
Understanding Microglia in Mesocorticolimbic Circuits: Implications for the Study of Chronic Stress and Substance Use Disorders

 , ,
, ,  , and
, and {kind=link}
{kind=link}
Abstract
1. Introduction
2. What Is the Role of Microglia in the Adult Brain?
3. Comparing Histological Methods for Interrogating Microglia Function After Pharmacological and Environmental Exposure
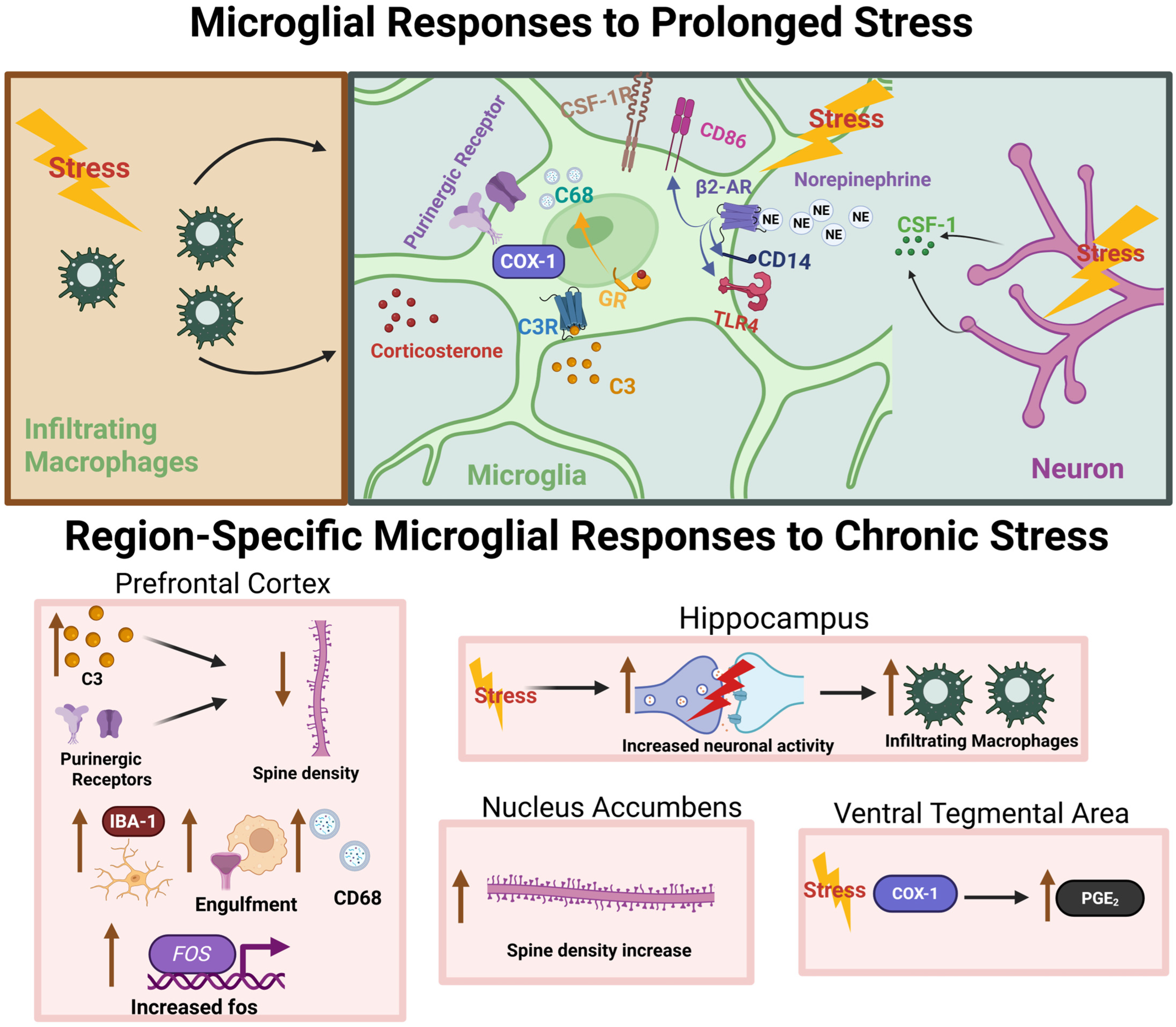
4. Microglia Mediate the Effects of Chronic Stress on the Brain
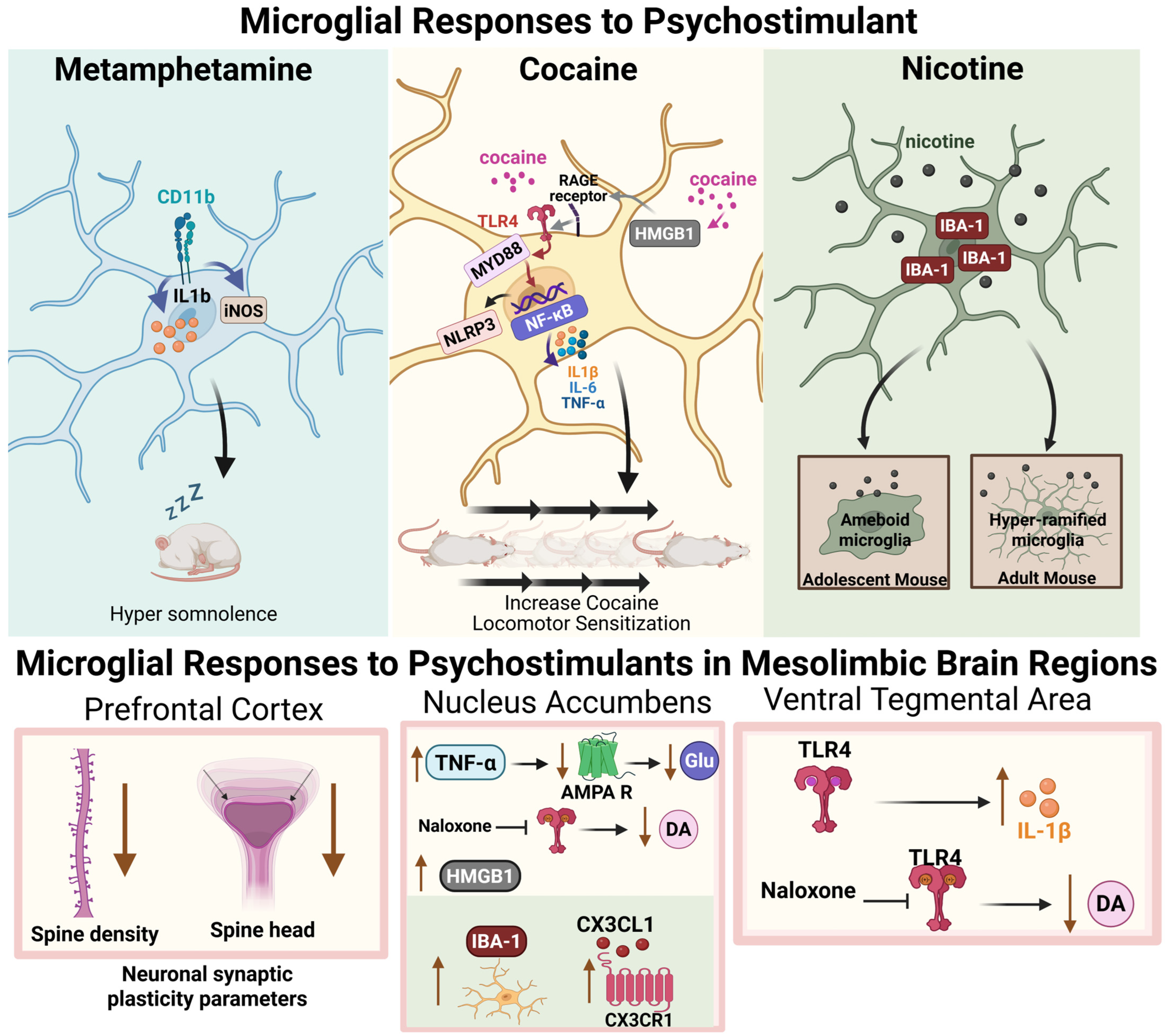
5. Microglia Respond to Psychostimulant Drug Exposure
6. Chronic Stress Effects on Drug Seeking: Potential Microglia Involvement
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ross, S.; Peselow, E. Co-occurring psychotic and addictive disorders: Neurobiology and diagnosis. Clin. Neuropharmacol. 2012, 35, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R. Chronic Stress, Drug Use, and Vulnerability to Addiction. In Annals of the New York Academy of Sciences; New York Academy of Sciences: New York, NY, USA, 2008; Volume 1141, pp. 105–130. [Google Scholar]
- Turner, R.J.; Lloyd, D.A. Cumulative Adversity and Drug Dependence in Young Adults: Racial/Ethnic Contrasts; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; Volume 98, pp. 305–315. [Google Scholar]
- Mantsch, J.R.; Baker, D.A.; Funk, D.; Lê, A.D.; Shaham, Y. Stress-Induced Reinstatement of Drug Seeking: 20 Years of Progress. Neuropsychopharmacology 2016, 41, 335–356. [Google Scholar] [CrossRef]
- Shaham, Y.; de Wit, H. Lost in Translation: CRF1 Receptor Antagonists and Addiction Treatment. Neuropsychopharmacology 2016, 41, 2795–2797. [Google Scholar] [CrossRef] [PubMed]
- Kwako, L.E.; Spagnolo, P.A.; Schwandt, M.L.; Thorsell, A.; George, D.T.; Momenan, R.; Rio, D.E.; Huestis, M.; Anizan, S.; Concheiro, M.; et al. The corticotropin releasing hormone-1 (CRH1) receptor antagonist pexacerfont in alcohol dependence: A randomized controlled experimental medicine study. Neuropsychopharmacology 2015, 40, 1053–1063. [Google Scholar] [CrossRef]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial Interactions with Synapses Are Modulated by Visual Experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Franklin, T.C.; Xu, C.; Duman, R.S. Depression and sterile inflammation: Essential role of danger associated molecular patterns. Brain Behav. Immun. 2018, 72, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Hendrickx, D.A.E.; van Eden, C.G.; Schuurman, K.G.; Hamann, J.; Huitinga, I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 2017, 309, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Player, M.R. Colony-stimulating factor-1 receptor inhibitors for the treatment of cancer and inflammatory disease. Curr. Top. Med. Chem. 2009, 9, 599–610. [Google Scholar] [CrossRef]
- Chitu, V.; Biundo, F.; Shlager, G.G.L.; Park, E.S.; Wang, P.; Gulinello, M.E.; Gokhan, S.; Ketchum, H.C.; Saha, K.; DeTure, M.A.; et al. Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling. Cell Rep. 2020, 30, 3004–3019.e5. [Google Scholar] [CrossRef]
- Chitu, V.; Gokhan, S.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef]
- Arreola, M.A.; Soni, N.; Crapser, J.D.; Hohsfield, L.A.; Elmore, M.R.P.; Matheos, D.P.; Wood, M.A.; Swarup, V.; Mortazavi, A.; Green, K.N. Microglial dyshomeostasis drives perineuronal net and synaptic loss in a CSF1R(+/-) mouse model of ALSP, which can be rescued via CSF1R inhibitors. Sci. Adv. 2021, 7, eabg1601. [Google Scholar] [CrossRef] [PubMed]
- Wicherska-Pawlowska, K.; Wrobel, T.; Rybka, J. Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int. J. Mol. Sci. 2021, 22, 13397. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Kemp, G.M.; Altimimi, H.F.; Nho, Y.; Heir, R.; Klyczek, A.; Stellwagen, D. Sustained TNF signaling is required for the synaptic and anxiety-like behavioral response to acute stress. Mol. Psychiatry 2022, 27, 4474–4484. [Google Scholar] [CrossRef]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A.; et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020, 182, 388–403.e15. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Asrat, H.S.; Flurer, J.K.; Schwierling, H.C.; Bollinger, J.L.; Vollmer, L.L.; Wohleb, E.S. Depletion of microglial BDNF increases susceptibility to the behavioral and synaptic effects of chronic unpredictable stress. Brain Behav. Immun. 2023, 109, 127–138. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.-B.; Julius, D.; Haynes, S.E.; Hollopeter, G.; Yang, G.; et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Ma, C.; Li, B.; Silverman, D.; Ding, X.; Li, A.; Xiao, C.; Huang, G.; Worden, K.; Muroy, S.; Chen, W.; et al. Microglia regulate sleep through calcium-dependent modulation of norepinephrine transmission. Nat. Neurosci. 2024, 27, 249–258. [Google Scholar] [CrossRef]
- Lalo, U.; Pankratov, Y. ATP-mediated signalling in the central synapses. Neuropharmacology 2023, 229, 109477. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.N.; Pankratov, Y.; Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.N.; Pankratov, Y. ATP from synaptic terminals and astrocytes regulates NMDA receptors and synaptic plasticity through PSD-95 multi-protein complex. Sci. Rep. 2016, 6, srep33609. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Jobling, A.I.; Greferath, U.; Chuang, T.; Ramesh, A.; Fletcher, E.L.; Vessey, K.A. Vesicular expression and release of ATP from dopaminergic neurons of the mouse retina and midbrain. Front. Cell. Neurosci. 2015, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Witting, A.; Walter, L.; Wacker, J.; Möller, T.; Stella, N. P2X7 receptors control 2-arachidonoylglycerol production by microglial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 3214–3219. [Google Scholar] [CrossRef]
- Liu, H.; Leak, R.K.; Hu, X. Neurotransmitter receptors on microglia. Stroke Vasc. Neurol. 2016, 1, 52–58. [Google Scholar] [CrossRef]
- Acosta-Martinez, M. Shaping Microglial Phenotypes Through Estrogen Receptors: Relevance to Sex-Specific Neuroinflammatory Responses to Brain Injury and Disease. J. Pharmacol. Exp. Ther. 2020, 375, 223–236. [Google Scholar] [CrossRef]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef]
- Guneykaya, D.; Ivanov, A.; Hernandez, D.P.; Haage, V.; Wojtas, B.; Meyer, N.; Maricos, M.; Jordan, P.; Buonfiglioli, A.; Gielniewski, B.; et al. Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep. 2018, 24, 2773–2783.e6. [Google Scholar] [CrossRef]
- Ball, J.B.; Mcnulty, C.J.; Green-Fulgham, S.M.; Dragavon, J.M.; Correia Rocha, I.R.; Finch, M.R.; Prévost, E.D.; Siddique, I.I.; Woodall, B.J.; Watkins, L.R.; et al. Combining RNAscope and immunohistochemistry to visualize inflammatory gene products in neurons and microglia. Front. Mol. Neurosci. 2023, 16, 1225847. [Google Scholar] [CrossRef]
- Schmid, C.D.; Melchior, B.; Masek, K.; Puntambekar, S.S.; Danielson, P.E.; Lo, D.D.; Gregor Sutcliffe, J.; Carson, M.J. Differential gene expression in LPS/IFNγ activated microglia and macrophages: In vitro versus in vivo. J. Neurochem. 2009, 109, 117–125. [Google Scholar] [CrossRef]
- Liu, Q.-R.; Canseco-Alba, A.; Liang, Y.; Ishiguro, H.; Onaivi, E.S. Low Basal CB2R in Dopamine Neurons and Microglia Influences Cannabinoid Tetrad Effects. Int. J. Mol. Sci. 2020, 21, 9763. [Google Scholar] [CrossRef]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef]
- Dissing-Olesen, L.; Walker, A.J.; Feng, Q.; Barr, H.J.; Walker, A.C.; Xie, L.; Wilton, D.K.; Das, I.; Benowitz, L.I.; Stevens, B. FEAST: A flow cytometry-based toolkit for interrogating microglial engulfment of synaptic and myelin proteins. Nat. Commun. 2023, 14, 6015. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; El-Behi, M.; Fontaine, B.; Delarasse, C. Analysis of Microglia and Monocyte-derived Macrophages from the Central Nervous System by Flow Cytometry. J. Vis. Exp. 2017, e55781. [Google Scholar] [CrossRef]
- Walker, D.; Lue, L.-F.; Beach, T.; Tooyama, I. Microglial Phenotyping in Neurodegenerative Disease Brains: Identification of Reactive Microglia with an Antibody to Variant of CD105/Endoglin. Cells 2019, 8, 766. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.; Glass, M. The Cannabinoid CB2 Receptor as a Target for Inflammation-Dependent Neurodegeneration. Curr. Neuropharmacol. 2007, 5, 73–80. [Google Scholar] [CrossRef]
- Bollinger, J.L.; Dadosky, D.T.; Flurer, J.K.; Rainer, I.L.; Woodburn, S.C.; Wohleb, E.S. Microglial P2Y12 mediates chronic stress-induced synapse loss in the prefrontal cortex and associated behavioral consequences. Neuropsychopharmacology 2023, 48, 1347–1357. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of Fractalkine Receptor CX3CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef]
- Kaiser, T.; Feng, G. Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 2019, 6, ENEURO.0448-0418. [Google Scholar] [CrossRef]
- Buttgereit, A.; Lelios, I.; Yu, X.; Vrohlings, M.; Krakoski, N.R.; Gautier, E.L.; Nishinakamura, R.; Becher, B.; Greter, M. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 2016, 17, 1397–1406. [Google Scholar] [CrossRef]
- Sharma, K.; Bisht, K.; Eyo, U.B. A Comparative Biology of Microglia Across Species. Front. Cell Dev. Biol. 2021, 9, 652748. [Google Scholar] [CrossRef] [PubMed]
- Chaure, F.J.; CGKLaboratory; Taborda-Bejarano, J.P.; York, E. CGK-Laboratory/CellSelect-3DMorph: Release v2.0.0. Zenodo. 2024. Available online: https://zenodo.org/records/14159877 (accessed on 14 November 2024).
- Radley, J.J.; Herman, J.P. Preclinical Models of Chronic Stress: Adaptation or Pathology? Biol. Psychiatry 2023, 94, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; Miller, M.M.; Goldwater, D.S.; Radley, J.J.; Rocher, A.B.; Hof, P.R.; Morrison, J.H.; McEwen, B.S. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006, 26, 7870–7874. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Powell, N.D.; Godbout, J.P.; Sheridan, J.F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 2013, 33, 13820–13833. [Google Scholar] [CrossRef] [PubMed]
- Brevet, M.; Kojima, H.; Asakawa, A.; Atsuchi, K.; Ushikai, M.; Ataka, K.; Inui, A.; Kimura, H.; Sevestre, H.; Fujimiya, M. Chronic foot-shock stress potentiates the influx of bone marrow-derived microglia into hippocampus. J. Neurosci. Res. 2010, 88, 1890–1897. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Terwilliger, R.; Duman, C.H.; Duman, R.S. Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biol. Psychiatry 2018, 83, 38–49. [Google Scholar] [CrossRef]
- Tillmon, H.; Soteros, B.M.; Shen, L.; Cong, Q.; Wollet, M.; General, J.; Chin, H.; Lee, J.B.; Carreno, F.R.; Morilak, D.A.; et al. Complement and microglia activation mediate stress-induced synapse loss in layer 2/3 of the medial prefrontal cortex in male mice. Nat. Commun. 2024, 15, 9803. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.-S.; Li, H.-H.; Wang, H.-J.; Zou, R.-S.; Lu, X.-J.; Wang, J.; Nie, B.-B.; Wu, J.-F.; Li, S.; et al. Microglia-dependent excessive synaptic pruning leads to cortical underconnectivity and behavioral abnormality following chronic social defeat stress in mice. Brain Behav. Immun. 2023, 109, 23–36. [Google Scholar] [CrossRef]
- Tanaka, K.; Furuyashiki, T.; Kitaoka, S.; Senzai, Y.; Imoto, Y.; Segi-Nishida, E.; Deguchi, Y.; Breyer, R.M.; Breyer, M.D.; Narumiya, S. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J. Neurosci. 2012, 32, 4319–4329. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Campisi, J.; Sharkey, C.M.; Kennedy, S.L.; Nickerson, M.; Greenwood, B.N.; Fleshner, M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 2005, 135, 1295–1307. [Google Scholar] [CrossRef]
- Maier, S.F. Bi-directional immune–brain communication: Implications for understanding stress, pain, and cognition. Brain Behav. Immun. 2003, 17, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Baratta, M.V.; Sprunger, D.B.; Watkins, L.R.; Maier, S.F. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav. Immun. 2007, 21, 47–59. [Google Scholar] [CrossRef]
- Schramm, E.; Waisman, A. Microglia as Central Protagonists in the Chronic Stress Response. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e200023. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Franklin, T.; Iwata, M.; Duman, R.S. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016, 17, 497–511. [Google Scholar] [CrossRef]
- Bollinger, J.L.; Collins, K.E.; Patel, R.; Wellman, C.L. Behavioral stress alters corticolimbic microglia in a sex- and brain region-specific manner. PLoS ONE 2017, 12, e0187631. [Google Scholar] [CrossRef]
- Wang, H.; He, Y.; Sun, Z.; Ren, S.; Liu, M.; Wang, G.; Yang, J. Microglia in depression: An overview of microglia in the pathogenesis and treatment of depression. J. Neuroinflamm. 2022, 19, 132. [Google Scholar] [CrossRef]
- Afridi, R.; Suk, K. Microglial Responses to Stress-Induced Depression: Causes and Consequences. Cells 2023, 12, 1521. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. Synaptic and behavioral effects of chronic stress are linked to dynamic and sex-specific changes in microglia function and astrocyte dystrophy. Neurobiol. Stress 2021, 14, 100312. [Google Scholar] [CrossRef]
- Yuan, T.; Orock, A.; Greenwood-Van Meerveld, B. Amygdala microglia modify neuronal plasticity via complement C1q/C3-CR3 signaling and contribute to visceral pain in a rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G1081–G1092. [Google Scholar] [CrossRef] [PubMed]
- Biltz, R.G.; Swanson, S.P.; Draime, N.; Davis, A.C.; Yin, W.; Goodman, E.J.; Gallagher, N.R.; Bhattacharya, A.; Sheridan, J.F.; Godbout, J.P. Antagonism of the brain P2X7 ion channel attenuates repeated social defeat induced microglia reactivity, monocyte recruitment and anxiety-like behavior in male mice. Brain Behav. Immun. 2024, 115, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Hinwood, M.; Morandini, J.; Day, T.A.; Walker, F.R. Evidence that Microglia Mediate the Neurobiological Effects of Chronic Psychological Stress on the Medial Prefrontal Cortex. Cereb. Cortex 2012, 22, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Milior, G.; Lecours, C.; Samson, L.; Bisht, K.; Poggini, S.; Pagani, F.; Deflorio, C.; Lauro, C.; Alboni, S.; Limatola, C.; et al. Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav. Immun. 2016, 55, 114–125. [Google Scholar] [CrossRef]
- Horchar, M.J.; Wohleb, E.S. Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain Behav. Immun. 2019, 81, 329–340. [Google Scholar] [CrossRef]
- Chen, H.; Fu, S.; Li, X.; Shi, M.; Qian, J.; Zhao, S.; Yuan, P.; Ding, L.; Xia, X.; Zheng, J.C. Microglial glutaminase 1 mediates chronic restraint stress-induced depression-like behaviors and synaptic damages. Signal Transduct. Target. Ther. 2023, 8, 452. [Google Scholar] [CrossRef]
- Han, Q.-Q.; Shen, S.-Y.; Chen, X.-R.; Pilot, A.; Liang, L.-F.; Zhang, J.-R.; Li, W.-H.; Fu, Y.; Le, J.-M.; Chen, P.-Q.; et al. Minocycline alleviates abnormal microglial phagocytosis of synapses in a mouse model of depression. Neuropharmacology 2022, 220, 109249. [Google Scholar] [CrossRef]
- Bassett, B.; Subramaniyam, S.; Fan, Y.; Varney, S.; Pan, H.; Carneiro, A.M.D.; Chung, C.Y. Minocycline alleviates depression-like symptoms by rescuing decrease in neurogenesis in dorsal hippocampus via blocking microglia activation/phagocytosis. Brain Behav. Immun. 2021, 91, 519–530. [Google Scholar] [CrossRef]
- Avalos, M.P.; Guzman, A.S.; Rigoni, D.; Gorostiza, E.A.; Sanchez, M.A.; Mongi-Bragato, B.; Garcia-Keller, C.; Perassi, E.M.; Virgolini, M.B.; Peralta Ramos, J.M.; et al. Minocycline prevents chronic restraint stress-induced vulnerability to developing cocaine self-administration and associated glutamatergic mechanisms: A potential role of microglia. Brain Behav. Immun. 2022, 101, 359–376. [Google Scholar] [CrossRef]
- Wang, B.; Huang, X.; Pan, X.; Zhang, T.; Hou, C.; Su, W.-J.; Liu, L.-L.; Li, J.-M.; Wang, Y.-X. Minocycline prevents the depressive-like behavior through inhibiting the release of HMGB1 from microglia and neurons. Brain Behav. Immun. 2020, 88, 132–143. [Google Scholar] [CrossRef]
- Qiao, H.; Li, M.-X.; Xu, C.; Chen, H.-B.; An, S.-C.; Ma, X.-M. Dendritic Spines in Depression: What We Learned from Animal Models. Neural Plast. 2016, 8056370. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Berglund, R.; Cheng, Y.; Piket, E.; Adzemovic, M.Z.; Zeitelhofer, M.; Olsson, T.; Guerreiro-Cacais, A.O.; Jagodic, M. The aging mouse CNS is protected by an autophagy-dependent microglia population promoted by IL-34. Nat. Commun. 2024, 15, 383. [Google Scholar] [CrossRef]
- Eyo, U.; Molofsky, A.V. Defining microglial-synapse interactions. Science 2023, 381, 1155–1156. [Google Scholar] [CrossRef]
- Klawonn, A.M.; Malenka, R.C. Nucleus Accumbens Modulation in Reward and Aversion. Cold Spring Harb. Symp. Quant. Biol. 2018, 83, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Bessa, J.M.; Morais, M.; Marques, F.; Pinto, L.; Palha, J.A.; Almeida, O.F.; Sousa, N. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl. Psychiatry 2013, 3, e266. [Google Scholar] [CrossRef]
- Gaspar, R.; Soares-Cunha, C.; Domingues, A.V.; Coimbra, B.; Baptista, F.I.; Pinto, L.; Ambrosio, A.F.; Rodrigues, A.J.; Gomes, C.A. The Duration of Stress Determines Sex Specificities in the Vulnerability to Depression and in the Morphologic Remodeling of Neurons and Microglia. Front. Behav. Neurosci. 2022, 16, 834821. [Google Scholar] [CrossRef]
- Douma, E.H.; de Kloet, E.R. Stress-induced plasticity and functioning of ventral tegmental dopamine neurons. Neurosci. Biobehav. Rev. 2020, 108, 48–77. [Google Scholar] [CrossRef]
- Hill, A.S.; Sahay, A.; Hen, R. Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology 2015, 40, 2368–2378. [Google Scholar] [CrossRef]
- Du Preez, A.; Onorato, D.; Eiben, I.; Musaelyan, K.; Egeland, M.; Zunszain, P.A.; Fernandes, C.; Thuret, S.; Pariante, C.M. Chronic stress followed by social isolation promotes depressive-like behaviour, alters microglial and astrocyte biology and reduces hippocampal neurogenesis in male mice. Brain Behav. Immun. 2021, 91, 24–47. [Google Scholar] [CrossRef]
- Fang, S.; Wu, Z.; Guo, Y.; Zhu, W.; Wan, C.; Yuan, N.; Chen, J.; Hao, W.; Mo, X.; Guo, X.; et al. Roles of microglia in adult hippocampal neurogenesis in depression and their therapeutics. Front. Immunol. 2023, 14, 1193053. [Google Scholar] [CrossRef]
- Poggini, S.; Lopez, M.B.; Albanese, N.C.; Golia, M.T.; Ibanez, F.G.; Limatola, C.; Furhmann, M.; Lalowski, M.; Tremblay, M.E.; Maggi, L.; et al. Minocycline treatment improves cognitive and functional plasticity in a preclinical mouse model of major depressive disorder. Behav. Brain Res. 2023, 441, 114295. [Google Scholar] [CrossRef]
- da Silva, M.C.M.; Iglesias, L.P.; Candelario-Jalil, E.; Khoshbouei, H.; Moreira, F.A.; de Oliveira, A.C.P. Role of Microglia in Psychostimulant Addiction. Curr. Neuropharmacol. 2023, 21, 235–259. [Google Scholar] [CrossRef]
- Ahearn, O.C.; Watson, M.N.; Rawls, S.M. Chemokines, cytokines and substance use disorders. Drug Alcohol. Depend. 2021, 220, 108511. [Google Scholar] [CrossRef]
- Kohno, M.; Link, J.; Dennis, L.E.; McCready, H.; Huckans, M.; Hoffman, W.F.; Loftis, J.M. Neuroinflammation in addiction: A review of neuroimaging studies and potential immunotherapies. Pharmacol. Biochem. Behav. 2019, 179, 34–42. [Google Scholar] [CrossRef]
- Lacagnina, M.J.; Rivera, P.D.; Bilbo, S.D. Glial and Neuroimmune Mechanisms as Critical Modulators of Drug Use and Abuse. Neuropsychopharmacology 2017, 42, 156–177. [Google Scholar] [CrossRef]
- Kays, J.S.; Yamamoto, B.K. Evaluation of Microglia/Macrophage Cells from Rat Striatum and Prefrontal Cortex Reveals Differential Expression of Inflammatory-Related mRNA after Methamphetamine. Brain Sci. 2019, 9, 340. [Google Scholar] [CrossRef]
- Wisor, J.P.; Schmidt, M.A.; Clegern, W.C. Cerebral microglia mediate sleep/wake and neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 2011, 25, 767–776. [Google Scholar] [CrossRef]
- Ye, J.; Gao, S.Q.; Liu, Z.C.; Chen, X.; He, J.G.; Hu, Z.L. The HMGB1-RAGE axis in nucleus accumbens facilitates cocaine-induced conditioned place preference via modulating microglial activation. Brain Behav. 2024, 14, e3457. [Google Scholar] [CrossRef]
- Chen, H.; Uz, T.; Manev, H. Minocycline affects cocaine sensitization in mice. Neurosci. Lett. 2009, 452, 258–261. [Google Scholar] [CrossRef]
- Alajaji, M.; Lazenka, M.F.; Kota, D.; Wise, L.E.; Younis, R.M.; Carroll, F.I.; Levine, A.; Selley, D.E.; Sim-Selley, L.J.; Damaj, M.I. Early adolescent nicotine exposure affects later-life cocaine reward in mice. Neuropharmacology 2016, 105, 308–317. [Google Scholar] [CrossRef]
- Siemsen, B.M.; Giannotti, G.; McFaddin, J.A.; Scofield, M.D.; McGinty, J.F. Biphasic effect of abstinence duration following cocaine self-administration on spine morphology and plasticity-related proteins in prelimbic cortical neurons projecting to the nucleus accumbens core. Brain Struct. Funct. 2019, 224, 741–758. [Google Scholar] [CrossRef]
- Lewitus, G.M.; Konefal, S.C.; Greenhalgh, A.D.; Pribiag, H.; Augereau, K.; Stellwagen, D. Microglial TNF-α Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron 2016, 90, 483–491. [Google Scholar] [CrossRef]
- Lewitus, G.M.; Pribiag, H.; Duseja, R.; St-Hilaire, M.; Stellwagen, D. An Adaptive Role of TNFα in the Regulation of Striatal Synapses. J. Neurosci. 2014, 34, 6146–6155. [Google Scholar] [CrossRef]
- Linker, K.E.; Gad, M.; Tawadrous, P.; Cano, M.; Green, K.N.; Wood, M.A.; Leslie, F.M. Microglial activation increases cocaine self-administration following adolescent nicotine exposure. Nat. Commun. 2020, 11, 306. [Google Scholar] [CrossRef]
- Goldstein, R.Z.; Volkow, N.D. Dysfunction of the prefrontal cortex in addiction: Neuroimaging findings and clinical implications. Nat. Rev. Neurosci. 2011, 12, 652–669. [Google Scholar] [CrossRef]
- Angarita, G.A.; Worhunsky, P.D.; Naganawa, M.; Toyonaga, T.; Nabulsi, N.B.; Li, C.R.; Esterlis, I.; Skosnik, P.D.; Radhakrishnan, R.; Pittman, B.; et al. Lower prefrontal cortical synaptic vesicle binding in cocaine use disorder: An exploratory (11) C-UCB-J positron emission tomography study in humans. Addict. Biol. 2022, 27, e13123. [Google Scholar] [CrossRef]
- Golden, S.A.; Russo, S.J. Mechanisms of Psychostimulant-Induced Structural Plasticity. Cold Spring Harb. Perspect. Med. 2012, 2, a011957. [Google Scholar] [CrossRef]
- DePoy, L.M.; Gourley, S.L. Synaptic Cytoskeletal Plasticity in the Prefrontal Cortex Following Psychostimulant Exposure. Traffic 2015, 16, 919–940. [Google Scholar] [CrossRef]
- Radley, J.J.; Anderson, R.M.; Cosme, C.V.; Glanz, R.M.; Miller, M.C.; Romig-Martin, S.A.; LaLumiere, R.T. The Contingency of Cocaine Administration Accounts for Structural and Functional Medial Prefrontal Deficits and Increased Adrenocortical Activation. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 11897–11910. [Google Scholar] [CrossRef]
- Selemon, L.D.; Begović, A.; Goldman-Rakic, P.S.; Castner, S.A. Amphetamine sensitization alters dendritic morphology in prefrontal cortical pyramidal neurons in the non-human primate. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 919–931. [Google Scholar] [CrossRef]
- Anderson, E.M.; Self, D.W. It’s only a matter of time: Longevity of cocaine-induced changes in dendritic spine density in the nucleus accumbens. Curr. Opin. Behav. Sci. 2017, 13, 117–123. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Zhang, Y.; Brown, K.; Coats, B.D.; Shridhar, M.; Sholar, P.W.; Patel, S.J.; Crysdale, N.Y.; Harrison, J.A.; Maier, S.F.; et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: Involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 2008, 28, 20–29. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Zhang, Y.; Shridhar, M.; Evans, J.H.; Buchanan, M.M.; Zhao, T.X.; Slivka, P.F.; Coats, B.D.; Rezvani, N.; Wieseler, J.; et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav. Immun. 2010, 24, 83–95. [Google Scholar] [CrossRef]
- Wang, X.; Loram, L.C.; Ramos, K.; de Jesus, A.J.; Thomas, J.; Cheng, K.; Reddy, A.; Somogyi, A.A.; Hutchinson, M.R.; Watkins, L.R.; et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. USA 2012, 109, 6325–6330. [Google Scholar] [CrossRef]
- Brown, K.T.; Levis, S.C.; O’Neill, C.E.; Northcutt, A.L.; Fabisiak, T.J.; Watkins, L.R.; Bachtell, R.K. Innate immune signaling in the ventral tegmental area contributes to drug-primed reinstatement of cocaine seeking. Brain Behav. Immun. 2018, 67, 130–138. [Google Scholar] [CrossRef]
- Northcutt, A.L.; Hutchinson, M.R.; Wang, X.; Baratta, M.V.; Hiranita, T.; Cochran, T.A.; Pomrenze, M.B.; Galer, E.L.; Kopajtic, T.A.; Li, C.M.; et al. DAT isn’t all that: Cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol. Psychiatry 2015, 20, 1525–1537. [Google Scholar] [CrossRef]
- Chivero, E.T.; Thangaraj, A.; Tripathi, A.; Periyasamy, P.; Guo, M.L.; Buch, S. NLRP3 Inflammasome Blockade Reduces Cocaine-Induced Microglial Activation and Neuroinflammation. Mol. Neurobiol. 2021, 58, 2215–2230. [Google Scholar] [CrossRef]
- Cotto, B.; Li, H.; Tuma, R.F.; Ward, S.J.; Langford, D. Cocaine-mediated activation of microglia and microglial MeCP2 and BDNF production. Neurobiol. Dis. 2018, 117, 28–41. [Google Scholar] [CrossRef]
- Burkovetskaya, M.E.; Small, R.; Guo, L.; Buch, S.; Guo, M.L. Cocaine self-administration differentially activates microglia in the mouse brain. Neurosci. Lett. 2020, 728, 134951. [Google Scholar] [CrossRef]
- Brown, K.T.; Levis, S.C.; O’Neill, C.E.; Levy, C.; Rice, K.C.; Watkins, L.R.; Bachtell, R.K. Toll-like receptor 4 antagonists reduce cocaine-primed reinstatement of drug seeking. Psychopharmacology 2023, 240, 1587–1600. [Google Scholar] [CrossRef]
- Sinha, R. How does stress increase risk of drug abuse and relapse? Psychopharmacology 2001, 158, 343–359. [Google Scholar] [CrossRef]
- Antelman, S.M.; Eichler, A.J.; Black, C.A.; Kocan, D. Interchangeability of stress and amphetamine in sensitization. Science 1980, 207, 329–331. [Google Scholar] [CrossRef]
- Piazza, P.V.; Deminiere, J.M.; Le Moal, M.; Simon, H. Factors that predict individual vulnerability to amphetamine self-administration. Science 1989, 245, 1511–1513. [Google Scholar] [CrossRef]
- Piazza, P.V.; Deminiere, J.M.; le Moal, M.; Simon, H. Stress- and pharmacologically-induced behavioral sensitization increases vulnerability to acquisition of amphetamine self-administration. Brain Res. 1990, 514, 22–26. [Google Scholar] [CrossRef]
- Piazza, P.V.; Maccari, S.; Deminiere, J.M.; Le Moal, M.; Mormede, P.; Simon, H. Corticosterone levels determine individual vulnerability to amphetamine self-administration. Proc. Natl. Acad. Sci. USA 1991, 88, 2088–2092. [Google Scholar] [CrossRef]
- Mantsch, J.R.; Ho, A.; Schlussman, S.D.; Kreek, M.J. Predictable individual differences in the initiation of cocaine self-administration by rats under extended-access conditions are dose-dependent. Psychopharmacology 2001, 157, 31–39. [Google Scholar] [CrossRef]
- Deroche, V.; Piazza, P.V.; Casolini, P.; Maccari, S.; Le Moal, M.; Simon, H. Stress-induced sensitization to amphetamine and morphine psychomotor effects depend on stress-induced corticosterone secretion. Brain Res. 1992, 598, 343–348. [Google Scholar] [CrossRef]
- Deroche, V.; Piazza, P.V.; Maccari, S.; Le Moal, M.; Simon, H. Repeated corticosterone administration sensitizes the locomotor response to amphetamine. Brain Res. 1992, 584, 309–313. [Google Scholar] [CrossRef]
- Goeders, N.E.; Guerin, G.F. Non-contingent electric footshock facilitates the acquisition of intravenous cocaine self-administration in rats. Psychopharmacology 1994, 114, 63–70. [Google Scholar] [CrossRef] [PubMed]
- De Vry, J.; Donselaar, I.; Van Ree, J.M. Food deprivation and acquisition of intravenous cocaine self-administration in rats: Effect of naltrexone and haloperidol. J. Pharmacol. Exp. Ther. 1989, 251, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.; Maccari, S.; Le Moal, M.; Simon, H.; Piazza, P.V. Social stress increases the acquisition of cocaine self-administration in male and female rats. Brain Res. 1995, 698, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, N.F.; Van Ree, J.M. Emotional but not physical stress enhances intravenous cocaine self-administration in drug-naive rats. Brain Res. 1993, 608, 216–222. [Google Scholar] [CrossRef]
- Rigoni, D.; Avalos, M.P.; Boezio, M.J.; Guzmán, A.S.; Calfa, G.D.; Perassi, E.M.; Pierotti, S.M.; Bisbal, M.; Garcia-Keller, C.; Cancela, L.M.; et al. Stress-induced vulnerability to develop cocaine addiction depends on cofilin modulation. Neurobiol. Stress 2021, 15, 100349. [Google Scholar] [CrossRef]
- Schenk, S.; Lacelle, G.; Gorman, K.; Amit, Z. Cocaine self-administration in rats influenced by environmental conditions: Implications for the etiology of drug abuse. Neurosci. Lett. 1987, 81, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Mantsch, J.R.; Saphier, D.; Goeders, N.E. Corticosterone facilitates the acquisition of cocaine self-administration in rats: Opposite effects of the type II glucocorticoid receptor agonist dexamethasone. J. Pharmacol. Exp. Ther. 1998, 287, 72–80. [Google Scholar] [CrossRef]
- Covington, H.E., 3rd; Miczek, K.A. Repeated social-defeat stress, cocaine or morphine. Effects on behavioral sensitization and intravenous cocaine self-administration “binges”. Psychopharmacology 2001, 158, 388–398. [Google Scholar] [CrossRef]
- Miczek, K.A.; Yap, J.J.; Covington, H.E., 3rd. Social stress, therapeutics and drug abuse: Preclinical models of escalated and depressed intake. Pharmacol. Ther. 2008, 120, 102–128. [Google Scholar] [CrossRef]
- Engeln, M.; Fox, M.E.; Lobo, M.K. Housing conditions during self-administration determine motivation for cocaine in mice following chronic social defeat stress. Psychopharmacology 2021, 238, 41–54. [Google Scholar] [CrossRef]
- Mantsch, J.R.; Katz, E.S. Elevation of glucocorticoids is necessary but not sufficient for the escalation of cocaine self-administration by chronic electric footshock stress in rats. Neuropsychopharmacology 2007, 32, 367–376. [Google Scholar] [CrossRef]
- McReynolds, J.R.; Wolf, C.P.; Starck, D.M.; Mathy, J.C.; Schaps, R.; Krause, L.A.; Hillard, C.J.; Mantsch, J.R. Role of mesolimbic cannabinoid receptor 1 in stress-driven increases in cocaine self-administration in male rats. Neuropsychopharmacology 2023, 48, 1121–1132. [Google Scholar] [CrossRef]
- Ahmed, S.H.; Koob, G.F. Transition from moderate to excessive drug intake: Change in hedonic set point. Science 1998, 282, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Mantsch, J.R.; Yuferov, V.; Mathieu-Kia, A.M.; Ho, A.; Kreek, M.J. Neuroendocrine alterations in a high-dose, extended-access rat self-administration model of escalating cocaine use. Psychoneuroendocrinology 2003, 28, 836–862. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, H.; Sharp, B.M. Amplified reacquisition of nicotine self-administration in rats by repeated stress during abstinence. Psychopharmacology 2014, 231, 3189–3195. [Google Scholar] [CrossRef]
- Glynn, R.M.; Rosenkranz, J.A.; Wolf, M.E.; Caccamise, A.; Shroff, F.; Smith, A.B.; Loweth, J.A. Repeated restraint stress exposure during early withdrawal accelerates incubation of cue-induced cocaine craving. Addict. Biol. 2018, 23, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Munshi, S.; Rosenkranz, J.A.; Caccamise, A.; Wolf, M.E.; Corbett, C.M.; Loweth, J.A. Cocaine and chronic stress exposure produce an additive increase in neuronal activity in the basolateral amygdala. Addict. Biol. 2021, 26, e12848. [Google Scholar] [CrossRef]
- Reverte, I.; Marchetti, C.; Pezza, S.; Zenoni, S.F.; Scaringi, G.; Ferrucci, L.; D’Ottavio, G.; Pignataro, A.; Andolina, D.; Raspa, M.; et al. Microglia-mediated calcium-permeable AMPAR accumulation in the nucleus accumbens drives hyperlocomotion during cocaine withdrawal. Brain Behav. Immun. 2024, 115, 535–542. [Google Scholar] [CrossRef]
- Kelly, K.A.; Miller, D.B.; Bowyer, J.F.; O’Callaghan, J.P. Chronic exposure to corticosterone enhances the neuroinflammatory and neurotoxic responses to methamphetamine. J. Neurochem. 2012, 122, 995–1009. [Google Scholar] [CrossRef]
- Lo Iacono, L.; Catale, C.; Martini, A.; Valzania, A.; Viscomi, M.T.; Chiurchiu, V.; Guatteo, E.; Bussone, S.; Perrone, F.; Di Sabato, P.; et al. From Traumatic Childhood to Cocaine Abuse: The Critical Function of the Immune System. Biol. Psychiatry 2018, 84, 905–916. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowak, D.B.; Taborda-Bejarano, J.P.; Chaure, F.J.; Mantsch, J.R.; Garcia-Keller, C. Understanding Microglia in Mesocorticolimbic Circuits: Implications for the Study of Chronic Stress and Substance Use Disorders. Cells 2025, 14, 1014. https://doi.org/10.3390/cells14131014
Nowak DB, Taborda-Bejarano JP, Chaure FJ, Mantsch JR, Garcia-Keller C. Understanding Microglia in Mesocorticolimbic Circuits: Implications for the Study of Chronic Stress and Substance Use Disorders. Cells. 2025; 14(13):1014. https://doi.org/10.3390/cells14131014
Chicago/Turabian StyleNowak, David B., Juan Pablo Taborda-Bejarano, Fernando J. Chaure, John R. Mantsch, and Constanza Garcia-Keller. 2025. "Understanding Microglia in Mesocorticolimbic Circuits: Implications for the Study of Chronic Stress and Substance Use Disorders" Cells 14, no. 13: 1014. https://doi.org/10.3390/cells14131014
APA StyleNowak, D. B., Taborda-Bejarano, J. P., Chaure, F. J., Mantsch, J. R., & Garcia-Keller, C. (2025). Understanding Microglia in Mesocorticolimbic Circuits: Implications for the Study of Chronic Stress and Substance Use Disorders. Cells, 14(13), 1014. https://doi.org/10.3390/cells14131014