
The Role of Protein Ubiquitination in the Onset and Progression of Sepsis
Abstract
1. Introduction
2. Mechanisms of Ubiquitination
3. Ubiquitination in Sepsis and the Inflammatory Response
3.1. Regulation of the Production of Inflammatory Cytokines
3.2. Regulation of the Activation of Inflammasomes
4. Ubiquitination in Sepsis and Immune Cell Functions
4.1. Activation of Neutrophils
4.2. Macrophage Polarization
4.3. Regulation of T Cell Functions
5. Ubiquitination in Sepsis and Organ Protection
5.1. Lung Protection
5.2. Liver Protection
5.3. Cardiac Function
5.4. Renal Function
5.5. Intestinal Function
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Term |
| AIM2 | Absent in melanoma 2 |
| AKI | Acute kidney injury |
| ASC | Apoptosis-associated speck-like protein containing a card |
| ATG5 | Autophagy-related 5 |
| BMDM | Bone marrow-derived macrophage |
| Cbl-b | Casitas B lymphoma-b |
| cIAPs | Cellular inhibitors of apoptosis proteins |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DUB | Deubiquitinase |
| E1 | Ubiquitin-activating enzyme |
| E2 | Ubiquitin-conjugating enzyme |
| E3 | Ubiquitin ligase |
| GSDMD | Gasdermin d |
| HUWE1 | HECT domain-containing ubiquitin E3 ligase HUWE1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IKK | IκB kinase |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| IRAK1/4 | IL-1 receptor-associated kinase 1/4 |
| IκB-α | Inhibitor of nuclear factor kappa-b alpha |
| JNK | c-Jun N-terminal kinase |
| LCK | Lymphocyte-specific tyrosine kinase |
| LPS | Lipopolysaccharide |
| LRR-PYD | Leucine-rich repeat and pyrin domain-containing protein |
| LUBAC | Linear ubiquitin assembly complex |
| M1/M2 | Met-1/Met-2 |
| MFHAS1 | Malignant fibrous histiocytoma amplified sequence 1 |
| MMR | Macrophage mannose receptor |
| MODS | Multiple organ dysfunction syndrome |
| MyD88 | Myeloid differentiation primary response 88 |
| NDP52 | Nuclear dot protein 52 kDa |
| NEMO | NF-κB essential modulator |
| NEK7 | NIMA-related kinase 7 |
| NF-κB | Nuclear factor kappa B |
| NLRP3 | NLR family pyrin domain-containing 3 |
| NLRC4 | NLR family card domain-containing protein 4 |
| NOX4 | NADPH oxidase 4 |
| OTU | Ovarian tumor deubiquitinases |
| OTULIN | OTU DUB with linear linkage specificity |
| PARP1 | Poly (ADP-ribose) polymerase 1 |
| PDLIM2 | Pdz and lim domain protein 2 |
| PP2Ac | Protein phosphatase 2Ac |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| Praja2 | E3 ubiquitin ligase praja ring finger 2 |
| RBR | RING-between-RING |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| RNF146 | Ring finger protein 146 |
| SIMD | Sepsis-induced myocardial dysfunction |
| SLP76 | SH2 domain-containing leukocyte protein of 76kD |
| TAK1 | Transforming growth factor-β-activated kinase 1 |
| TLRs | Toll-like receptors |
| TNFR | Tumor necrosis factor receptor |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TNF-α | Tumor necrosis factor-alpha |
| TRAF2/6 | Tumor necrosis factor receptor-associated factor 2/6 |
| TRIM27 | Tripartite motif-containing protein 27 |
| Ubc13 | Ubiquitin-conjugating enzyme E2 N |
| UBE2M | Ubiquitin-conjugating enzyme E2 M |
| UPS | Ubiquitin–proteasome system |
| USP22 | Ubiquitin-specific peptidase 22 |
| USP5 | Ubiquitin-specific peptidase 5 |
| VANGL2 | Van gogh-like protein 2 |
| WWP1 | WW domain-containing E3 ubiquitin-protein ligase 1 |
| ZAP70 | Zeta-chain-associated protein kinase 70 |
References
- Wang, Z.; Sun, S.; Huang, L.; Chen, X.; Xu, H.; Ma, H.; Xiao, M.; Wang, L. Pathological roles of ubiquitination and deubiquitination systems in sepsis-induced myocardial dysfunction. Biomol. Biomed. 2025, 7, 1444–1458. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Wang, G.; Xie, J. Immune dysregulation in sepsis: Experiences, lessons and perspectives. Cell Death Discov. 2023, 9, 465. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Aschenbrenner, A.C.; Bauer, M.; Bock, C.; Calandra, T.; Gat-Viks, I.; Kyriazopoulou, E.; Lupse, M.; Monneret, G.; Pickkers, P.; et al. The pathophysiology of sepsis and precision-medicine-based immunotherapy. Nat. Immunol. 2024, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Barichello, T.; Generoso, J.S.; Singer, M.; Dal-Pizzol, F. Biomarkers for sepsis: More than just fever and leukocytosis-a narrative review. Crit. Care. 2022, 26, 14. [Google Scholar] [CrossRef]
- Willmann, K.; Moita, L.F. Physiologic disruption and metabolic reprogramming in infection and sepsis. Cell Metab. 2024, 36, 927–946. [Google Scholar] [CrossRef]
- Srdic, T.; Durasevic, S.; Lakic, I.; Ruzicic, A.; Vujovic, P.; Jevdovic, T.; Dakic, T.; Dordevic, J.; Tosti, T.; Glumac, S.; et al. From molecular mechanisms to clinical therapy: Understanding sepsis-induced multiple organ dysfunction. Int. J. Mol. Sci. 2024, 25, 7770. [Google Scholar] [CrossRef]
- Hao, Y.; Liu, R.; Wang, H.; Rui, T.; Guo, J. Research progress on mechanisms and treatment of sepsis-induced myocardial dysfunction. Int. J. Gen. Med. 2024, 17, 3387–3393. [Google Scholar] [CrossRef]
- Shvilkina, T.; Shapiro, N. Sepsis-Induced myocardial dysfunction: Heterogeneity of functional effects and clinical significance. Front. Cardiovasc. Med. 2023, 10, 1200441. [Google Scholar] [CrossRef]
- Lin, H.; Wang, W.; Lee, M.; Meng, Q.; Ren, H. Current Status of Septic Cardiomyopathy: Basic Science and Clinical Progress. Front. Pharmacol. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.K.; Yu, J.C.; Li, R.B.; Zhou, H.; Chang, X. New insights into the role of mitochondrial metabolic dysregulation and immune infiltration in septic cardiomyopathy by integrated bioinformatics analysis and experimental validation. Cell Mol. Biol. Lett. 2024, 29, 21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Zhang, W.; Zhou, M.; Zhu, C.; Zou, Z. Ubiquitination in pyroptosis pathway: A potential therapeutic target for sepsis. Cytokine Growth Factor Rev. 2024, 80, 72–86. [Google Scholar] [CrossRef]
- Kelsall, I.R. Non-lysine ubiquitylation: Doing things differently. Front. Mol. Biosci. 2022, 9, 1008175. [Google Scholar] [CrossRef] [PubMed]
- Suryadinata, R.; Roesley, S.N.; Yang, G.; Sarcevic, B. Mechanisms of generating polyubiquitin chains of different topology. Cells 2014, 3, 674–689. [Google Scholar] [CrossRef]
- Tang, J.; Tu, S.; Lin, G.; Guo, H.; Yan, C.; Liu, Q.; Huang, L.; Tang, N.; Xiao, Y.; Pope, R.M.; et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. 2020, 217, e20182091. [Google Scholar] [CrossRef]
- Wu, G.; Li, D.; Liang, W.; Sun, W.; Xie, X.; Tong, Y.; Shan, B.; Zhang, M.; Lu, X.; Yuan, J.; et al. PP6 negatively modulates LUBAC-mediated M1-ubiquitination of RIPK1 and c-FLIP(L) to promote TNFalpha-mediated cell death. Cell Death Dis. 2022, 13, 773. [Google Scholar] [CrossRef]
- Sun, W.; Lu, H.; Ma, L.; Ding, C.; Wang, H.; Chu, Y. Deubiquitinase USP5 regulates RIPK1 driven pyroptosis in response to myocardial ischemic reperfusion injury. Cell Commun. Signal. 2024, 22, 466. [Google Scholar] [CrossRef]
- Ning, M.; Liu, Y.; Wang, D.; Wei, J.; Hu, G.; Xing, P. Knockdown of TRIM27 alleviated sepsis-induced inflammation, apoptosis, and oxidative stress via suppressing ubiquitination of PPARgamma and reducing NOX4 expression. Inflamm. Res. 2022, 71, 1315–1325. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Tang, S.; Geng, Y.; Wang, Y.; Lin, Q.; Yu, Y.; Li, H. The roles of ubiquitination and deubiquitination of NLRP3 inflammasome in inflammation-related diseases: A review. Biomol. Biomed. 2024, 24, 708–721. [Google Scholar] [CrossRef]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, S. Structural mechanisms of HECT-type ubiquitin ligases. Biol. Chem. 2018, 399, 127–145. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Renz, C.; Asimaki, E.; Meister, C.; Albanese, V.; Petriukov, K.; Krapoth, N.C.; Wegmann, S.; Wollscheid, H.P.; Wong, R.P.; Fulzele, A.; et al. Ubiquiton-An inducible, linkage-specific polyubiquitylation tool. Mol. Cell. 2024, 84, 386–400.e11. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef]
- Kim, H.T.; Kim, K.P.; Lledias, F.; Kisselev, A.F.; Scaglione, K.M.; Skowyra, D.; Gygi, S.P.; Goldberg, A.L. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 2007, 282, 17375–17386. [Google Scholar] [CrossRef]
- Hayakawa, M. Role of K63-linked polyubiquitination in NF-kappaB signalling: Which ligase catalyzes and what molecule is targeted? J. Biochem. 2012, 151, 115–118. [Google Scholar] [CrossRef]
- Malynn, B.A.; Ma, A. Ubiquitin makes its mark on immune regulation. Immunity 2010, 33, 843–852. [Google Scholar] [CrossRef]
- Guo, Y.; Li, L.; Xu, T.; Guo, X.; Wang, C.; Li, Y.; Yang, Y.; Yang, D.; Sun, B.; Zhao, X.; et al. HUWE1 mediates inflammasome activation and promotes host defense against bacterial infection. J. Clin. Investig. 2020, 130, 6301–6316. [Google Scholar] [CrossRef]
- Cockram, P.E.; Kist, M.; Prakash, S.; Chen, S.H.; Wertz, I.E.; Vucic, D. Ubiquitination in the regulation of inflammatory cell death and cancer. Cell Death Differ. 2021, 28, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Wayson, S.M.; Dixit, V.M.; Fairbrother, W.J.; Vucic, D. The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-kappaB activation independently of TRAF1 AND TRAF2. J. Biol. Chem. 2006, 281, 29022–29029. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.B.; Takeuchi, M.; Goeddel, D.V. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc. Natl. Acad. Sci. USA 1996, 93, 13973–13978. [Google Scholar] [CrossRef]
- Vince, J.E.; Pantaki, D.; Feltham, R.; Mace, P.D.; Cordier, S.M.; Schmukle, A.C.; Davidson, A.J.; Callus, B.A.; Wong, W.W.; Gentle, I.E.; et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J. Biol. Chem. 2009, 284, 35906–35915. [Google Scholar] [CrossRef]
- Fuseya, Y.; Fujita, H.; Kim, M.; Ohtake, F.; Nishide, A.; Sasaki, K.; Saeki, Y.; Tanaka, K.; Takahashi, R.; Iwai, K. The HOIL-1L ligase modulates immune signalling and cell death via monoubiquitination of LUBAC. Nat. Cell Biol. 2020, 22, 663–673. [Google Scholar] [CrossRef]
- Chen, S.; Deng, Y.; Huang, C.; Xie, X.; Long, Z.; Lao, S.; Gao, X.; Wang, K.; Wang, S.; Li, X.; et al. BSRF1 modulates IFN-beta-mediated antiviral responses by inhibiting NF-kappaB activity via an IKK-dependent mechanism in Epstein-Barr virus infection. Int. J. Biol. Macromol. 2025, 306, 141600. [Google Scholar] [CrossRef]
- Huyghe, J.; Priem, D.; Bertrand, M.J.M. Cell death checkpoints in the TNF pathway. Trends Immunol. 2023, 44, 628–643. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The K48-K63 branched ubiquitin chain regulates NF-kappaB signaling. Mol. Cell. 2016, 64, 251–266. [Google Scholar] [CrossRef]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. Regulation of NF-κB by ubiquitination. Curr. Opin. Immunol. 2013, 25, 4–12. [Google Scholar] [CrossRef]
- Griewahn, L.; Koser, A.; Maurer, U. Keeping cell death in check: Ubiquitylation-dependent control of TNFR1 and TLR signaling. Front. Cell Dev. Biol. 2019, 7, 117. [Google Scholar] [CrossRef]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.L.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, C.H.; Bakshi, S.; Kelsall, I.R.; Ortiz-Guerrero, J.; Shpiro, N.; Cohen, P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem. Biophys. Res. Commun. 2016, 474, 452–461. [Google Scholar] [CrossRef]
- Zhu, B.; Zhu, L.; Xia, L.; Xiong, Y.; Yin, Q.; Rui, K. Roles of ubiquitination and deubiquitination in regulating dendritic cell maturation and function. Front. Immunol. 2020, 11, 586613. [Google Scholar] [CrossRef] [PubMed]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Spit, M.; Rieser, E.; Walczak, H. Linear ubiquitination at a glance. J. Cell Sci. 2019, 132, jcs208512. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Yan, X.; Li, Q.; Wang, P.; Sun, Y.; Xu, T. hnRNPub inhibits LPS-induced NF-kappaB pathway by targeting TRAF6 for K48-linked ubiquitination in miiuy croaker (Miichthys miiuy). Fish. Shellfish. Immunol. 2022, 121, 498–504. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, J.; Jiang, H.; Hu, Z.; Zhang, Y.; He, L.; Yang, J.; Xie, Y.; Wu, D.; Li, H.; et al. Vangl2 suppresses NF-kappaB signaling and ameliorates sepsis by targeting p65 for NDP52-mediated autophagic degradation. eLife 2024, 12, RP87935. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Xu, T.; Guo, Y.; Qi, X. Ubiquitination-mediated inflammasome activation during bacterial infection. Int. J. Mol. Sci. 2019, 20, 2110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guan, X.; Liu, W.; Zhu, Z.; Jin, H.; Zhu, Y.; Chen, Y.; Zhang, M.; Xu, C.; Tang, X.; et al. YTHDF1 alleviates sepsis by upregulating WWP1 to induce NLRP3 ubiquitination and inhibit caspase-1-dependent pyroptosis. Cell Death Discov. 2022, 8, 244. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hoshino, S.; Abe, T.; Okita, N.; Tagawa, R.; Nagai, W.; Konno, R.; Suzuki, Y.; Furuya, K.; Ishikawa, N.; et al. Identification of WWP1 as an obesity-associated E3 ubiquitin ligase with a protective role against oxidative stress in adipocytes. Biochem. Biophys. Res. Commun. 2019, 508, 117–122. [Google Scholar] [CrossRef]
- Feng, S.Y.; Tuipulotu, D.E.; Pandey, A.; Jing, W.D.; Shen, C.; Ngo, C.; Tessema, M.B.; Li, F.J.; Fox, D.; Mathur, A.; et al. Pathogen-selective killing by guanylate-binding proteins as a molecular mechanism leading to inflammasome signaling. Nat. Commun. 2022, 13, 4395. [Google Scholar] [CrossRef]
- Lin, X.W.; Xu, W.C.; Luo, J.G.; Guo, X.J.; Sun, T.; Zhao, X.L.; Fu, Z.J. WW domain containing E3 ubiquitin protein ligase 1 (WWP1) negatively regulates TLR4-mediated TNF-alpha and IL-6 production by proteasomal degradation of TNF receptor associated factor 6 (TRAF6). PLoS ONE 2013, 8, e67633. [Google Scholar] [CrossRef]
- Moretti, J.; Blander, J.M. Increasing complexity of NLRP3 inflammasome regulation. J. Leukoc. Biol. 2021, 109, 561–571. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Shin, H.J.; Park, E.-J.; Kim, I.S.; Jo, E.-K. Updated insights into the molecular networks for NLRP3 inflammasome activation. Cell. Mol. Immunol. 2025, 22, 563–596. [Google Scholar] [CrossRef] [PubMed]
- Di, Q.; Zhao, X.; Tang, H.; Li, X.; Xiao, Y.; Wu, H.; Wu, Z.; Quan, J.; Chen, W. USP22 suppresses the NLRP3 inflammasome by degrading NLRP3 via ATG5-dependent autophagy. Autophagy 2023, 19, 873–885. [Google Scholar] [CrossRef]
- Yu, M.C.; Li, X.L.; Ning, M.L.; Yan, Z.Z.; Yu, W.T. USP22 inhibits microglial M1 polarization by regulating the PU.1/NLRP3 inflammasome pathway. Brain Res. Bull. 2025, 220, 111157. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R.A.-O. NLRP3 inflammasome activation and cell death. Cell Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Barnett, K.C.; Li, S.; Liang, K.; Ting, J.P.Y. A 360° view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell 2023, 186, 2288–2312. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, Y.; Shu, W.; Dong, S.; Sun, Y.; Ma, X. Neutrophil-derived heparin-binding protein increases endothelial permeability in acute lung injury by promoting TRIM21 and the ubiquitination of P65. Cell Biol. Toxicol. 2025, 41, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Wang, H.; Chen, W.; Sun, Z.; Chen, J.; Xu, Y.; Weng, M.; Shi, Q.; Ma, D.; Miao, C. Ubiquitylation of MFHAS1 by the ubiquitin ligase praja2 promotes M1 macrophage polarization by activating JNK and p38 pathways. Cell Death Dis. 2017, 8, e2763. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Feng, L.L.; Song, P.P.; Xu, F.; Li, A.; Wang, Y.B.; Shen, Y.; Wu, X.F.; Luo, Q.; Wu, X.X.; et al. Isomeranzin suppresses inflammation by inhibiting M1 macrophage polarization through the NF-κB and ERK pathway. Int. Immunopharmacol. 2016, 38, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.W.; Holdbrooks, A.T.; Liu, Y.D.; Reynolds, S.L.; Yanagisawa, L.L.; Benveniste, E.N. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 2012, 189, 3439–3448. [Google Scholar] [CrossRef]
- Li, H.W.; Wu, Y.Y.; Xiang, L.S.; Zhao, Q.; Liu, L.; Zhu, Z.X.; Lin, W.M.; Li, Z.; Yang, Y.; Ze, Y.T.; et al. A20 attenuates oxidized self-DNA-mediated inflammation in acute kidney injury. Signal Transduct. Tar. 2025, 10, 154. [Google Scholar] [CrossRef]
- Yu, J.; Li, H.; Wu, Y.; Luo, M.; Chen, S.; Shen, G.; Wei, X.; Shao, B. Inhibition of NLRP3 inflammasome activation by A20 through modulation of NEK7. Proc. Natl. Acad. Sci. USA 2024, 121, e2316551121. [Google Scholar] [CrossRef]
- Wen, X.; Bai, S.; Xiong, G.; Xiu, H.; Li, J.; Yang, J.; Yu, Q.; Li, B.; Hu, R.; Cao, L.; et al. Inhibition of the neddylation E2 enzyme UBE2M in macrophages protects against E. coli-induced sepsis. J. Biol. Chem. 2025, 301, 108085. [Google Scholar] [CrossRef]
- Augustin, R.C.; Bao, R.; Luke, J.J. Targeting Cbl-b in cancer immunotherapy. J. Immunother. Cancer 2023, 11, e006007. [Google Scholar] [CrossRef]
- Lutz-Nicoladoni, C.; Wolf, D.; Sopper, S. Modulation of immune cell functions by the E3 ligase Cbl-b. Front. Oncol. 2015, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Gavali, S.; Liu, J.; Li, X.; Paolino, M. Ubiquitination in T-Cell activation and checkpoint inhibition: New avenues for targeted cancer immunotherapy. Int. J. Mol. Sci. 2021, 22, 10800. [Google Scholar] [CrossRef] [PubMed]
- Schanz, O.; Cornez, I.; Yajnanarayana, S.P.; David, F.S.; Peer, S.; Gruber, T.; Krawitz, P.; Brossart, P.; Heine, A.; Landsberg, J.; et al. Tumor rejection in Cblb−/− mice depends on IL-9 and Th9 cells. J. Immunother. Cancer 2021, 9, e002889. [Google Scholar] [CrossRef]
- Thell, K.; Urban, M.; Harrauer, J.; Haslinger, I.; Kuttke, M.; Brunner, J.S.; Vogel, A.; Schabbauer, G.; Penninger, J.; Gaweco, A. Master checkpoint Cbl-b inhibition: Anti-tumour efficacy in a murine colorectal cancer model following siRNA-based cell therapy. Ann. Oncol. 2019, 30, v503–v504. [Google Scholar] [CrossRef]
- Yi, X.M.; Li, M.; Chen, Y.D.; Shu, H.B.; Li, S. Reciprocal regulation of IL-33 receptor-mediated inflammatory response and pulmonary fibrosis by TRAF6 and USP38. Proc. Natl. Acad. Sci. USA 2022, 119, e2116279119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, Z.; Li, J.; Huang, K.; Ding, Z.; Chen, B.; Ren, T.; Xu, P.; Wang, G.; Zhang, H.; et al. The deubiquitinase OTUD1 stabilizes NRF2 to alleviate hepatic ischemia/reperfusion injury. Redox Biol. 2024, 75, 103287. [Google Scholar] [CrossRef]
- Yang, L.Y.; Du, M.; Liu, K.Y.; Wang, P.C.; Zhu, J.B.; Li, F.C.; Wang, Z.; Huang, K.; Liang, M.L. Pimpinellin ameliorates macrophage inflammation by promoting RNF146-mediated PARP1 ubiquitination. Phytother. Res. 2024, 38, 1783–1798. [Google Scholar] [CrossRef]
- Huang, D.A.-O.; Wang, J.; Chen, L.; Jiang, W.; Inuzuka, H.; Simon, D.A.-O.; Wei, W. Targeting the PARylation-dependent ubiquitination signaling pathway for cancer therapies. Biomol. Biomed. 2025, 15, 237. [Google Scholar] [CrossRef]
- Gong, X.; Li, Y.; He, Y.; Zhou, F. USP7-SOX9-miR-96-5p-NLRP3 Network Regulates Myocardial Injury and Cardiomyocyte Pyroptosis in Sepsis. Hum. Gene Ther. 2022, 33, 1073–1090. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; Liu, Z.; Shu, S.; Fu, Y.; Liu, Y.; Cai, J.; Tang, C.; Liu, Y.; Yin, X.; et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021, 38, 101767. [Google Scholar] [CrossRef]
- Lei, H.; Yang, L.; Xu, H.; Wang, Z.; Li, X.; Liu, M.; Wu, Y. Ubiquitin-specific protease 47 regulates intestinal inflammation through deubiquitination of TRAF6 in epithelial cells. Sci. China Life Sci. 2022, 65, 1624–1635. [Google Scholar] [CrossRef]
- Chen, R.R.; Pang, X.B.; Li, L.; Zeng, Z.R.; Chen, M.H.; Zhang, S.H. Ubiquitin-specific proteases in inflammatory bowel disease-related signalling pathway regulation. Cell Death Dis. 2022, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, H.; Li, L.K.; Li, J.S.; Xie, J.; Weng, J.; Tan, H.; Liu, Y.J.; Guo, T.L.; Wang, M.Y. Roles of ubiquitin-specific proteases in inflammatory diseases. Front. Immunol. 2024, 15, 1392734. [Google Scholar] [CrossRef]
- Yang, F.; Feng, C.; Zhang, X.D.; Lu, J.; Zhao, Y. The diverse biological functions of neutrophils, beyond the defense against infections. Inflammation 2017, 40, 311–323. [Google Scholar] [CrossRef]
- Gupta, S.; Lee, C.M.; Wang, J.F.; Parodo, J.; Jia, S.H.; Hu, J.; Marshall, J.C. Heat-shock protein-90 prolongs septic neutrophil survival by protecting c-Src kinase and caspase-8 from proteasomal degradation. J. Leukoc. Biol. 2018, 103, 933–944. [Google Scholar] [CrossRef]
- Galbas, T.; Raymond, M.; Sabourin, A.; Bourgeois-Daigneault, M.C.; Guimont-Desrochers, F.; Yun, T.J.; Cailhier, J.F.; Ishido, S.; Lesage, S.; Cheong, C.; et al. MARCH1 E3 ubiquitin ligase dampens the innate inflammatory response by modulating monocyte functions in mice. J. Immunol. 2017, 198, 852–861. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Arabpour, M.; Saghazadeh, A.; Rezaei, N. Anti-inflammatory and M2 macrophage polarization-promoting effect of mesenchymal stem cell-derived exosomes. Int. Immunopharmacol. 2021, 97, 107823. [Google Scholar] [CrossRef]
- Chen, X.X.; Tang, J.; Shuai, W.Z.; Meng, J.G.; Feng, J.; Han, Z.H. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. 2020, 69, 883–895. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, L.; Li, X. Advances in Mesenchymal stem cells regulating macrophage polarization and treatment of sepsis-induced liver injury. Front. Immunol. 2023, 14, 1238972. [Google Scholar] [CrossRef]
- Xin, Y.; Gao, X.; Wang, W.; Xu, X.; Yu, L.; Ju, X.; Li, A. Circulating cell-free DNA indicates M1/M2 responses during septic peritonitis. Biochem. Biophys. Res. Commun. 2016, 477, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Shi, Q.Q.; Zhu, M.M.; Shen, J.; Wang, H.H.; Ma, D.; Miao, C.H. MFHAS1 is associated with sepsis and stimulates TLR2/NF-κB signaling pathway following negative regulation. PLoS ONE 2015, 10, e0143662. [Google Scholar] [CrossRef]
- Kumar, V. T cells and their immunometabolism: A novel way to understanding sepsis immunopathogenesis and future therapeutics. Eur. J. Cell. Biol. 2018, 97, 379–392. [Google Scholar] [CrossRef]
- Huang, S.; Liu, D.; Han, L.; Deng, J.; Wang, Z.; Jiang, J.; Zeng, L. Decoding the potential role of regulatory T cells in sepsis-induced immunosuppression. Eur. J. Immunol. 2024, 54, e2350730. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Chen, J.; Tong, Y.; Zhang, Y.; Feng, Q.; Tang, Z. USP9x promotes CD8 (+) T-cell dysfunction in association with autophagy inhibition in septic liver injury. Acta Biochim. Biophys. Sin. 2022, 54, 1765–1774. [Google Scholar] [CrossRef]
- Shen, J.; Jiang, Y.; Bu, W.; Yu, M.; Huang, R.; Tang, C.; Yang, Z.; Gao, H.; Su, L.; Cheng, D.; et al. Protein ubiquitination modification in pulmonary fibrosis. Compr. Physiol. 2025, 15, e70013. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, J.; Wang, X.; Ji, F.; Ronco, C.; Tian, J.; Yin, Y. Gut-liver crosstalk in sepsis-induced liver injury. Crit. Care. 2020, 24, 614. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Chen, W.; Wang, X.; Zhao, Z.; Li, Y.; Zhang, L.; Jiao, J.; Yang, Q.; Ding, Q.; et al. Hepatocyte CD36 modulates UBQLN1-mediated proteasomal degradation of autophagic SNARE proteins contributing to septic liver injury. Autophagy 2023, 19, 2504–2519. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Wu, D.M.; He, M.; Zhang, F.; Zhang, T.; Liu, T.; Li, J.; Li, L.; Xu, Y. Samotolisib attenuates acute liver injury through inhibiting Caspase-11-mediated pyroptosis via regulating E3 ubiquitin ligase Nedd4. Front. Pharmacol. 2021, 12, 726198. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, S.; Sun, Z.; Guo, X.; Zhou, H. E3 ubiquitin ligase Nedd4 is a key negative regulator for non-canonical inflammasome activation. Cell Death Differ. 2019, 26, 2386–2399. [Google Scholar] [CrossRef]
- Pazzaglia, S.; Pioli, C. Multifaceted role of PARP-1 in DNA repair and inflammation: Pathological and therapeutic implications in cancer and non-cancer diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Qiu, T.; Wang, T.; Chen, Z.; Ma, X.; Zhang, L.; Zou, J. USP4 deficiency exacerbates hepatic ischaemia/reperfusion injury via TAK1 signalling. Clin. Sci. 2019, 133, 335–349. [Google Scholar] [CrossRef]
- Zhu, J.; Luo, Z.; Pan, Y.; Zheng, W.; Li, W.; Zhang, Z.; Xiong, P.; Xu, D.; Du, M.; Wang, B.; et al. H19/miR-148a/USP4 axis facilitates liver fibrosis by enhancing TGF-beta signaling in both hepatic stellate cells and hepatocytes. J. Cell Physiol. 2019, 234, 9698–9710. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, J. Ubiquitination-deubiquitination in the Hippo signaling pathway (Review). Oncol. Rep. 2019, 41, 1455–1475. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Lerman, L.O.; Lerman, A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circul. Res. 2007, 100, 1276–1291. [Google Scholar] [CrossRef]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. Iubmb Life 2015, 67, 544–555. [Google Scholar] [CrossRef]
- Caraballo, C.; Jaimes, F. Organ dysfunction in sepsis: An ominous trajectory from infection to death. Yale J. Biol. Med. 2019, 92, 629–640. [Google Scholar]
- Cao, Q.; Wang, Y.; Harris, D.C. Pathogenic and protective role of macrophages in kidney disease. Am. J. Physiol. Renal. Physiol. 2013, 305, F3–F11. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Wang, L.; Diao, Z.; Liu, W. The role of autophagy in kidney inflammatory injury via the NF-kappaB route induced by LPS. Int. J. Med. Sci. 2015, 12, 655–667. [Google Scholar] [CrossRef]
- Huang, X.R.; Ye, L.; An, N.; Wu, C.Y.; Wu, H.L.; Li, H.Y.; Huang, Y.H.; Ye, Q.R.; Liu, M.D.; Yang, L.W.; et al. Macrophage autophagy protects against acute kidney injury by inhibiting renal inflammation through the degradation of TARM1. Autophagy 2025, 21, 120–140. [Google Scholar] [CrossRef]
- Niu, M.W.; Chen, P. Crosstalk between gut microbiota and sepsis. Burn. Trauma. 2021, 9, tkab036. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, N.; Su, X.; Gao, Y.; Yang, R. Gut-Microbiota-Derived metabolites maintain gut and systemic immune homeostasis. Cells 2023, 12, 793. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Coopersmith, C.M. Redefining the gut as the motor of critical illness. Trends Mol. Med. 2014, 20, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Ren, H.; Li, G.; Wang, D.; Zhou, Q.; Wu, J.; Zheng, J.; Huang, J.; Slade, D.A.; Wu, X.; et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine 2019, 41, 497–508. [Google Scholar] [CrossRef]
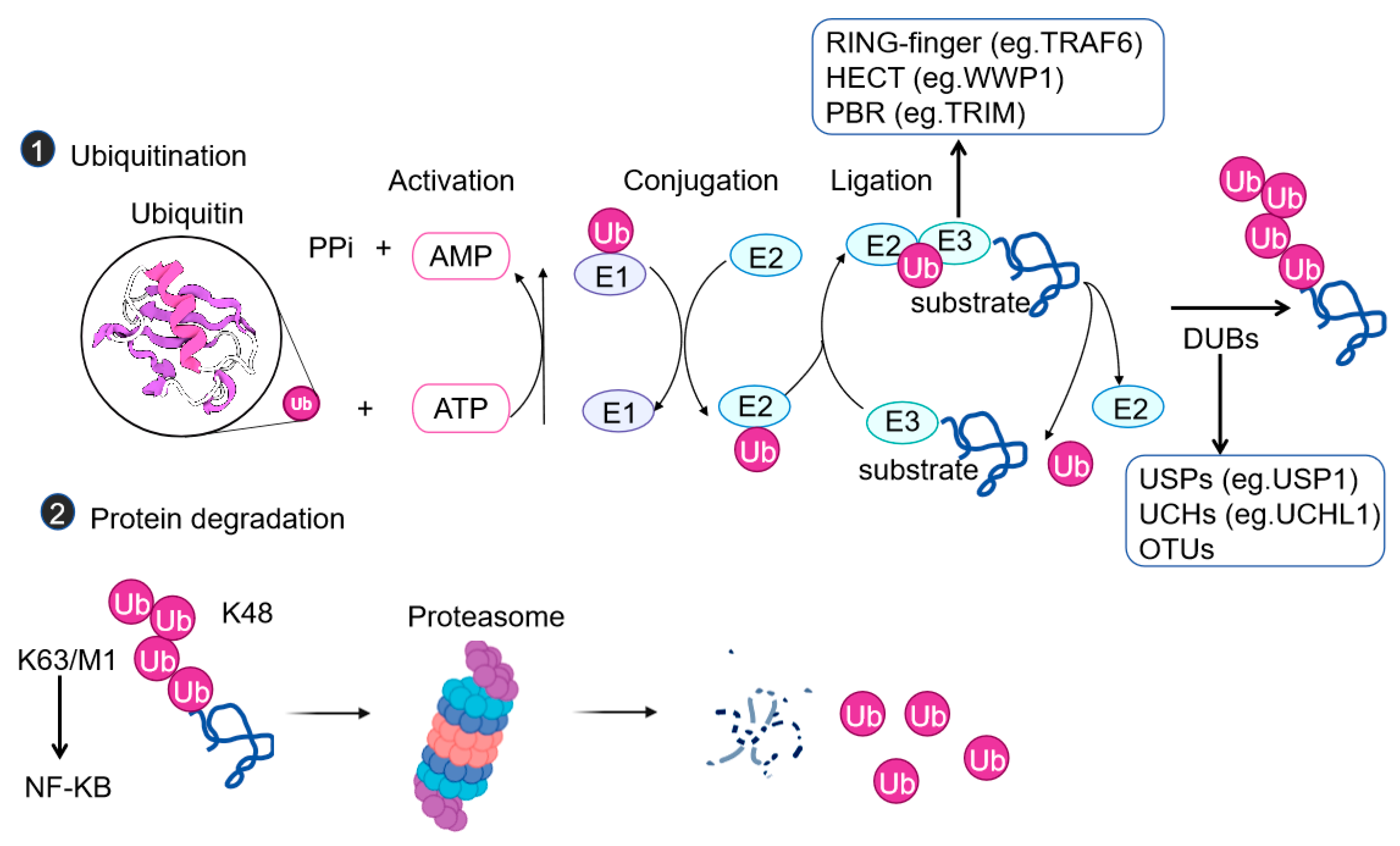

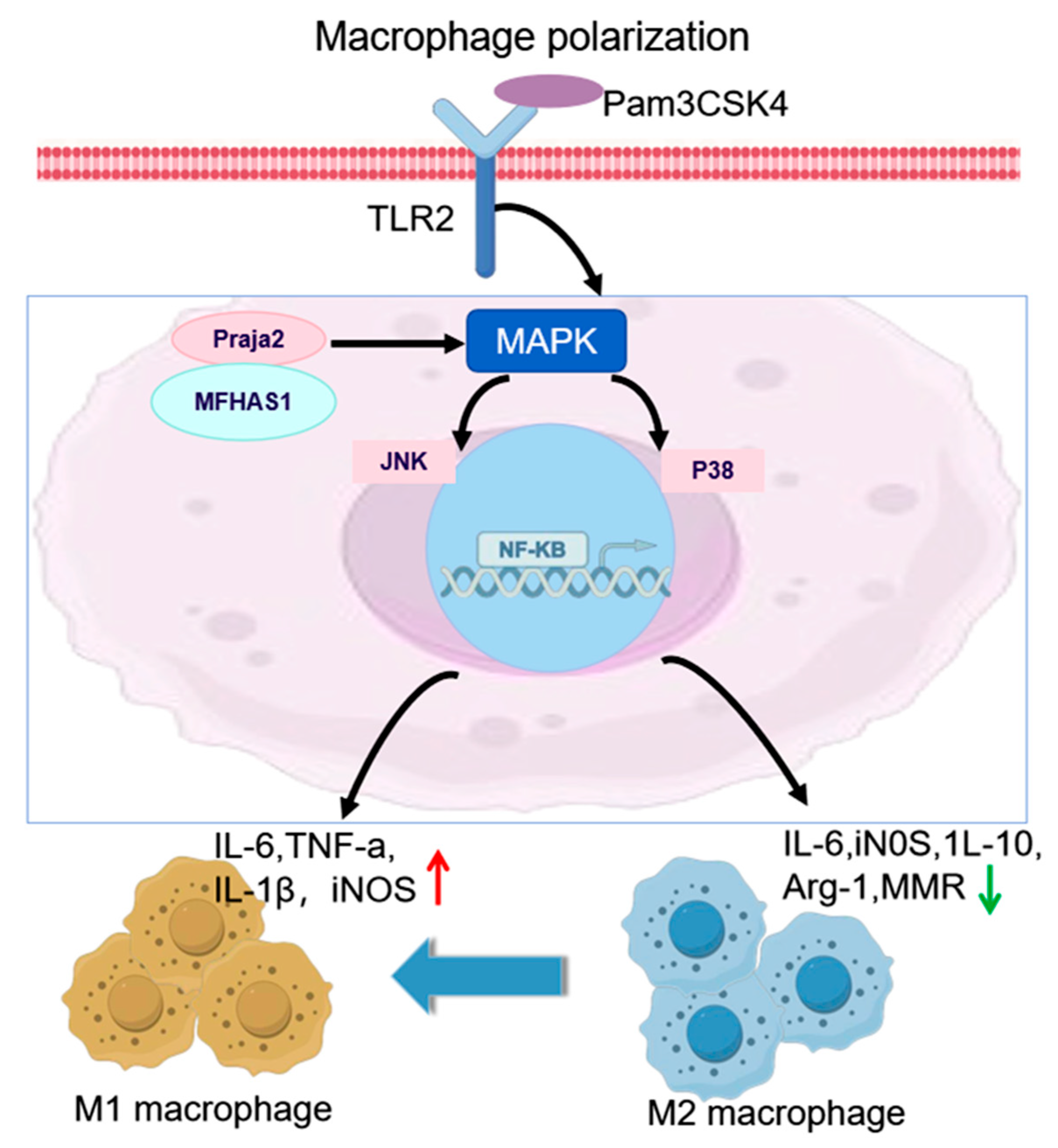

{kind=link}
{kind=link}
{kind=link}
| Pathological Process | Key Ubiquitination Event | Enzyme/Complex Involved | Mechanism | Functional Impact in Sepsis | Refs. |
|---|---|---|---|---|---|
| Inflammatory response | NF-κB non-degradative activation | TRAF6, LUBAC, Ubc13 | K63/M1 polyubiquitination of TRAF6/RIPK1 recruits the TAK1/IKK complex, promoting NF-κB nuclear translocation without protein degradation | Enhances TNF-α/IL-1β production, driving systemic inflammation | [41,42,43,44,45,46,47] |
| NLRP3 inflammasome assembly | HUWE1, WWP1, USP22 | HUWE1-mediated K27 ubiquitination induces NLRP3 conformational change for inflammasome assembly; WWP1/USP22 regulates via degradation or autophagy | Promotes IL-1β release (HUWE1) or inhibits pyroptosis (USP22) | [30,52,53,54,55,56,57] | |
| Immune cell functions | Activation of neutrophils | TRIM21, HBP | Inhibition of K48-linked ubiquitination of TRIM21; promotion of K63-linked ubiquitination of p65 | Contributes to acute lung injury (ALI) pathogenesis in sepsis | [63] |
| Macrophage M1/M2 polarization | Praja2, A20, UBE2M | Praja2 enhances MFHAS1 accumulation via non-degradative ubiquitination, driving M1 polarization; A20 degrades NEK7 to inhibit NLRP3 | M1-dominated inflammation (Praja2) or reduced pyroptosis (A20) | [64,65,66,67,68,69] | |
| T cell activation inhibition | Cbl-b, Itch | Cbl-b/Itch mediates K33 ubiquitination of TCR-ζ, blocking ZAP70 recruitment and T cell activation | Suppressed excessive T cell response, preventing immunopathology | [70,71,72,73,74] | |
| Organ protection | Lung injury regulation | TRIM27, TRAF6, USP38 | TRIM27 promotes PPARγ degradation via K48 ubiquitination, exacerbating NOX4-mediated oxidative stress; TRAF6/USP38 regulates IL-33R signaling | Enhanced oxidative stress (TRIM27) or fibrosis (TRAF6/USP38) | [19,75] |
| Liver anti-oxidative stress | OTUD1, RNF146 | OTUD1 deubiquitinates NRF2 to activate antioxidant pathways; RNF146 promotes PARP1 degradation via K48 ubiquitination | Reduced hepatic oxidative injury and parthanatos (RNF146) | [76,77,78] | |
| Cardiomyocyte pyroptosis | USP7, SOX9 | USP7 stabilizes SOX9 via deubiquitination, upregulating miR-96-5p and NLRP3 expression | Exacerbated myocardial pyroptosis and dysfunction | [79] | |
| Renal mitophagy activation | PINK1/PARK2 pathway | PARK2-mediated ubiquitination of damaged mitochondria promotes mitophagy and reduces tubular injury | Alleviated acute kidney injury (AKI) via mitochondrial clearance | [80] | |
| Intestinal barrier protection | USP47 | USP47 stabilizes TRAF6 via deubiquitination, enhancing NF-κB-driven intestinal inflammation | Disrupted mucosal barrier and enhanced gut inflammation | [81,82,83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.-Y.; Liu, Y.; Fang, M. The Role of Protein Ubiquitination in the Onset and Progression of Sepsis. Cells 2025, 14, 1012. https://doi.org/10.3390/cells14131012
Chen M-Y, Liu Y, Fang M. The Role of Protein Ubiquitination in the Onset and Progression of Sepsis. Cells. 2025; 14(13):1012. https://doi.org/10.3390/cells14131012
Chicago/Turabian StyleChen, Meng-Yan, Yang Liu, and Min Fang. 2025. "The Role of Protein Ubiquitination in the Onset and Progression of Sepsis" Cells 14, no. 13: 1012. https://doi.org/10.3390/cells14131012
APA StyleChen, M.-Y., Liu, Y., & Fang, M. (2025). The Role of Protein Ubiquitination in the Onset and Progression of Sepsis. Cells, 14(13), 1012. https://doi.org/10.3390/cells14131012