

Identification of Transcriptomic Differences in Induced Pluripotent Stem Cells and Neural Progenitors from Amyotrophic Lateral Sclerosis Patients Carrying Different Mutations: A Pilot Study

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. CD34+ Isolation
2.3. iPSCs Reprogramming
2.4. iPSC Culture
2.5. Alkaline Phosphatase Staining
2.6. RNA Isolation and RT-PCR
2.7. Immunofluorescence Staining
2.8. Embryoid Body Generation
2.9. Neurosphere Generation
2.10. Western Blot
2.11. Mutational Analysis of iPSCs
2.12. RNA Sequencing
2.13. Data Availability
3. Results
3.1. Generation of iPSCs from CD34+ Cells Isolated from ALS Patients and Healthy Donors
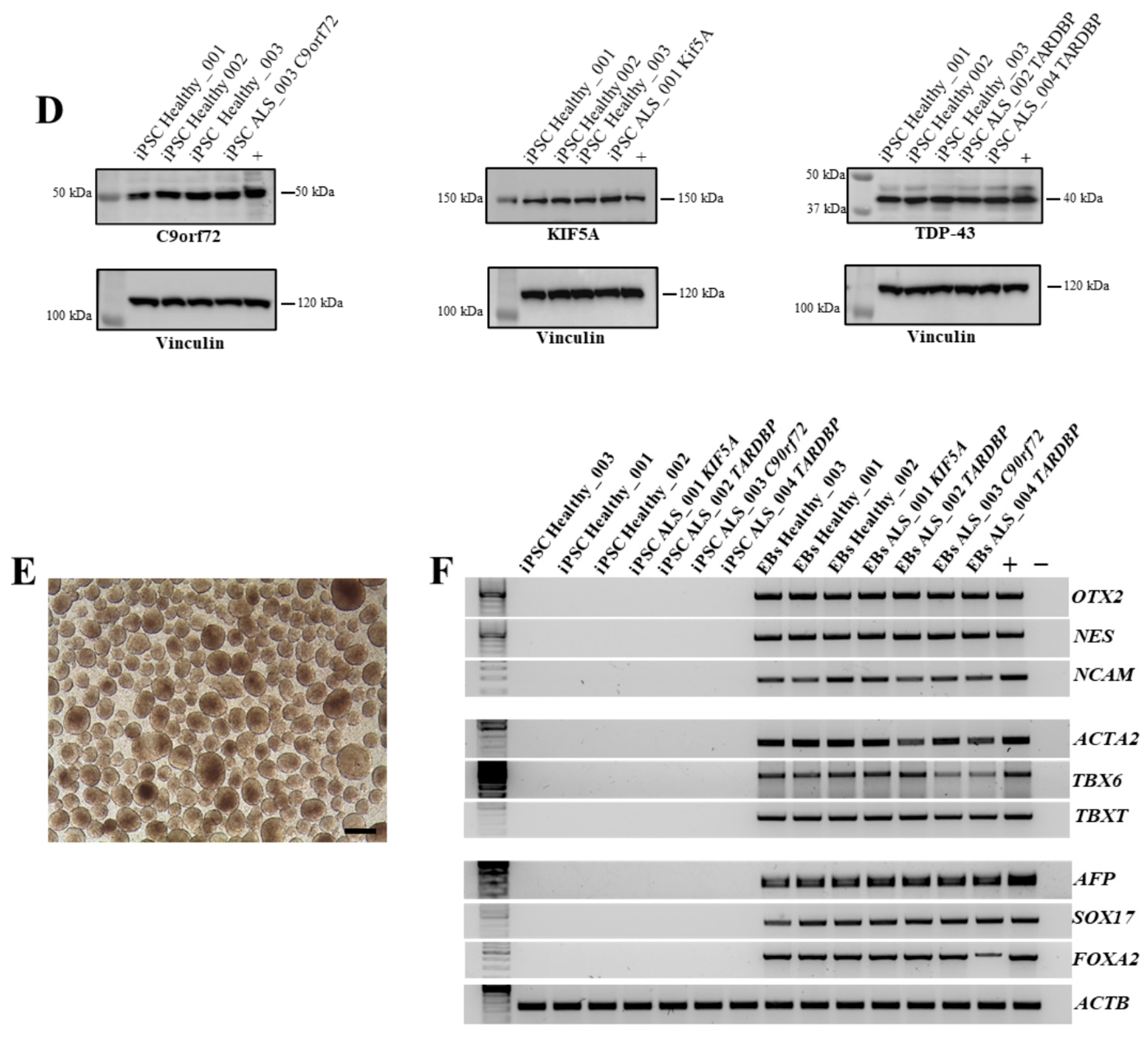
3.2. Characterization of iPSCs from CD34+ Cells Isolated from ALS Patients and Healthy Donors
3.3. Neurosphere Generation and Neuroectoderm Marker Expression
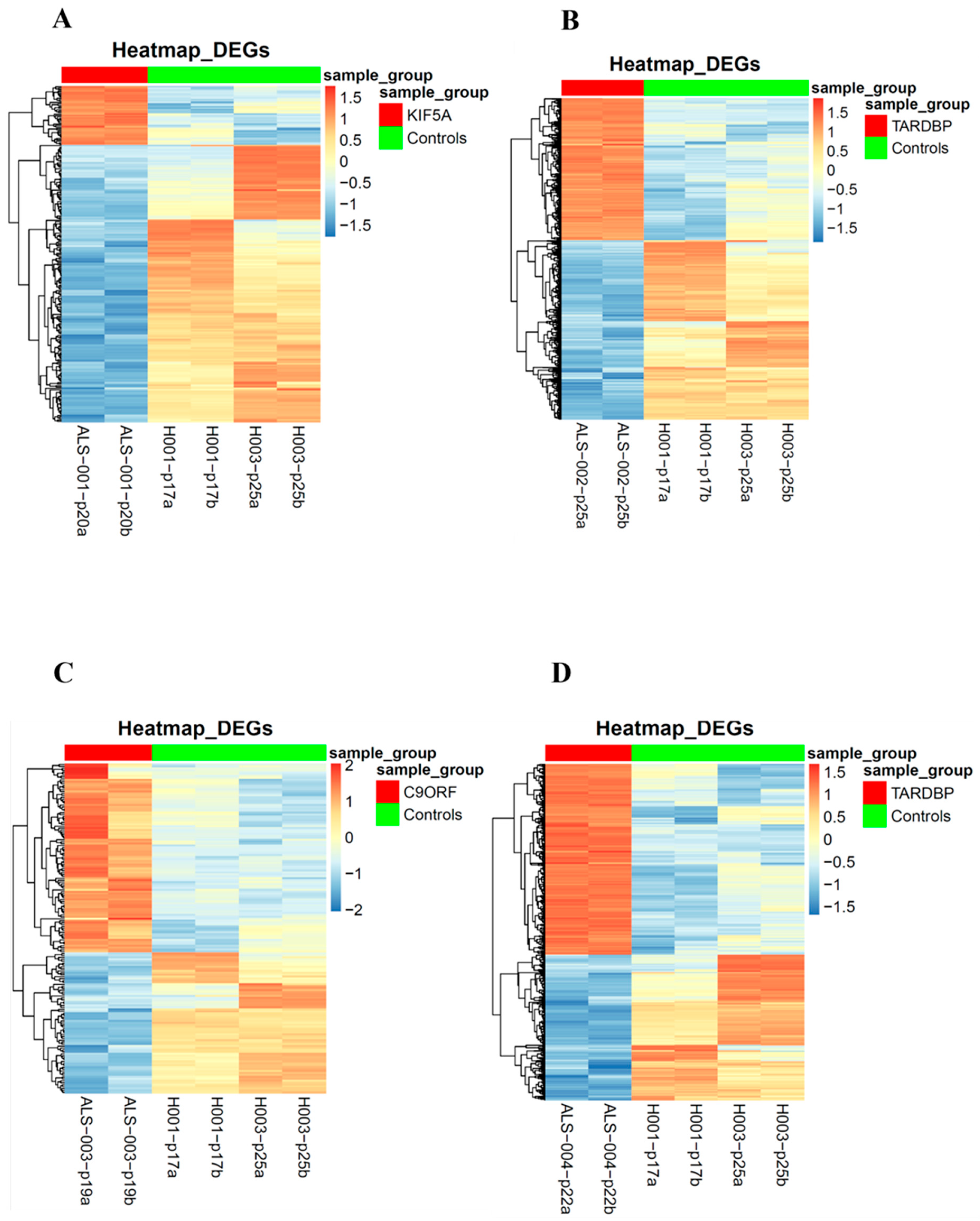
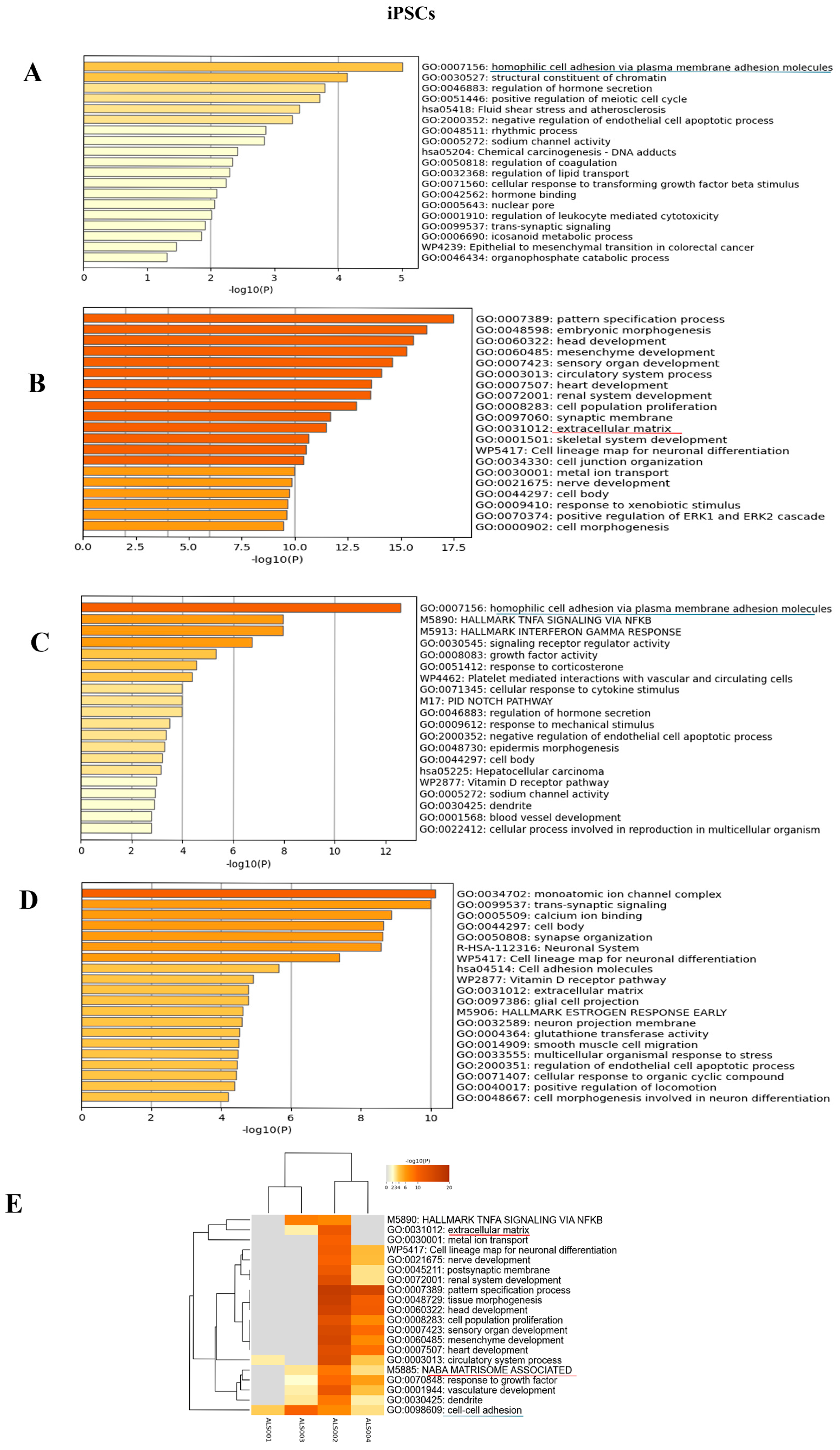
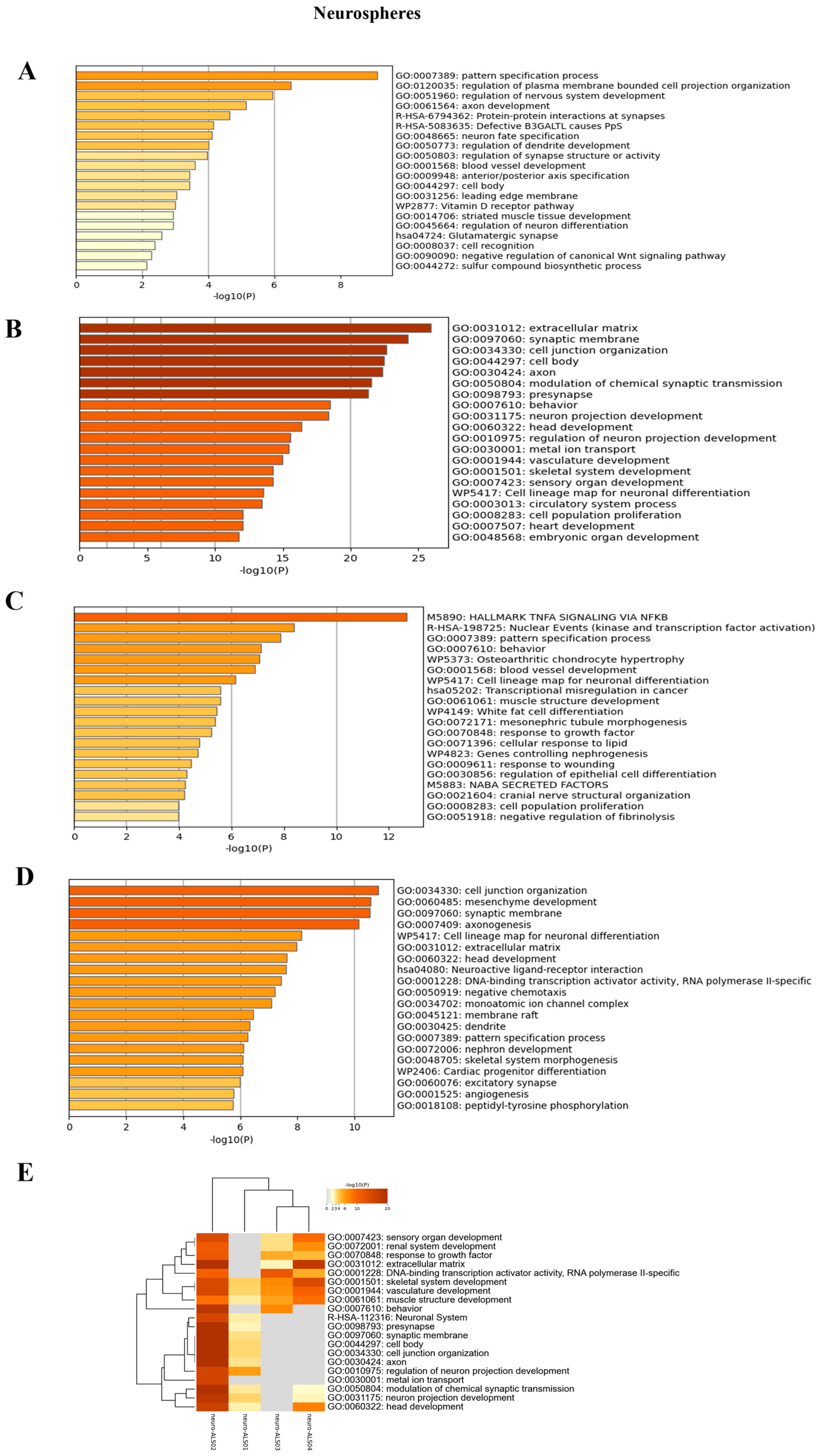
3.4. Transcriptomic Analysis of iPSCs and Neurospheres from ALS and Healthy Controls
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic Lateral Sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging Insights into the Complex Genetics and Pathophysiology of Amyotrophic Lateral Sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.; Hu, F. Cellular and Physiological Functions of C9ORF72 and Implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef]
- Frick, P.; Sellier, C.; Mackenzie, I.R.A.; Cheng, C.-Y.; Tahraoui-Bories, J.; Martinat, C.; Pasterkamp, R.J.; Prudlo, J.; Edbauer, D.; Oulad-Abdelghani, M.; et al. Novel Antibodies Reveal Presynaptic Localization of C9orf72 Protein and Reduced Protein Levels in C9orf72 Mutation Carriers. Acta Neuropathol. Commun. 2018, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Amyotrophic Lateral Sclerosis: Disease State Overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-Wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1267–1288. [Google Scholar] [CrossRef]
- Pant, D.C.; Parameswaran, J.; Rao, L.; Loss, I.; Chilukuri, G.; Parlato, R.; Shi, L.; Glass, J.D.; Bassell, G.J.; Koch, P.; et al. ALS-Linked KIF5A ΔExon27 Mutant Causes Neuronal Toxicity through Gain-of-Function. EMBO Rep. 2022, 23, e54234. [Google Scholar] [CrossRef]
- Wong, C.-O.; Venkatachalam, K. Motor Neurons from ALS Patients with Mutations in C9ORF72 and SOD1 Exhibit Distinct Transcriptional Landscapes. Hum. Mol. Genet. 2019, 28, 2799–2810. [Google Scholar] [CrossRef]
- Popescu, I.R.; Nicaise, C.; Liu, S.; Bisch, G.; Knippenberg, S.; Daubie, V.; Bohl, D.; Pochet, R. Neural Progenitors Derived from Human Induced Pluripotent Stem Cells Survive and Differentiate upon Transplantation into a Rat Model of Amyotrophic Lateral Sclerosis. Stem Cells Transl. Med. 2013, 2, 167–174. [Google Scholar] [CrossRef]
- Mazzini, L.; De Marchi, F.; Buzanska, L.; Follenzi, A.; Glover, J.C.; Gelati, M.; Lombardi, I.; Maioli, M.; Mesa-Herrera, F.; Mitrečić, D.; et al. Current Status and New Avenues of Stem Cell-Based Preclinical and Therapeutic Approaches in Amyotrophic Lateral Sclerosis. Expert Opin. Biol. Ther. 2024, 24, 933–954. [Google Scholar] [CrossRef]
- Agosta, F.; Al-Chalabi, A.; Filippi, M.; Hardiman, O.; Kaji, R.; Meininger, V.; Nakano, I.; Shaw, P.; Shefner, J.; van den Berg, L.H.; et al. The El Escorial Criteria: Strengths and Weaknesses. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J. Revisiting the Concept of Amyotrophic Lateral Sclerosis as a Multisystems Disorder of Limited Phenotypic Expression. Curr. Opin. Neurol. 2017, 30, 599–607. [Google Scholar] [CrossRef]
- Olgasi, C.; Talmon, M.; Merlin, S.; Cucci, A.; Richaud-Patin, Y.; Ranaldo, G.; Colangelo, D.; Di Scipio, F.; Berta, G.N.; Borsotti, C.; et al. Patient-Specific IPSC-Derived Endothelial Cells Provide Long-Term Phenotypic Correction of Hemophilia A. Stem Cell Rep. 2018, 11, 1391–1406. [Google Scholar] [CrossRef]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.-L.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and Expansion of Highly Pure Motor Neuron Progenitors from Human Pluripotent Stem Cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef]
- Workman, M.J.; Lim, R.G.; Wu, J.; Frank, A.; Ornelas, L.; Panther, L.; Galvez, E.; Perez, D.; Meepe, I.; Lei, S.; et al. Large-Scale Differentiation of IPSC-Derived Motor Neurons from ALS and Control Subjects. Neuron 2023, 111, 1191–1204.e5. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 14 March 2025).
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Ghaffari, L.T.; Trotti, D.; Haeusler, A.R.; Jensen, B.K. Breakdown of the Central Synapses in C9orf72-Linked ALS/FTD. Front. Mol. Neurosci. 2022, 15, 1005112. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Wang, C.; Zhang, D.; Yao, X.; Zhao, Q.; Huang, X.; Lin, F.; Xue, C.; Wang, Y.; He, R.; et al. PCDHA9 as a Candidate Gene for Amyotrophic Lateral Sclerosis. Nat. Commun. 2024, 15, 2189. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Van Den Bosch, L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 12175. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Zaepfel, B.L.; Hayes, L.; Fitchman, B.; Salzberg, Y.; Luo, E.C.; Bowen, K.; Trost, H.; Aigner, S.; Rigo, F.; et al. G4C2 Repeat RNA Initiates a POM121-Mediated Reduction in Specific Nucleoporins in C9orf72 ALS/FTD. Neuron 2020, 107, 1124–1140.e11. [Google Scholar] [CrossRef]
- Ziff, O.J.; Harley, J.; Wang, Y.; Neeves, J.; Tyzack, G.; Ibrahim, F.; Skehel, M.; Chakrabarti, A.M.; Kelly, G.; Patani, R. Nucleocytoplasmic MRNA Redistribution Accompanies RNA Binding Protein Mislocalization in ALS Motor Neurons and Is Restored by VCP ATPase Inhibition. Neuron 2023, 111, 3011–3027.e7. [Google Scholar] [CrossRef]
- Baron, D.M.; Fenton, A.R.; Saez-Atienzar, S.; Giampetruzzi, A.; Sreeram, A.; Shankaracharya; Keagle, P.J.; Doocy, V.R.; Smith, N.J.; Danielson, E.W.; et al. ALS-Associated KIF5A Mutations Abolish Autoinhibition Resulting in a Toxic Gain of Function. Cell Rep. 2022, 39, 110598. [Google Scholar] [CrossRef]
- Pintér, P.; Alpár, A. The Role of Extracellular Matrix in Human Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 11085. [Google Scholar] [CrossRef]
- Bosiacki, M.; Gąssowska-Dobrowolska, M.; Kojder, K.; Fabiańska, M.; Jeżewski, D.; Gutowska, I.; Lubkowska, A. Perineuronal Nets and Their Role in Synaptic Homeostasis. Int. J. Mol. Sci. 2019, 20, 4108. [Google Scholar] [CrossRef]
- Gómez-Pinedo, U.; Galán, L.; Matías-Guiu, J.A.; Pytel, V.; Moreno, T.; Guerrero-Sola, A.; Matías-Guiu, J. Notch Signalling in the Hippocampus of Patients with Motor Neuron Disease. Front. Neurosci. 2019, 13, 302. [Google Scholar] [CrossRef]
- Liu, C.; Li, D.; Lv, C.; Gao, Z.; Qi, Y.; Wu, H.; Tian, Y.; Guo, Y. Activation of the Notch Signaling Pathway and Cellular Localization of Notch Signaling Molecules in the Spinal Cord of SOD1-G93A ALS Model Mice. Neuroscience 2020, 432, 84–93. [Google Scholar] [CrossRef]
- Moreland, T.; Poulain, F.E. To Stick or Not to Stick: The Multiple Roles of Cell Adhesion Molecules in Neural Circuit Assembly. Front. Neurosci. 2022, 16, 889155. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Michalopoulou, M.; Nikolaou, C.; Cambouri, C.; Rombos, A.; Dimitrakopoulos, A.; Vassilopoulos, D. The Role of Soluble Intercellular Adhesion Molecules in Neurodegenerative Disorders. J. Neurol. Sci. 2005, 228, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, G.; Busdraghi, L.M.; Chece, G.; Menini, A.; Ceccanti, M.; Libonati, L.; Cambieri, C.; Fiorentino, F.; Rotili, D.; Scavizzi, F.; et al. GDF15-GFRAL Signaling Drives Weight Loss and Lipid Metabolism in Mouse Model of Amyotrophic Lateral Sclerosis. Brain Behav. Immun. 2025, 124, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Isik, F.I.; Thomson, S.; Cueto, J.F.; Spathos, J.; Breit, S.N.; Tsai, V.W.W.; Brown, D.A.; Finney, C.A. A Systematic Review of the Neuroprotective Role and Biomarker Potential of GDF15 in Neurodegeneration. Front. Immunol. 2024, 15, 1514518. [Google Scholar] [CrossRef]
- Soustelle, L.; Aimond, F.; López Andrés, C.; Brugioti, V.; Raoul, C.; Layalle, S. ALS-Associated KIF5A Mutation Causes Locomotor Deficits Associated with Cytoplasmic Inclusions, Alterations of Neuromuscular Junctions, and Motor Neuron Loss. J. Neurosci. 2023, 43, 8058–8072. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.M.; Kim, S.; Kwon, Y.; Kim, H.J. The Role of TDP-43 Propagation in Neurodegenerative Diseases: Integrating Insights from Clinical and Experimental Studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Apolloni, S.; D’ambrosi, N. Fibrosis as a Common Trait in Amyotrophic Lateral Sclerosis Tissues. Neural Regen. Res. 2022, 17, 97–98. [Google Scholar] [CrossRef]
- Schmidt, E.R.E.; Pasterkamp, R.J.; van den Berg, L.H. Axon Guidance Proteins: Novel Therapeutic Targets for ALS? Prog. Neurobiol. 2009, 88, 286–301. [Google Scholar] [CrossRef]
- Ye, L.; Dittlau, K.S.; Sicart, A.; Janky, R.; Van Damme, P.; Van Den Bosch, L. Sporadic ALS HiPSC-Derived Motor Neurons Show Axonal Defects Linked to Altered Axon Guidance Pathways. Neurobiol. Dis. 2025, 206, 106815. [Google Scholar] [CrossRef]
- Baxi, E.G.; Thompson, T.; Li, J.; Kaye, J.A.; Lim, R.G.; Wu, J.; Ramamoorthy, D.; Lima, L.; Vaibhav, V.; Matlock, A.; et al. Answer ALS, a Large-Scale Resource for Sporadic and Familial ALS Combining Clinical and Multi-Omics Data from Induced Pluripotent Cell Lines. Nat. Neurosci. 2022, 25, 226–237. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Mutation | Age at Disease Onset | Age at Diagnosis | Gender | Family History | Phenotype | Cognition | Age at Death | Disease Duration (Months from Onset to Death) |
|---|---|---|---|---|---|---|---|---|---|
| ALS_001 | KIF5A (exon 27 3’ splice junction variant, c.3020+1G>A) | 61 | 62 | Male | Mother, maternal grandfather, great-grandfather, and three first degree cousins | Prevalent UMN (spinal) | ALS-ci | 66 | 60 * |
| ALS_002 | TARDBP (c.1144G>A, p.A382T) | 63 | 64 | Male | Brother | Prevalent UMN (bulbar) | ALS-ci | 67 | 50 |
| ALS_003 | C9orf72 (intron 1 GGGCC repeat pathogenetic expansion) | 57 | 58 | Male | Father | Classic (spinal) | ALS-ci | 60 | 30 |
| ALS_004 | TARDBP (c.1144G>A, p.A382T) | 61 | 65 | Male | Father | Prevalent UMN (spinal) | Normal | - | 84 (alive) |
| H_001 | - | 43 | - | Male | - | - | - | - | - |
| H_002 | - | 58 | - | Female | - | - | - | - | - |
| H_003 | - | 60 | - | Male | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgromo, C.; Tosi, M.; Olgasi, C.; De Marchi, F.; Favero, F.; Venturin, G.; Piola, B.; Cucci, A.; Corrado, L.; Mazzini, L.; et al. Identification of Transcriptomic Differences in Induced Pluripotent Stem Cells and Neural Progenitors from Amyotrophic Lateral Sclerosis Patients Carrying Different Mutations: A Pilot Study. Cells 2025, 14, 958. https://doi.org/10.3390/cells14130958
Sgromo C, Tosi M, Olgasi C, De Marchi F, Favero F, Venturin G, Piola B, Cucci A, Corrado L, Mazzini L, et al. Identification of Transcriptomic Differences in Induced Pluripotent Stem Cells and Neural Progenitors from Amyotrophic Lateral Sclerosis Patients Carrying Different Mutations: A Pilot Study. Cells. 2025; 14(13):958. https://doi.org/10.3390/cells14130958
Chicago/Turabian StyleSgromo, Chiara, Martina Tosi, Cristina Olgasi, Fabiola De Marchi, Francesco Favero, Giorgia Venturin, Beatrice Piola, Alessia Cucci, Lucia Corrado, Letizia Mazzini, and et al. 2025. "Identification of Transcriptomic Differences in Induced Pluripotent Stem Cells and Neural Progenitors from Amyotrophic Lateral Sclerosis Patients Carrying Different Mutations: A Pilot Study" Cells 14, no. 13: 958. https://doi.org/10.3390/cells14130958
APA StyleSgromo, C., Tosi, M., Olgasi, C., De Marchi, F., Favero, F., Venturin, G., Piola, B., Cucci, A., Corrado, L., Mazzini, L., D’Alfonso, S., & Follenzi, A. (2025). Identification of Transcriptomic Differences in Induced Pluripotent Stem Cells and Neural Progenitors from Amyotrophic Lateral Sclerosis Patients Carrying Different Mutations: A Pilot Study. Cells, 14(13), 958. https://doi.org/10.3390/cells14130958