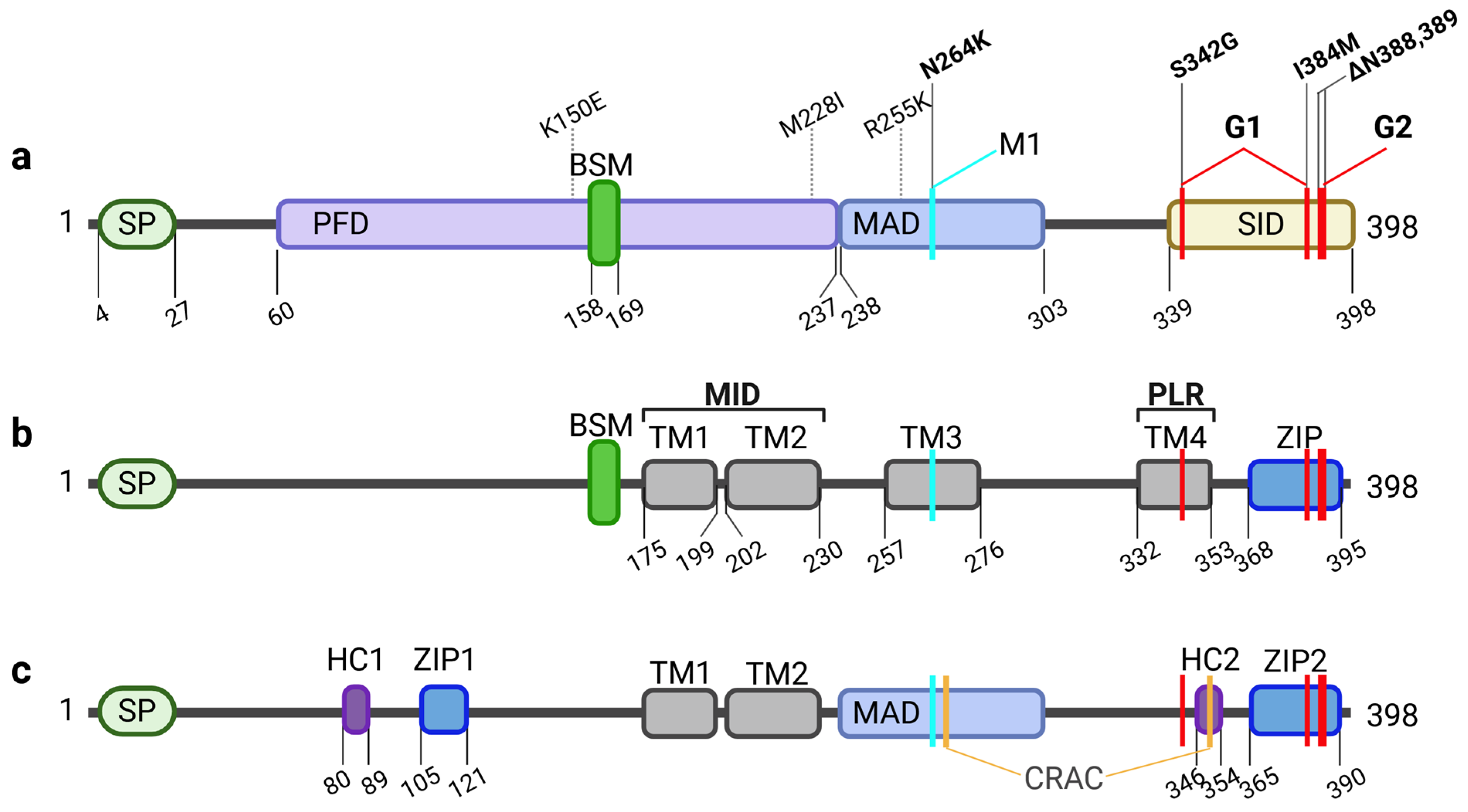
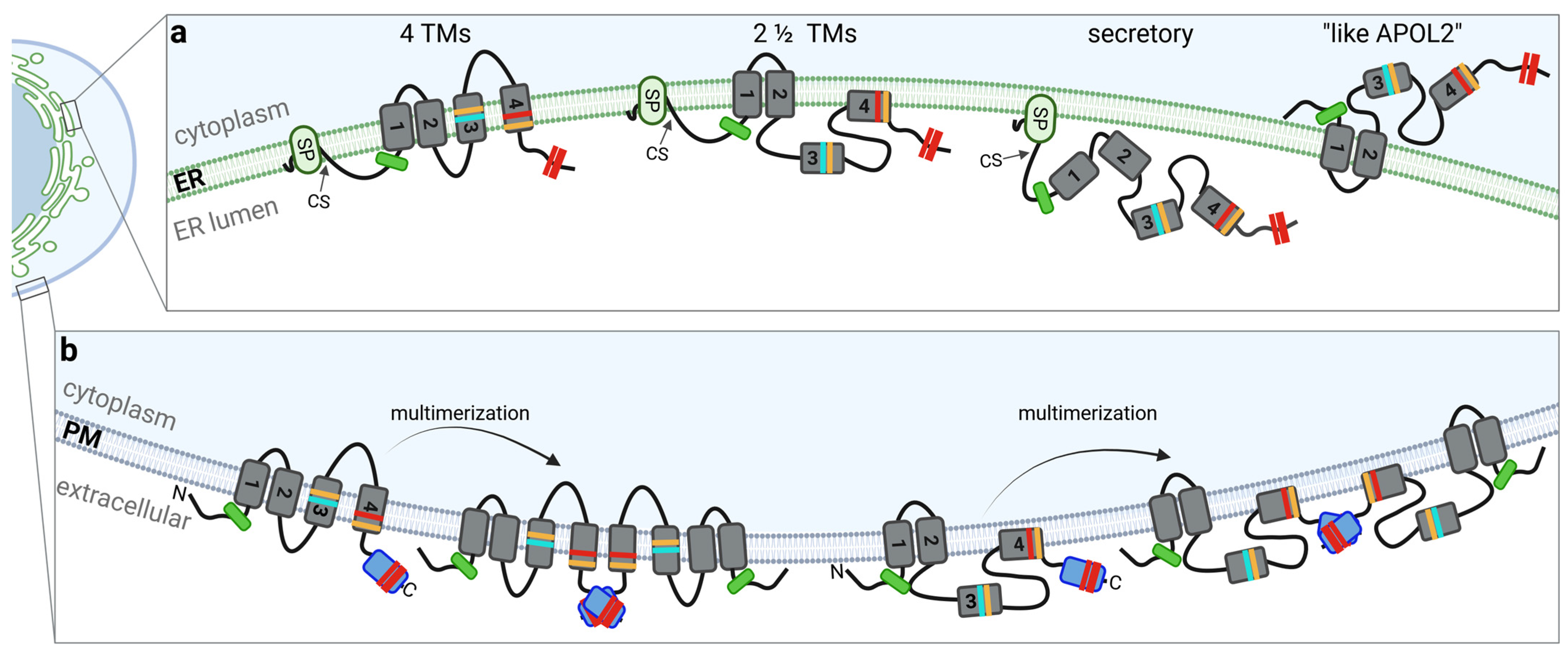
3. APOL1: Splice Variants and Their Membrane Topology
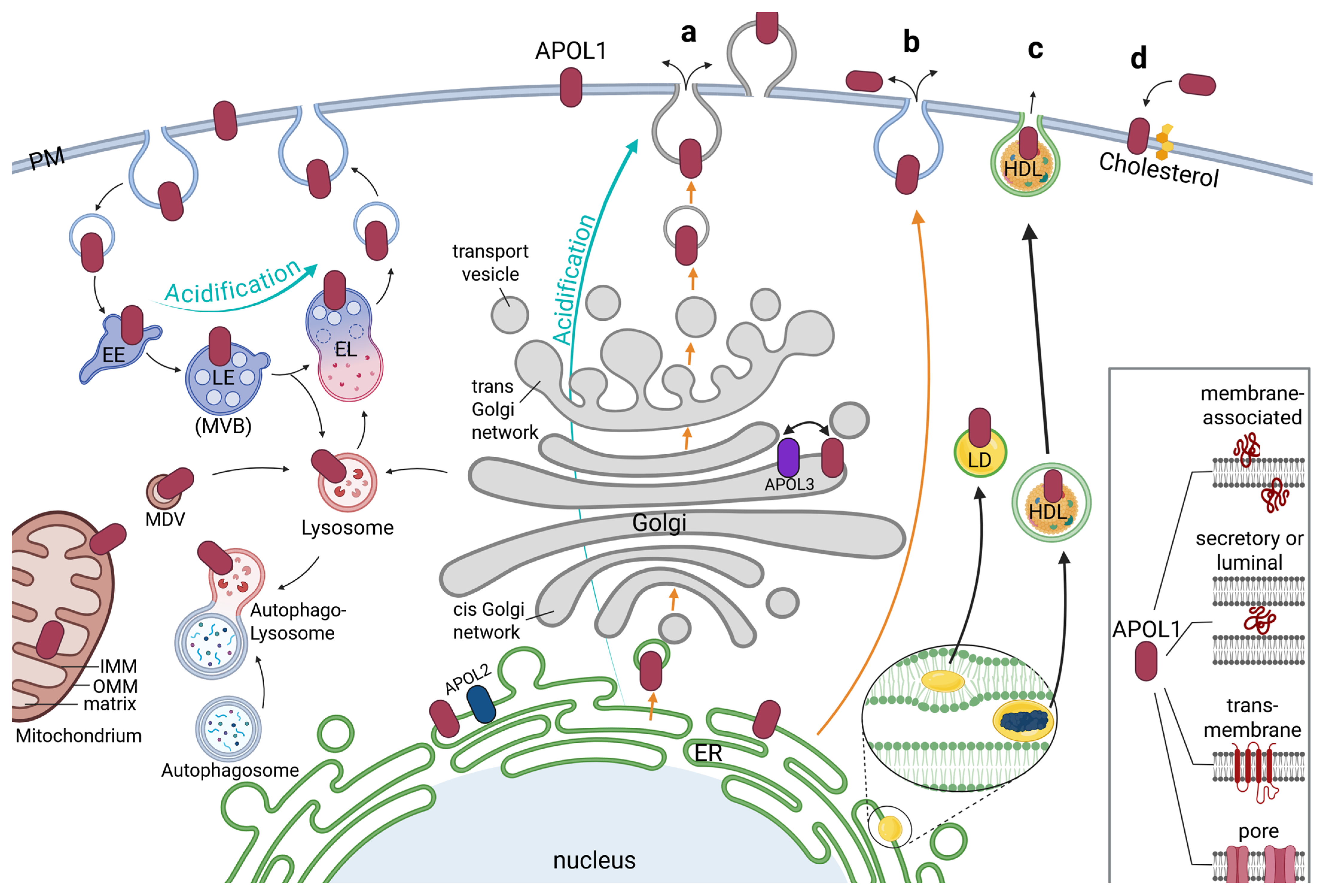
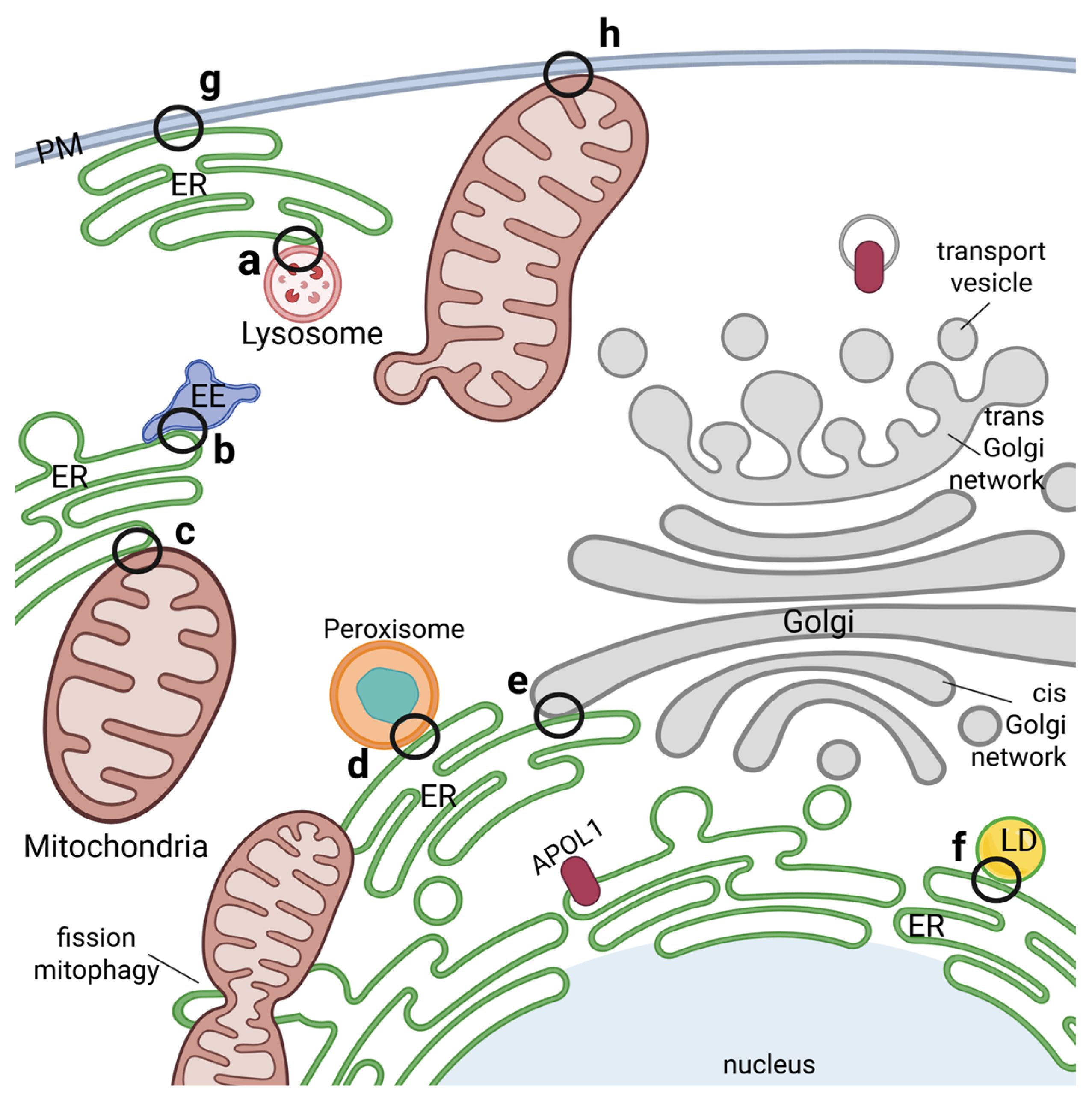
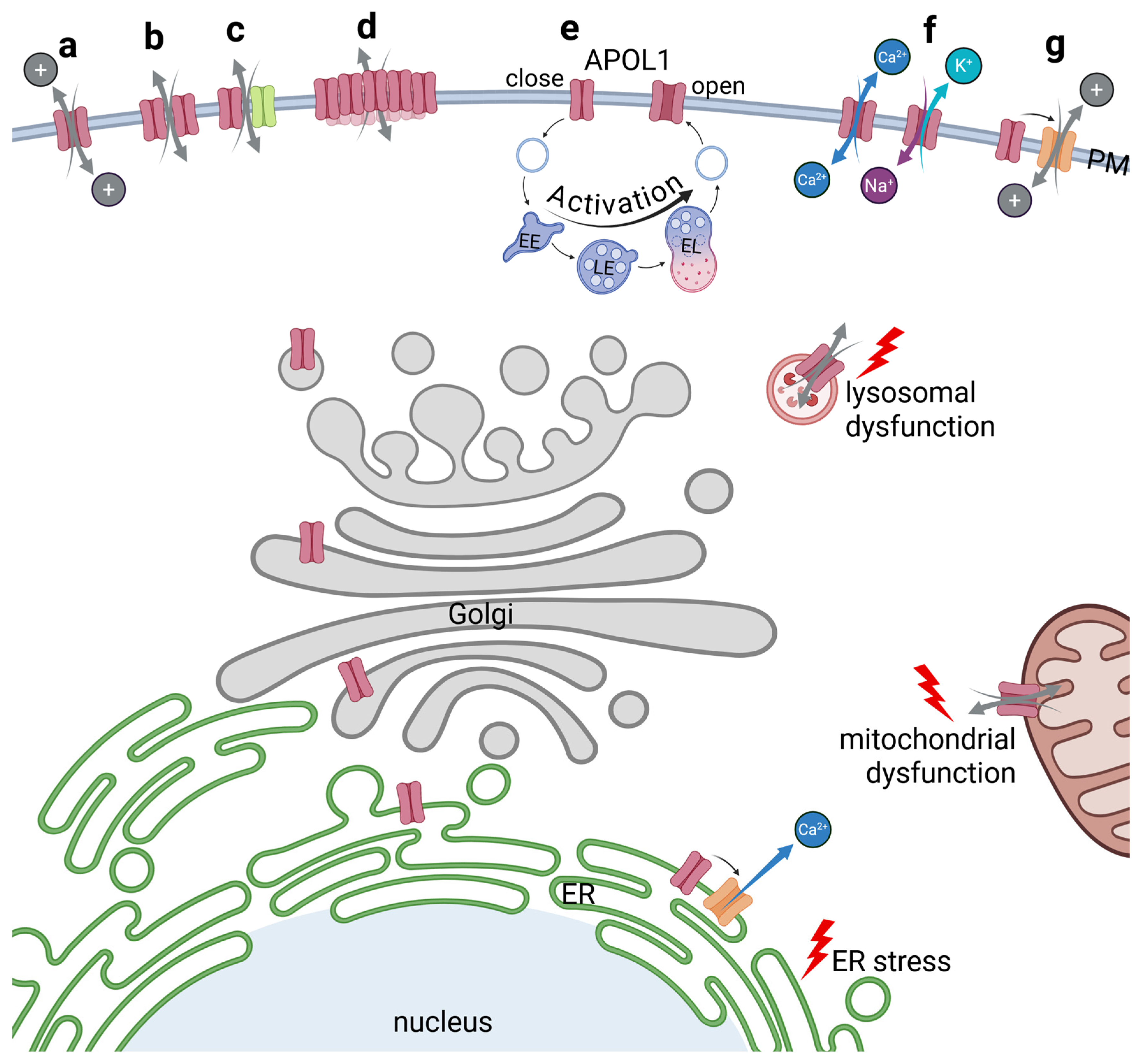
4. Intracellular Localization of APOL1
5. APOL1 and Cellular Injury
5.1. APOL1 Cytoxicity: Loss-of-Protection or Gain-of-Cytotoxicity?
5.2. APOL1 Cytoxicity Due to Disturbances of the Endolysosomal and Autophagic System
5.3. APOL1-Mediated Impairment of Mitochondrial Functions
5.4. APOL1 Mediated Disturbances of the ER Homeostasis
5.5. APOL1 as a Regulator in Inflammation
5.6. APOL1-Linked Cytotoxicity Due to Altered Lipid Biding
5.7. APOL1’s Role as a Membrane Pore or Ion Channel
6. APOL1-Associated Cytotoxicity in Podocytes and the Role of Second Hits
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKI | acute kidney injury |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| APOL1-5 | Apolipoprotein L1-6 |
| APOLD1 | Apolipoprotein L domain containing 1 |
| ATF4 | activating transcription factor 4 |
| BK | big potassium |
| COVAN | COVID-19-associated nephropathy |
| eIF1 | eukaryotic translation initiation factor 1 |
| FSGS | focal segmental glomerulosclerosis |
| GPCR | G protein-coupled receptor |
| HAT | human African trypanosomiasis |
| HDL | high density lipoproteins |
| HIVAN | HIV-associated nephropathy |
| IP3 | inositol 1,4,5-trisphosphate |
| JNK | c-Jun-N-terminal kinase |
| PCD | programmed cell death |
| SERCA | sarcoplasmic/endoplasmic reticulum calcium ATPase |
| suPAR | soluble urokinase plasminogen activator receptor |
| TRPC6 | transient receptor potential cation channel subfamily C member 6 |
| UBD | Ubiquitin D |
| UPR | unfolded protein response |
References
- Duchateau, P.N.; Pullinger, C.R.; Cho, M.H.; Eng, C.; Kane, J.P. Apolipoprotein L Gene Family: Tissue-Specific Expression, Splicing, Promoter Regions; Discovery of a New Gene. J. Lipid Res. 2001, 42, 620–630. [Google Scholar] [CrossRef]
- Smith, E.E.; Malik, H.S. The Apolipoprotein L Family of Programmed Cell Death and Immunity Genes Rapidly Evolved in Primates at Discrete Sites of Host-Pathogen Interactions. Genome Res. 2009, 19, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Schmitz, J.; Fischer, K.; Granado, D.; Groh, A.-C.; Krausel, V.; Lüttgenau, S.M.; Amelung, T.M.; Pavenstädt, H.; Weide, T. Evolution of Renal-Disease Factor APOL1 Results in Cis and Trans Orientations at the Endoplasmic Reticulum That Both Show Cytotoxic Effects. Mol. Biol. Evol. 2021, 38, 4962–4976. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Scheek, S.; Borbiev, T.; Lanahan, A.A.; Schneider, A.; Demetriades, A.M.; Hiemisch, H.; Barnes, C.A.; Verin, A.D.; Worley, P.F. Verge: A Novel Vascular Early Response Gene. J. Neurosci. 2004, 24, 4092–4103. [Google Scholar] [CrossRef]
- Stritt, S.; Nurden, P.; Nurden, A.T.; Schved, J.F.; Bordet, J.C.; Roux, M.; Alessi, M.C.; Trégouët, D.A.; Mäkinen, T.; Giansily-Blaizot, M. APOLD1 Loss Causes Endothelial Dysfunction Involving Cell Junctions, Cytoskeletal Architecture, and Weibel-Palade Bodies, While Disrupting Hemostasis. Haematologica 2023, 108, 772. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cañestro, C.; Bonetti, N.R.; Wüst, P.; Nageswaran, V.; Liberale, L.; Beer, J.H.; Montecucco, F.; Lüscher, T.F.; Bohacek, J.; Camici, G.G. Apold1 Deficiency Associates with Increased Arterial Thrombosis In Vivo. Eur. J. Clin. Investig. 2020, 50, e13191. [Google Scholar] [CrossRef]
- Fan, Z.; Ardicoglu, R.; Batavia, A.A.; Rust, R.; von Ziegler, L.; Waag, R.; Zhang, J.; Desgeorges, T.; Sturman, O.; Dang, H.; et al. The Vascular Gene Apold1 Is Dispensable for Normal Development but Controls Angiogenesis under Pathological Conditions. Angiogenesis 2023, 26, 385–407. [Google Scholar] [CrossRef]
- Freson, K. Loss of APOLD1: A New Vascular Bleeding Disorder? Haematologica 2023, 108, 665. [Google Scholar] [CrossRef]
- Liao, Y.; Xie, J.; Qu, B. Apolipoprotein L Domain Containing 1 Inhibits Tissue Factor to Impede Thrombus Formation in a Rat Model of Deep Vein Thrombosis via Activating PI3K/Akt Pathway. Ann. Vasc. Surg. 2023, 89, 312–321. [Google Scholar] [CrossRef]
- Vanhamme, L.; Paturiaux-Hanocq, F.; Poelvoorde, P.; Nolan, D.P.; Lins, L.; Van Den Abbeele, J.; Pays, A.; Tebabi, P.; Van Xong, H.; Jacquet, A.; et al. Apolipoprotein L-I Is the Trypanosome Lytic Factor of Human Serum. Nature 2003, 422, 83–87. [Google Scholar] [CrossRef]
- Perez-Morga, D.; Vanhollebeke, B.; Paturiaux-Hanocq, F.; Nolan, D.P.; Lins, L.; Homblé, F.; Vanhamme, L.; Tebabi, P.; Pays, A.; Poelvoorde, P.; et al. Microbiology: Apolipoprotein L-I Promotes Trypanosome Lysis by Forming Pores in Lysosomal Membranes. Science 2005, 309, 469–472. [Google Scholar] [CrossRef]
- Pays, E.; Radwanska, M.; Magez, S. The Pathogenesis of African Trypanosomiasis. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 19–45. [Google Scholar] [CrossRef] [PubMed]
- Vanhollebeke, B.; Pays, E. The Function of Apolipoproteins L. Cell. Mol. Life Sci. 2006, 63, 1937–1944. [Google Scholar] [CrossRef]
- Van Xong, H.; Vanhamme, L.; Chamekh, M.; Chimfwembe, C.E.; Van Den Abbeele, J.; Pays, A.; Van Melrvenne, N.; Hamers, R.; De Baetselier, P.; Pays, E. A VSG Expression Site-Associated Gene Confers Resistance to Human Serum in Trypanosoma Rhodesiense. Cell 1998, 95, 839–846. [Google Scholar] [CrossRef]
- Uzureau, P.; Uzureau, S.; Lecordier, L.; Fontaine, F.; Tebabi, P.; Homblé, F.; Grélard, A.; Zhendre, V.; Nolan, D.P.; Lins, L.; et al. Mechanism of Trypanosoma Brucei Gambiense Resistance to Human Serum. Nature 2013, 501, 430–434. [Google Scholar] [CrossRef]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of Trypanolytic ApoL1 Variants with Kidney Disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.R.; Friedman, D.J. APOL1 and APOL1-Associated Kidney Disease: A Common Disease, an Unusual Disease Gene—Proceedings of the Henry Shavelle Professorship. Glomerular Dis. 2023, 3, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Pollak, M.R. Apol1 Nephropathy: From Genetics to Clinical Applications. Clin. J. Am. Soc. Nephrol. 2021, 16, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Khatua, A.K.; Cheatham, A.M.; Kruzel, E.D.; Singhal, P.C.; Skorecki, K.; Popik, W. Exon 4-Encoded Sequence Is a Major Determinant of Cytotoxicity of Apolipoprotein L1. Am. J. Physiol.—Cell Physiol. 2015, 309, C22–C37. [Google Scholar] [CrossRef]
- Cheatham, A.M.; Davis, S.E.; Khatua, A.K.; Popik, W. Blocking the 5′ Splice Site of Exon 4 by a Morpholino Oligomer Triggers APOL1 Protein Isoform Switch. Sci. Rep. 2018, 8, 8739. [Google Scholar] [CrossRef]
- Nichols, B.; Jog, P.; Lee, J.H.; Blackler, D.; Wilmot, M.; D’Agati, V.; Markowitz, G.; Kopp, J.B.; Alper, S.L.; Pollak, M.R.; et al. Innate Immunity Pathways Regulate the Nephropathy Gene Apolipoprotein L1. Kidney Int. 2014, 87, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; Gupta, N.; de Mazière, A.M.; Posthuma, G.; Chiu, C.P.; Pierce, A.A.; Hötzel, K.; Tao, J.; Foreman, O.; Koukos, G.; et al. Apolipoprotein L1-Specific Antibodies Detect Endogenous APOL1 inside the Endoplasmic Reticulum and on the Plasma Membrane of Podocytes. J. Am. Soc. Nephrol. 2020, 31, 2044–2064. [Google Scholar] [CrossRef]
- Giovinazzo, J.A.; Thomson, R.P.; Khalizova, N.; Zager, P.; Malani, N.; Rodriguez-Boulan, E.; Raper, J.; Schreiner, R. Apolipoprotein L-1 Renal Risk Variants Form Active Channels at the Plasma Membrane Driving Cytotoxicity. Elife 2020, 9, e51185. [Google Scholar] [CrossRef] [PubMed]
- Olabisi, O.A.; Zhang, J.-Y.; VerPlank, L.; Zahler, N.; DiBartolo, S.; Heneghan, J.F.; Schlöndorff, J.S.; Suh, J.H.; Yan, P.; Alper, S.L.; et al. APOL1 Kidney Disease Risk Variants Cause Cytotoxicity by Depleting Cellular Potassium and Inducing Stress-Activated Protein Kinases. Proc. Natl. Acad. Sci. USA 2016, 113, 830–837. [Google Scholar] [CrossRef]
- Schaub, C.; Verdi, J.; Lee, P.; Terra, N.; Limon, G.; Raper, J.; Thomson, R. Cation Channel Conductance and PH Gating of the Innate Immunity Factor APOL1 Are Governed by Pore-Lining Residues within the C-Terminal Domain. J. Biol. Chem. 2020, 295, 13138–13149. [Google Scholar] [CrossRef]
- Thomson, R.; Finkelstein, A. Human Trypanolytic Factor APOL1 Forms PH-Gated Cation-Selective Channels in Planar Lipid Bilayers: Relevance to Trypanosome Lysis. Proc. Natl. Acad. Sci. USA 2015, 112, 2894–2899. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wang, X.; Wen, X.; Moran, P.; Paluch, M.; Hass, P.E.; Heidersbach, A.; Haley, B.; Kirchhofer, D.; Brezski, R.J.; et al. Domain-Specific Antibodies Reveal Differences in the Membrane Topologies of Apolipoprotein L1 in Serum and Podocytes. J. Am. Soc. Nephrol. 2020, 31, 2065–2082. [Google Scholar] [CrossRef]
- Datta, S.; Antonio, B.M.; Zahler, N.H.; Theile, J.W.; Krafte, D.; Zhang, H.; Rosenberg, P.B.; Chaves, A.B.; Muoio, D.M.; Zhang, G.; et al. APOL1-Mediated Monovalent Cation Transport Contributes to APOL1-Mediated Podocytopathy in Kidney Disease. J. Clin. Investig. 2024, 134, e172262. [Google Scholar] [CrossRef]
- Kruzel-Davila, E.; Bavli-Kertselli, I.; Ofir, A.; Cheatham, A.M.; Shemer, R.; Zaknoun, E.; Chornyy, S.; Tabachnikov, O.; Davis, S.E.; Khatua, A.K.; et al. Endoplasmic Reticulum-Translocation Is Essential for APOL1 Cellular Toxicity. iScience 2022, 25, 103717. [Google Scholar] [CrossRef]
- Gupta, N.; Waas, B.; Austin, D.; De Mazière, A.M.; Kujala, P.; Stockwell, A.D.; Li, T.; Yaspan, B.L.; Klumperman, J.; Scales, S.J. Apolipoprotein L1 (APOL1) Renal Risk Variant-Mediated Podocyte Cytotoxicity Depends on African Haplotype and Surface Expression. Sci. Rep. 2024, 14, 3765. [Google Scholar] [CrossRef]
- Granado, D.; Müller, D.; Krausel, V.; Kruzel-Davila, E.; Schuberth, C.; Eschborn, M.; Wedlich-Söldner, R.; Skorecki, K.; Pavenstädt, H.; Michgehl, U.; et al. Intracellular APOL1 Risk Variants Cause Cytotoxicity Accompanied by Energy Depletion. J. Am. Soc. Nephrol. 2017, 28, 3227–3238. [Google Scholar] [CrossRef] [PubMed]
- Wakashin, H.; Heymann, J.; Roshanravan, H.; Daneshpajouhnejad, P.; Rosenberg, A.; Shin, M.K.; Hoek, M.; Kopp, J.B. APOL1 Renal Risk Variants Exacerbate Podocyte Injury by Increasing Inflammatory Stress. BMC Nephrol. 2020, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Wen, H.; Saleem, M.A.; Mikulak, J.; Malhotra, A.; Skorecki, K.; Singhal, P.C. Vascular Smooth Muscle Cells Contribute to APOL1-Induced Podocyte Injury in HIV Milieu. Exp. Mol. Pathol. 2015, 98, 491–501. [Google Scholar] [CrossRef]
- Pays, E. The Two Levels of Podocyte Dysfunctions Induced by Apolipoprotein L1 Risk Variants. Kidney Dial. 2024, 4, 126–143. [Google Scholar] [CrossRef]
- Uzureau, S.; Lecordier, L.; Uzureau, P.; Hennig, D.; Graversen, J.H.; Homblé, F.; Mfutu, P.E.; Oliveira Arcolino, F.; Ramos, A.R.; La Rovere, R.M.; et al. APOL1 C-Terminal Variants May Trigger Kidney Disease through Interference with APOL3 Control of Actomyosin. Cell Rep. 2020, 30, 3821–3836.e13. [Google Scholar] [CrossRef]
- Wan, G.; Zhaorigetu, S.; Liu, Z.; Kaini, R.; Jiang, Z.; Hu, C.A. Apolipoprotein L1, a Novel Bcl-2 Homology Domain 3-Only Lipid-Binding Protein, Induces Autophagic Cell Death. J. Biol. Chem. 2008, 283, 21540–21549. [Google Scholar] [CrossRef]
- Chun, J.; Zhang, J.Y.; Wilkins, M.S.; Subramanian, B.; Riella, C.; Magraner, J.M.; Alper, S.L.; Friedman, D.J.; Pollak, M.R. Recruitment of APOL1 Kidney Disease Risk Variants to Lipid Droplets Attenuates Cell Toxicity. Proc. Natl. Acad. Sci. USA 2019, 116, 3712–3721. [Google Scholar] [CrossRef]
- Ma, L.; Chou, J.W.; Snipes, J.A.; Bharadwaj, M.S.; Craddock, A.L.; Cheng, D.; Weckerle, A.; Petrovic, S.; Hicks, P.J.; Hemal, A.K.; et al. APOL1 Renal-Risk Variants Induce Mitochondrial Dysfunction. J. Am. Soc. Nephrol. 2016, 116, 3712–3721. [Google Scholar] [CrossRef]
- Madhavan, S.M.; O’Toole, J.F.; Konieczkowski, M.; Barisoni, L.; Thomas, D.B.; Ganesan, S.; Bruggeman, L.A.; Buck, M.; Sedor, J.R. APOL1 Variants Change C-Terminal Conformational Dynamics and Binding to SNARE Protein VAMP8. JCI Insight 2017, 2, e92581. [Google Scholar] [CrossRef]
- Lecordier, L.; Heo, P.; Graversen, J.H.; Hennig, D.; Skytthe, M.K.; Cornet d’Elzius, A.; Pincet, F.; Pérez-Morga, D.; Pays, E. Apolipoproteins L1 and L3 Control Mitochondrial Membrane Dynamics. Cell Rep. 2023, 42, 113528. [Google Scholar] [CrossRef]
- Shah, S.S.; Lannon, H.; Dias, L.; Zhang, J.Y.; Alper, S.L.; Pollak, M.R.; Friedman, D.J. APOL1 Kidney Risk Variants Induce Cell Death via Mitochondrial Translocation and Opening of the Mitochondrial Permeability Transition Pore. J. Am. Soc. Nephrol. 2019, 30, 2355–2368. [Google Scholar] [CrossRef] [PubMed]
- Limou, S.; Nelson, G.W.; Kopp, J.B.; Winkler, C.A. APOL1 Kidney Risk Alleles: Population Genetics and Disease Associations. Adv. Chronic Kidney Dis. 2014, 21, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Zhaorigetu, S.; Wan, G.; Kaini, R.; Jiang, Z.; C. A.A. ApoL1, a BH3-Only Lipid-Binding Protein, Induces Autophagic Cell Death. Autophagy 2008, 4, 1079–1082. [Google Scholar] [CrossRef]
- Beckerman, P.; Bi-Karchin, J.; Park, A.S.D.; Qiu, C.; Dummer, P.D.; Soomro, I.; Boustany-Kari, C.M.; Pullen, S.S.; Miner, J.H.; Hu, C.-A.A.; et al. Transgenic Expression of Human APOL1 Risk Variants in Podocytes Induces Kidney Disease in Mice. Nat. Med. 2017, 23, 429–438. [Google Scholar] [CrossRef]
- Ma, L.; Ainsworth, H.C.; Snipes, J.A.; Murea, M.; Choi, Y.A.; Langefeld, C.D.; Parks, J.S.; Bharadwaj, M.S.; Chou, J.W.; Hemal, A.K.; et al. APOL1 Kidney-Risk Variants Induce Mitochondrial Fission. Kidney Int. Rep. 2020, 5, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Riella, C.V.; Chung, H.; Shah, S.S.; Wang, M.; Magraner, J.M.; Ribas, G.T.; Ribas, H.T.; Zhang, J.Y.; Alper, S.L.; et al. DGAT2 Inhibition Potentiates Lipid Droplet Formation To Reduce Cytotoxicity in APOL1 Kidney Risk Variants. J. Am. Soc. Nephrol. 2022, 33, 889–907. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Zadoorian, A.; Du, X.; Yang, H. Lipid Droplet Biogenesis and Functions in Health and Disease. Nat. Rev. Endocrinol. 2023, 19, 443–459. [Google Scholar] [CrossRef]
- Phillips, M.J.; Voeltz, G.K. Structure and Function of ER Membrane Contact Sites with Other Organelles. Nat. Rev. Mol. Cell Biol. 2015, 17, 69–82. [Google Scholar] [CrossRef]
- Cheng, D.; Weckerle, A.; Yu, Y.; Ma, L.; Zhu, X.; Murea, M.; Freedman, B.I.; Parks, J.S.; Shelness, G.S. Biogenesis and Cytotoxicity of APOL1 Renal Risk Variant Proteins in Hepatocytes and Hepatoma Cells. J. Lipid Res. 2015, 56, 1583–1593. [Google Scholar] [CrossRef]
- Prinz, W.A.; Toulmay, A.; Balla, T. The Functional Universe of Membrane Contact Sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Shukha, K.; Mueller, J.L.; Chung, R.T.; Curry, M.P.; Friedman, D.J.; Pollak, M.R.; Berg, A.H. Most ApoL1 Is Secreted by the Liver. J. Am. Soc. Nephrol. 2017, 28, 1079–1083. [Google Scholar] [CrossRef]
- Hayek, S.S.; Koh, K.H.; Grams, M.E.; Wei, C.; Ko, Y.-A.; Li, J.; Samelko, B.; Lee, H.; Dande, R.R.; Lee, H.W.; et al. A Tripartite Complex of SuPAR, APOL1 Risk Variants and Avβ3 Integrin on Podocytes Mediates Chronic Kidney Disease. Nat. Med. 2017, 23, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.J.; Pollak, M.R.R. APOL1 and Kidney Disease: From Genetics to Biology. Annu. Rev. Physiol. 2020, 82, 323–342. [Google Scholar] [CrossRef]
- Daneshpajouhnejad, P.; Kopp, J.B.; Winkler, C.A.; Rosenberg, A.Z. The Evolving Story of Apolipoprotein L1 Nephropathy: The End of the Beginning. Nat. Rev. Nephrol. 2022, 18, 307–320. [Google Scholar] [CrossRef]
- Kruzel-Davila, E.; Shemer, R.; Ofir, A.; Bavli-Kertselli, I.; Darlyuk-Saadon, I.; Oren-Giladi, P.; Wasser, W.G.; Magen, D.; Zaknoun, E.; Schuldiner, M.; et al. APOL1-Mediated Cell Injury Involves Disruption of Conserved Trafficking Processes. J. Am. Soc. Nephrol. 2017, 28, 1117–1130. [Google Scholar] [CrossRef]
- Gerstner, L.; Chen, M.; Kampf, L.L.; Milosavljevic, J.; Lang, K.; Schneider, R.; Hildebrandt, F.; Helmstädter, M.; Walz, G.; Hermle, T. Inhibition of Endoplasmic Reticulum Stress Signaling Rescues Cytotoxicity of Human Apolipoprotein-L1 Risk Variants in Drosophila. Kidney Int. 2022, 101, 1216–1231. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Lee, J.G.; Fu, Y.; van de Leemput, J.; Ray, P.E.; Han, Z. APOL1-G2 Accelerates Nephrocyte Cell Death by Inhibiting the Autophagy Pathway. DMM Dis. Model. Mech. 2023, 16, dmm050223. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, L.A.; O’Toole, J.F.; Ross, M.D.; Madhavan, S.M.; Smurzynski, M.; Wu, K.; Bosch, R.J.; Gupta, S.; Pollak, M.R.; Sedor, J.R.; et al. Plasma Apolipoprotein L1 Levels Do Not Correlate with CKD. J. Am. Soc. Nephrol. 2014, 25, 634–644. [Google Scholar] [CrossRef]
- McCarthy, G.M.; Blasio, A.; Donovan, O.G.; Schaller, L.B.; Bock-Hughes, A.; Magraner, J.M.; Suh, J.H.; Tattersfield, C.F.; Stillman, I.E.; Shah, S.S.; et al. Recessive, Gain-of-Function Toxicity in an APOL1 BAC Transgenic Mouse Model Mirrors Human APOL1 Kidney Disease. DMM Dis. Model. Mech. 2021, 14, dmm048952. [Google Scholar] [CrossRef]
- O’Toole, J.F.; Schilling, W.; Kunze, D.; Madhavan, S.M.; Konieczkowski, M.; Gu, Y.; Luo, L.; Wu, Z.; Bruggeman, L.A.; Sedor, J.R. ApoL1 Overexpression Drives Variant-Independent Cytotoxicity. J. Am. Soc. Nephrol. 2018, 29, 869–879. [Google Scholar] [CrossRef]
- Lannon, H.; Shah, S.S.; Dias, L.; Blackler, D.; Alper, S.L.; Pollak, M.R.; Friedman, D.J. Apolipoprotein L1 (APOL1) Risk Variant Toxicity Depends on the Haplotype Background. Kidney Int. 2019, 96, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, D.B.; Shegokar, V.; Nihalani, D.; Rathore, Y.S.; Mallik, L.; Ashish; Zare, V.; Ikizler, H.O.; Powar, R.; Holzman, L.B. APOL1 Null Alleles from a Rural Village in India Do Not Correlate with Glomerulosclerosis. PLoS ONE 2012, 7, e51546. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Kataria, R.; Zhang, J.Y.; Moore, S.; Petitpas, K.; Mohamed, A.; Zahler, N.; Pollak, M.R.; Olabisi, O.A. Kidney Disease-Associated APOL1 Variants Have Dose-Dependent, Dominant Toxic Gain-of-Function. J. Am. Soc. Nephrol. 2020, 31, 2083–2096. [Google Scholar] [CrossRef]
- Lan, X.; Jhaveri, A.; Cheng, K.; Wen, H.; Saleem, M.A.; Mathieson, P.W.; Mikulak, J.; Aviram, S.; Malhotra, A.; Skorecki, K.; et al. APOL1 Risk Variants Enhance Podocyte Necrosis through Compromising Lysosomal Membrane Permeability. Am. J. Physiol. Renal Physiol. 2014, 307, F326–F336. [Google Scholar] [CrossRef]
- Vanwalleghem, G.; Fontaine, F.; Lecordier, L.; Tebabi, P.; Klewe, K.; Nolan, D.P.; Yamaryo-Botté, Y.; Botté, C.; Kremer, A.; Burkard, G.S.; et al. Coupling of Lysosomal and Mitochondrial Membrane Permeabilization in Trypanolysis by APOL1. Nat. Commun. 2015, 6, 8078. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 Arbiters of Apoptosis and Their Growing Role as Cancer Targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-Only Proteins: Orchestrators of Apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Dummer, P.D.; Limou, S.; Rosenberg, A.Z.; Heymann, J.; Nelson, G.; Winkler, C.A.; Kopp, J.B. APOL1 Kidney Disease Risk Variants: An Evolving Landscape. Semin. Nephrol. 2015, 35, 222–236. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Galindo-Moreno, J.; Iurlaro, R.; El Mjiyad, N.; Díez-Pérez, J.; Gabaldón, T.; Muñoz-Pinedo, C. Apolipoprotein L2 Contains a BH3-like Domain but It Does Not Behave as a BH3-Only Protein. Cell Death Dis. 2014, 5, e1275. [Google Scholar] [CrossRef]
- Heneghan, J.F.; Vandorpe, D.H.; Shmukler, B.E.; Giovinazzo, J.A.; Raper, J.; Friedman, D.J.; Pollak, M.R.; Alper, S.L.; Alper, S.L. BH3 Domain-Independent Apolipoprotein L1 Toxicity Rescued by BCL2 Prosurvival Proteins. Am. J. Physiol. Physiol. 2015, 309, C332–C347. [Google Scholar] [CrossRef]
- Lan, X.; Wen, H.; Lederman, R.; Malhotra, A.; Mikulak, J.; Popik, W.; Skorecki, K.; Singhal, P.C. Protein Domains of APOL1 and Its Risk Variants. Exp. Mol. Pathol. 2015, 99, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Kumar, V.; Lan, X.; Shoshtari, S.S.M.M.; Eng, J.M.M.; Zhou, X.; Wang, F.; Wang, H.; Skorecki, K.; Xing, G.; et al. APOL1 Risk Variants Cause Podocytes Injury through Enhancing Endoplasmic Reticulum Stress. Biosci. Rep. 2018, 38, BSR20171713. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Patil, G.; Mishra, A.; Lan, X.; Popik, W.; Malhotra, A.; Skorecki, K.; Singhal, P.C. Effect of APOL1 Disease Risk Variants on APOL1 Gene Product. Biosci. Rep. 2017, 37, BSR20160531. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xiao, W.; Lee, K.; Salem, F.; Wen, J.; He, L.; Zhang, J.; Fei, Y.; Cheng, D.; Bao, H.; et al. Inhibition of Reticulon-1a-Mediated Endoplasmic Reticulum Stress in Early Aki Attenuates Renal Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 2007–2021. [Google Scholar] [CrossRef]
- Andrade-Silva, M.; Dhillon, P.; Sanchez-Navarro, A.; Mukhi, D.; Hu, H.; Kolligundla, L.P.; Bergeson, A.; Abedini, A.; Levinsohn, J.; Dumoulin, B.; et al. The Critical Role of Endoplasmic Reticulum Stress and the Stimulator of Interferon Genes (STING) Pathway in Kidney Fibrosis. Kidney Int. 2025, 107, 302–316. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, J.; Xiao, W.; Lee, K.; Li, Z.; Wen, J.; He, L.; Gui, D.; Xue, R.; Jian, G.; et al. Rtn1a-Mediated Endoplasmic Reticulum Stress in Podocyte Injury and Diabetic Nephropathy. Sci. Rep. 2017, 7, 323. [Google Scholar] [CrossRef]
- Yu, S.; Gu, X.; Zheng, Q.; Liu, Y.; Suhas, T.; Du, W.; Xie, L.; Fang, Z.; Zhao, Y.; Yang, M.; et al. Tauroursodeoxycholic Acid Ameliorates Renal Injury Induced by COL4A3 Mutation. Kidney Int. 2024, 106, 433–449. [Google Scholar] [CrossRef]
- Zhu, F.; Li, S.; Gu, Q.; Xie, N.; Wu, Y. APOL1 Induces Pyroptosis of Fibroblasts Through NLRP3/Caspase-1/GSDMD Signaling Pathway in Ulcerative Colitis. J. Inflamm. Res. 2023, 16, 6385–6396. [Google Scholar] [CrossRef]
- Jha, A.; Kumar, V.; Haque, S.; Ayasolla, K.; Saha, S.; Lan, X.; Malhotra, A.; Saleem, M.A.; Skorecki, K.; Singhal, P.C. Alterations in Plasma Membrane Ion Channel Structures Stimulate NLRP3 Inflammasome Activation in APOL1 Risk Milieu. FEBS J. 2020, 287, 2000–2022. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Raman, A.; Coffey, N.J.; Sheng, X.; Wahba, J.; Seasock, M.J.; Ma, Z.; Beckerman, P.; Laczkó, D.; Palmer, M.B.; et al. The Key Role of NLRP3 and STING in APOL1-Associated Podocytopathy. J. Clin. Investig. 2021, 131, e136329. [Google Scholar] [CrossRef]
- Pays, E. Apolipoprotein-L Functions in Membrane Remodeling. Cells 2024, 13, 2115. [Google Scholar] [CrossRef]
- Kruzel-Davila, E.; Skorecki, K. Dilemmas and Challenges in Apolipoprotein L1 Nephropathy Research. Curr. Opin. Nephrol. Hypertens. 2019, 28, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Pant, J.; Giovinazzo, J.A.; Tuka, L.S.; Peña, D.; Raper, J.; Thomson, R. Apolipoproteins L1-6 Share Key Cation Channel-Regulating Residues but Have Different Membrane Insertion and Ion Conductance Properties. J. Biol. Chem. 2021, 297, 100951. [Google Scholar] [CrossRef]
- Bruno, J.; Pozzi, N.; Oliva, J.; Edwards, J.C. Apolipoprotein L1 Confers PH-Switchable Ion Permeability to Phospholipid Vesicles. J. Biol. Chem. 2017, 292, 18344–18353. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, B.; Dakin, L.A.; Fortier, A.; Nanou, E.; Blasio, A.; Mann, J.; Miller, H.; Fletcher, M.; Wang, T.; Nanthakumar, S.; et al. Small Molecule APOL1 Inhibitors as a Precision Medicine Approach for APOL1-Mediated Kidney Disease. Nat. Commun. 2025, 16, 167. [Google Scholar] [CrossRef]
- Egbuna, O.; Zimmerman, B.; Manos, G.; Fortier, A.; Chirieac, M.C.; Dakin, L.A.; Friedman, D.J.; Bramham, K.; Campbell, K.; Knebelmann, B.; et al. Inaxaplin for Proteinuric Kidney Disease in Persons with Two APOL1 Variants. N. Engl. J. Med. 2023, 388, 969–979. [Google Scholar] [CrossRef]
- Hung, A.M.; Assimon, V.A.; Chen, H.C.; Yu, Z.; Vlasschaert, C.; Triozzi, J.L.; Chan, H.; Wheless, L.; Wilson, O.; Shah, S.C.; et al. Genetic Inhibition of APOL1 Pore-Forming Function Prevents APOL1-Mediated Kidney Disease. J. Am. Soc. Nephrol. 2023, 34, 1889–1899. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Wang, M.; Tian, L.; Genovese, G.; Yan, P.; Wilson, J.G.; Thadhani, R.; Mottl, A.K.; Appel, G.B.; Bick, A.G.; et al. UBD Modifies APOL1-Induced Kidney Disease Risk. Proc. Natl. Acad. Sci. USA 2018, 115, 3446–3451. [Google Scholar] [CrossRef]
- Skorecki, K.L.; Lee, J.H.; Langefeld, C.D.; Rosset, S.; Tzur, S.; Wasser, W.G.; Shemer, R.; Hawkins, G.A.; Divers, J.; Parekh, R.S.; et al. A Null Variant in the Apolipoprotein L3 Gene Is Associated with Non-Diabetic Nephropathy. Nephrol. Dial. Transplant. 2018, 33, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Levin, M.G.; Duda, J.T.; Landry, L.G.; Witschey, W.R.; Damrauer, S.M.; Ritchie, M.D.; Rader, D.J. Protein-Truncating Variant in APOL3 Increases Chronic Kidney Disease Risk in Epistasis with APOL1 Risk Alleles. JCI Insight 2024, 9, e181238. [Google Scholar] [CrossRef]
- Grampp, S.; Krüger, R.; Lauer, V.; Uebel, S.; Knaup, K.X.; Naas, J.; Höffken, V.; Weide, T.; Schiffer, M.; Naas, S.; et al. Hypoxia Hits APOL1 in the Kidney. Kidney Int. 2023, 104, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Juliar, B.A.; Stanaway, I.B.; Sano, F.; Fu, H.; Smith, K.D.; Akilesh, S.; Scales, S.J.; El Saghir, J.; Bhatraju, P.K.; Liu, E.; et al. Interferon-γ Induces Combined Pyroptotic Angiopathy and APOL1 Expression in Human Kidney Disease. Cell Rep. 2024, 43, 114310. [Google Scholar] [CrossRef] [PubMed]
- Blazer, A.; Qian, Y.; Schlegel, M.P.; Algasas, H.; Buyon, J.P.; Cadwell, K.; Cammer, M.; Heffron, S.P.; Liang, F.X.; Mehta-Lee, S.; et al. APOL1 Variant-Expressing Endothelial Cells Exhibit Autophagic Dysfunction and Mitochondrial Stress. Front. Genet. 2022, 13, 769936. [Google Scholar] [CrossRef]
- Carracedo, M.; Ericson, E.; Ågren, R.; Forslöw, A.; Madeyski-Bengtson, K.; Svensson, A.; Riddle, R.; Christoffersson, J.; González-King Garibotti, H.; Lazovic, B.; et al. APOL1 Promotes Endothelial Cell Activation beyond the Glomerulus. iScience 2023, 26, 106830. [Google Scholar] [CrossRef]
- Wu, J.; Ma, Z.; Raman, A.; Beckerman, P.; Dhillon, P.; Mukhi, D.; Palmer, M.; Chen, H.C.; Cohen, C.R.; Dunn, T.; et al. APOL1 Risk Variants in Individuals of African Genetic Ancestry Drive Endothelial Cell Defects That Exacerbate Sepsis. Immunity 2021, 54, 2632–2649. [Google Scholar] [CrossRef]
- Pell, J.; Nagata, S.; Menon, M.C. Nonpodocyte Roles of APOL1 Variants: An Evolving Paradigm. Kidney360 2023, 4, e1325–e1331. [Google Scholar] [CrossRef]
- Pavenstädt, H.; Kriz, W.; Kretzler, M. Cell Biology of the Glomerular Podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef]
- Garg, P. A Review of Podocyte Biology. Am. J. Nephrol. 2018, 47, 3–13. [Google Scholar] [CrossRef]
- Grahammer, F.; Schell, C.; Huber, T.B. The Podocyte Slit Diaphragm—From a Thin Grey Line to a Complex Signalling Hub. Nat. Rev. Nephrol. 2013, 9, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, A.V. Endoplasmic Reticulum Stress, the Unfolded Protein Response and Autophagy in Kidney Diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Anders, H.J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Prim. 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höffken, V.; Braun, D.A.; Pavenstädt, H.; Weide, T. A Cell Biologist’s View on APOL1: What We Know and What We Still Need to Address. Cells 2025, 14, 960. https://doi.org/10.3390/cells14130960
Höffken V, Braun DA, Pavenstädt H, Weide T. A Cell Biologist’s View on APOL1: What We Know and What We Still Need to Address. Cells. 2025; 14(13):960. https://doi.org/10.3390/cells14130960
Chicago/Turabian StyleHöffken, Verena, Daniela Anne Braun, Hermann Pavenstädt, and Thomas Weide. 2025. "A Cell Biologist’s View on APOL1: What We Know and What We Still Need to Address" Cells 14, no. 13: 960. https://doi.org/10.3390/cells14130960
APA StyleHöffken, V., Braun, D. A., Pavenstädt, H., & Weide, T. (2025). A Cell Biologist’s View on APOL1: What We Know and What We Still Need to Address. Cells, 14(13), 960. https://doi.org/10.3390/cells14130960