1. Introduction
Global statistics show that colorectal cancer (CRC) is the third most common cancer and is ranked the second most common cause of cancer-related death [
1]. CRC treatments include radical surgery, chemotherapy, radiation therapy, concurrent chemoradiation therapy, and targeted therapy [
2]. In clinical settings, about 20% of patients with CRC are identified as advanced stage, and among them, 40% of patients with CRC display recurrence after the treatment of localized tumors. The prognosis of recurrent or metastatic CRC is not satisfactory, and the five-year survival rate is less than 20% [
3]. The development of new strategies for CRC treatment remains an ongoing issue.
Immunotherapy has brought cancer therapy into a new era, and it has been approved for several cancer types, including CRC [
4]. Two negative regulatory molecules, cytotoxic T lymphocyte-associated protein 4 and programmed cell death protein 1 (PD-1), are expressed on T cells that downregulate the immune response and prevent excessive inflammation. Immune checkpoint inhibitors block negative regulatory signals and restore T cell immune function to restore the anti-tumor effect. Despite the success of different cancer treatments, the response rate to immune checkpoint inhibitors is quite low, ranging from 20% to 40% [
5]. Accumulating evidence suggests that the tumor response to immune checkpoint inhibitors is highly dependent on tumor immunogenicity [
6]. The combination of immunogenic cell death-inducing drugs with immunotherapy may enhance anti-tumor efficacy and expand the benefits of immunotherapy.
The tumor microenvironment (TME) is a complex ecosystem that promotes tumor initiation, progression, invasion, and metastasis [
7]. It contains cancerous and non-cancerous cells, including various immune cell types, cancer-associated fibroblasts, surrounding blood vessels, extracellular matrices, vascular endothelial cells, and pericytes. The reciprocal interaction of tumor and non-tumor cells remodels the environment to support nutrients and provide an excellent space for tumor growth [
7]. Immune cells in the TME play different roles in exerting anti-tumor effects or escaping immunosurveillance effects through innate or adaptive immune responses. Several immune cell types, such as natural killer (NK) cells, dendritic cells (DCs), myeloid-derived suppressor cells, and CD8
+ cytotoxic T cells, function as tumor suppressors by secreting cytokines or chemokines. Another subpopulation of T cells is regulatory T cells (Tregs), which play a suppressive role in immune response and promoting tumor development. Macrophages are classified into two subtypes: those that polarize to M1 macrophages that exhibit anti-tumor activity and those that polarize to M2 macrophages that promote tumor growth [
8,
9].
The intracellular DNA-sensing signal cGAS-STING pathway is a defense system that is activated by the detection of foreign pathogens or bacterial invasion to mediate innate immune functions [
10]. DNA released from the nucleus damaged by chemotherapeutic agents or radiotherapy or self-DNA released from tumor cells is transferred into the cytosol and the formation of micronuclei occurs [
11]. DNA is exposed in the cytosol for cGAS surveillance when the nuclear envelope of the micronuclei ruptures. When cytosolic DNA containing double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA) is exposed, it binds to cGAS and is converted into the secondary messenger cyclic GMP-AMP (cGAMP). cGAMP binds to STING located in the endoplasmic reticulum and is translocated to the Golgi apparatus [
12,
13]. This subsequently recruits and activates TANK-binding kinase 1 (TBK1) and transcription factor IFN regulatory factor 3 (IRF3) and induces the production of type I interferon (IFN) or other cytokines [
14,
15]. This DNA-sensing signaling pathway contributes to tumorigenesis and metastasis by evading immune cell surveillance. Therefore, targeting the cGAS-STING pathway to enhance cancer immunotherapy may be an option for cancer treatment [
16]. However, their therapeutic efficacy in patients with colon cancer remains limited.
Ciprofloxacin (ciproxin) is a broad-spectrum antibiotic belonging to the fluoroquinolone class. Ciprofloxacin exerts antibacterial effects in the treatment of the urinary tract, the respiratory tract, the gastrointestinal tract, pneumonia, skin and soft tissue, sepsis, bone and joint infections, and sexually transmitted infections, and was approved by the U.S. Food and Drug Administration in 1987 [
17]. The mechanisms of action of ciprofloxacin entail the inhibition of the activity of DNA gyrase (topoisomerase II) and interference in bacterial DNA replication and repair processes, ultimately leading to bacterial death [
18]. The topoisomerase II inhibitor teniposide mediates the cGAS-STING axis and induces tumor immunogenicity by inducing the release of high-mobility group box 1 (HMGB1) and type I IFN signaling in tumor cells. Teniposide-treated tumor cells can activate the anti-tumor T cell response and DC maturation both in vitro and in vivo, and potentiate the anti-tumor efficacy of anti-PD1 in mouse multiple-type tumor models [
19].
However, whether ciprofloxacin mediates the cGAS-STING signaling pathway to suppress tumor growth remains unknown. Therefore, in this study, we focus on the effect of ciprofloxacin anti-tumor activity and analyze its potential therapeutic effect in augmenting anti-PD1 activity against colon cancer.
2. Materials and Methods
2.1. Colorectal CT26 Cell Culture
Murine colon adenocarcinoma CT26 cells, purchased from the American Type Culture Collection (Manassas, VA, USA), were maintained in a culture medium containing the Roswell Park Memorial Institute (RPMI)-1640 medium (GIBCO, Grand Island, NY, USA) replenished with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT, USA) and 2 mM L-glutamine (Merck, Darmstadt, Germany). CT26 cells were grown and maintained under specific conditions, i.e., at a temperature of 37 °C and in a humidified 5% CO2 incubator. The cells were maintained in a growth medium till the cells in their log phase reached 80% confluence; then, they were sub-cultured three times per week.
2.2. Cell Viability Assay
The 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay was used to detect the intracellular mitochondrial enzyme activity of cells, in which a formazan product was formed that indicated cell viability. CT26 cells were cultured in 24-well plates and incubated with different concentrations of ciprofloxacin for 24, 48, and 72 h. After this incubation with ciprofloxacin, cells were washed with phosphate-buffered saline (PBS) and placed in the MTT solution for 4 h at 37 °C. The solution was aspirated, and the formazan crystals deposited inside the cells were dissolved using dimethyl sulfoxide for 30 min. The absorbance of the samples was measured at 570/630 nm using an ELISA reader (Molecular Devices, San Jose, CA, USA).
2.3. Morphology Observation
Cells were grown in 6-well culture plates overnight and followed by incubation with 4 and 16 μM of ciprofloxacin for 24, 48, and 72 h. The cells were washed with PBS and Liu’s A solution was added for 45 s, then Liu’s B solution (Muto Pure Chemicals Co., Ltd., Bunkyou-Ku, Tokyo, Japan) was added for 90 s to stain the cells. The BX51 light microscope (Olympus, Tokyo, Japan) was applied to visualize the cell morphology, and the cell morphology was photographed using a CCD camera (SPOT Imaging, Sterling Heights, MI, USA).
2.4. Cell Cycle Analysis
CT26 cells were treated with ciprofloxacin for 24, 48, and 72 h. After treatment, cells were harvested and washed with PBS, and 70% ethanol was used to fix the cells for 10 min. The fixed cells were then washed with cold PBS, 10 mg/mL RNase was used to lyse the RNA, and the cells were stained with 1 mg/mL propidium iodide at 37 °C for 30 min in the dark. The DNA histogram of the cells was analyzed using flow cytometry (BD FACSCalibur, Becton Dickinson, Lincoln Park, NJ, USA). The percentages of G0/G1, S, and G2/M phase acquisition in 104 cells were analyzed using the software ModFit LT (version 4.0). The cell cycle experiments were performed in triplicate.
2.5. Detection of Cytosolic DNA Concentration
Cells treated with ciprofloxacin were washed with PBS, trypsin was used to detach the cells, and the cells were centrifuged at 1200 rpm for 5 min at 4 °C to assemble them. Commercial lysis buffer (Chemicon® Cytoplasmic Lysis Buffer, Merck Millipore, Burlington, MA, USA) was used to solubilize and break up the cells. The disrupted cells were then centrifuged at 1200 rpm for 5 min at 4 °C, and the supernatant containing cytosolic DNA (including mitochondrial DNA) was collected. The spectrophotometer (NanoDrop2000C, Thermo Fisher Scientific Inc., Waltham, MA, USA) was used to detect total and single-stranded DNA contents based on the absorbance at the wavelength of 260 nm. The concentration of dsDNA was determined by subtracting the quantity of ssDNA from that of the total DNA.
2.6. mRNA Expression Analysis
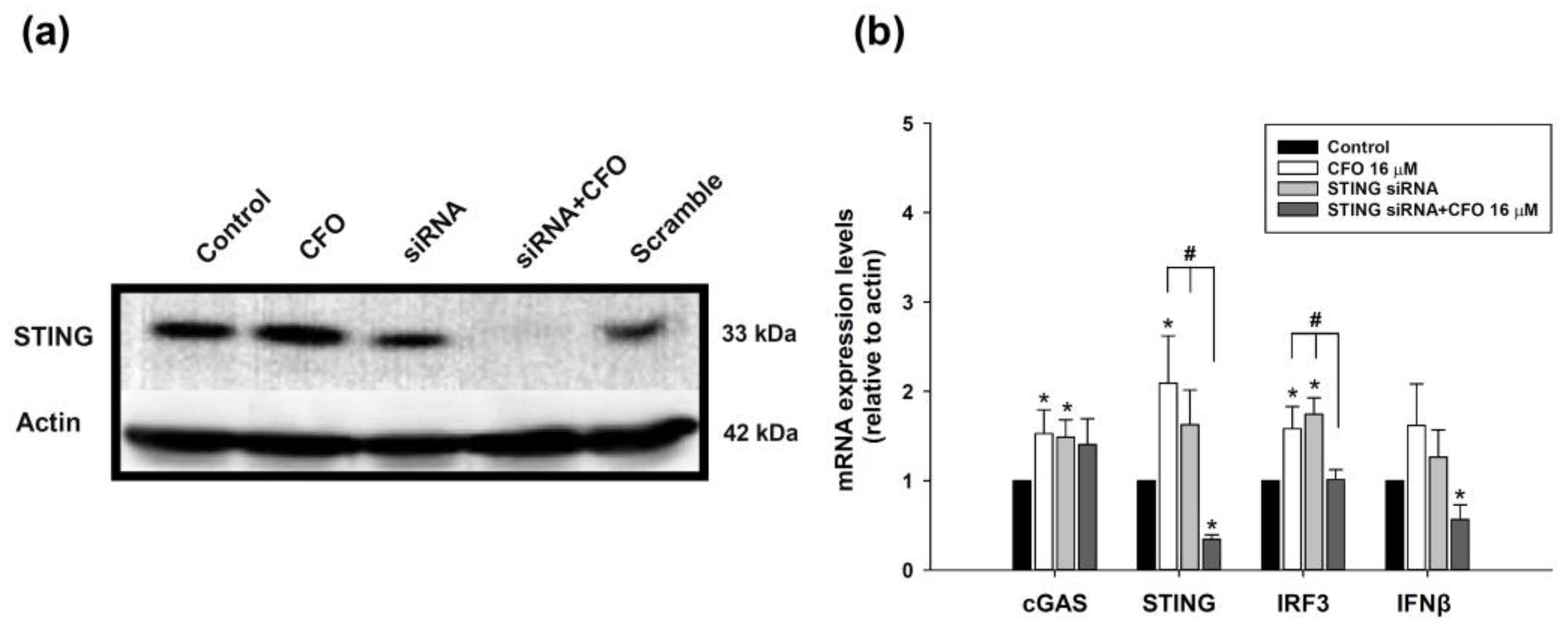
After ciprofloxacin treatment, the medium was removed, and RNAzol® RT reagent (Molecular Research Center, Inc., Cincinnati, OH, USA) was added for 15 min to disrupt cells, and total RNA was isolated from the samples. The RNA concentration was determined by measuring the absorbance at 260 nm using the NanoDrop2000C Spectrophotometer. Subsequently, 1 µg of RNA was used to synthesize cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). The cDNA was then used as the template to perform quantitative reverse transcription PCR (qPCR) using a LightCycler® 96 real-time PCR device (Roche, Penzberg, Upper Bavaria, Germany). qPCR programs were performed as follows: at 95 °C for 3 min for hot start; 40 cycles at 95 °C for 3 s, 65 °C for 30 s, and 72 °C for 3 min, and finally held at 4 °C. Primers used for the cGAS-STING pathway genes were as follows: cGAS, forward 5′-TGAACATGTGAAGATTTCTGCTCC-3′ and reverse 5′-TGACTCAGCGGATTTCCTCG-3′; STING, forward 5’-ACTGCCGCCTCATTGTCTAC-3′ and reverse 5′-ATGGGGGCATTCATGGTA-3′; IRF3, forward 5′-CGGAGGCTTAGCTGACAAAGA-3′ and reverse 5′-ATGCTCTAGCCAGGGGAGGA-3′; IFNβ, forward 5′-CGGAGGCTTAGCTGACAAAGA-3′ and reverse 5′-ATGCTCTAGCCAGGGGAGGA-3′; and actin, forward 5′-GCCAACCGTGAAAAGATGAC-3′ and reverse 5′-GAGGCATACAGGGACAGCAC-3′. The actin expression level was used as an internal control to normalize the expression of other mRNAs in the samples.
2.7. Western Blot
After treatment, cells were lysed with a lysis buffer for 30 min at 4 °C (Cell Signaling Technology, Danvers, MA, USA) to harvest whole-cell lysate and then centrifuged at 15,000× g for 15 min at 4 °C. The supernatants were collected, and the bicinchoninic acid protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA) was used to determine the protein concentrations. The lysates were boiled for 10 min at 100 °C to denature proteins, and an equal mass of denatured proteins was loaded onto sodium dodecyl sulfate–polyacrylamide gels for running, and subsequently, the proteins were transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were soaked with the blocking buffer (containing 5% defatted milk) at room temperature for 1 h with gentle agitation to prevent non-specific binding. Subsequently, the membranes were incubated with primary antibodies against the detected proteins cGAS (GeneTex, Irvine, CA, USA), STING (Cell Signaling Technology, Danvers, MA, USA), IFNβ (Abcam, Cambridge, UK), and actin (GeneTex) at 4 °C overnight. Then, PVDF membranes were incubated with a secondary antibody solution (Jackson ImmunoResearch, West Grove, PA, USA) conjugated with horseradish peroxidase for 1 h at room temperature. The membranes were applied with the enhanced chemiluminescence substrate (GeneTex), and signals were captured using a CCD camera-based image machine, i.e., the Multigel-21 Multi-Function Gel Image System (Top Bio Co., Ltd., Lin Kou, New Taipei City, Taiwan). The band intensity was analyzed using the software ImageJ Version 1.54i, and the internal control level was normalized. β-actin was used as the internal control.
2.8. Syngeneic Tumor Animal Model
Five-week-old male BALB/c mice were obtained from the National Laboratory Animal Center of Taiwan (Taipei, Taiwan) and kept under a standard 12 h/12 h light/dark cycle in a specific pathogen-free facility. The experimental protocols followed the rules and regulations of the Affidavit of Approval of Animal Use Protocol [
20], and were approved by the Institutional Animal Care and Use Committee (IACUC) of MacKay Memorial Hospital (IACUC number: MMH-A-S-106-01). Subsequently, 4 × 10
6 CT26 colon cells in saline were hypodermically implanted into the right hind limbs of the mice, and CT26 colon tumors were allowed to grow to a diameter of approximately 0.5 cm. The mice were randomly assigned to four treatment groups: Group I, control; Group II, anti-mouse PD-1 antibody (200 μg administered through an intraperitoneal injection every other day for a total of three times per mouse) clone RMP1-14 obtained from Bio X cell, Lebanon, NH, USA; Group III, ciprofloxacin (30 mg/kg/d administered through oral gavage for 10 consecutive days); and Group IV, a combination of ciprofloxacin and anti-PD1.
2.9. Isolation of Macrophages from Mouse Spleen
BALB/c mice were euthanized, and their spleen was isolated, ground, and filtered through a 70 μm cell strainer. The specimens were added to HISTOPAQUE 1083 (Sigma-Aldrich, St. Louis, MO, USA) and centrifuged at 2000 rpm for 20 min at room temperature. The mononuclear cell layer was isolated and collected in Hank’s balanced salt solution (HBSS). The cells were washed with HBSS and centrifuged at 1200 rpm for 10 min twice. The cells were cultured in the RPMI-1640 medium containing 10% FBS and 1% penicillin/streptomycin antibiotics in an incubator with 5% CO2 at 37 °C. After 7 d, the adherent cells were stimulated with lipopolysaccharide (100 ng/mL, Sigma-Aldrich) and IFN-γ (20 ng/mL, Bio-Techne, Minneapolis, MN, USA) to allow them to polarize to M1 macrophages.
2.10. Measurement of Tumor Size and Evaluation of Biochemical Toxicity
The tumor size and body weight of each mouse were recorded and calculated every other day. Tumor size was measured with an electronic caliper and calculated using the following formula: 0.5 × L × W2, where L denotes the length and W denotes the width of the tumor. White blood cell count and liver and kidney function were used as indicators of biochemical toxicity after the administration of ciprofloxacin or anti-PD1 antibodies. Blood samples were gathered from the retro-orbital sinus every week. The number of white blood cells in the blood was counted using an automatic Coulter counter (HEMAVET HV950; Drew Scientific Inc., Plantation, FL, USA). The plasma levels of alanine transferase and creatinine in the blood, representing liver and kidney function, were determined with plasma samples loaded on Fuji Dri Chem slides (FUJIFILM Corporation Asaka Technology Development Center, Minamiashigara-shi, Kanagawa, Japan) and detected using an analyzer machine (Fujifilm DryChem NX-500, FUJIFILM Corporation, Tokyo, Japan).
2.11. Flow Cytometry to Detect Immune Cell Expression in Tumor and Spleen
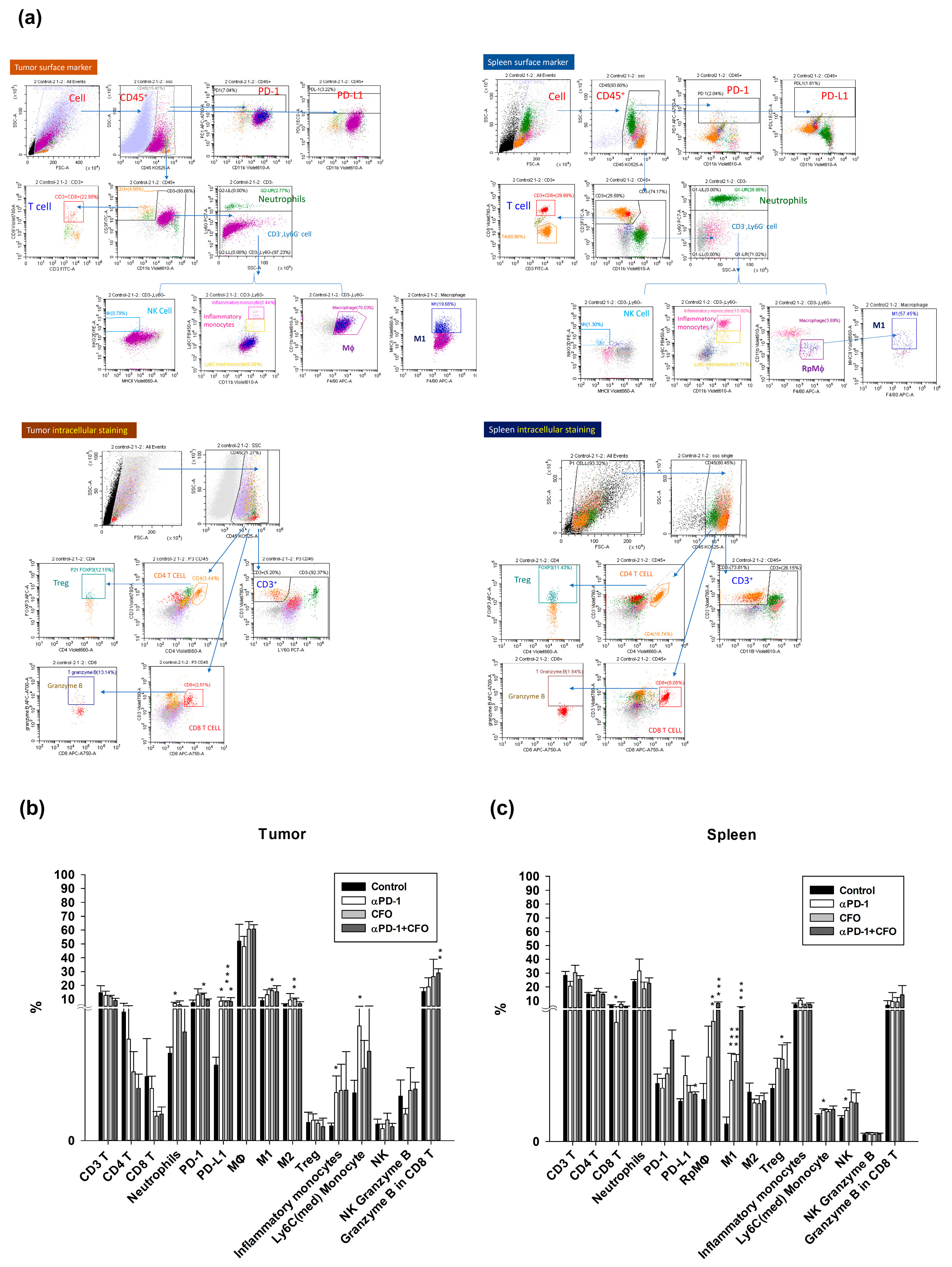
At the end of treatment, animals were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) and euthanatized. The tumor samples were removed, chopped into small species, and digested with DNase I (10 μg/mL) (Sigma-Aldrich) and Liberase TM Research Grade (25 μg/mL) (Roche, Basel, Switzerland) solution for 30 min at 37 °C. The suspension was sieved using a 70 μm cell strainer and then centrifuged at 1200 rpm for 5 min at 4 °C to collect single cells. Spleen tissues were harvested, ground, and filtered using a 120 μm cell strainer. To obtain a single-cell suspension, the samples were centrifuged at 1800 rpm for 5 min at 4 °C. The red blood cells of all samples were lysed using the ammonium–chloride–potassium lysis buffer (Invitrogen, Waltham, MA, USA). Subsequently, cells were incubated with rat IgG (1 μg/1 × 106 cells, Sigma-Aldrich) for 15 min at 37 °C to diminish the non-specific binding before dyeing the immune cells with specific surface markers. Fluorochrome-conjugated mouse antibodies were added and incubated for 30 min at 4 °C. The antibodies conjugated with fluorochromes included the following: anti-CD45-Brilliant Violet 510TM, anti-PD1-APC/Cyanine7, anti-PD-L1-PE/DazzleTM 594, anti-CD3-FITC, anti-CD11b-Brilliant Violet 605TM, anti-Ly6G-PE/Cyanine7, anti-NKG2D-PE, anti-MHCII-Brilliant Violet 650TM, anti-CD11b-Brilliant Violet 605TM, anti-Ly6C-Brilliant Violet 421TM, anti-F4/80-APC, anti-CD8-Brilliant Violet 785TM, anti-granzyme B-Alexa Fluor® 700, and anti-Foxp3-Alexa Fluor® 647 (BioLegend, San Diego, CA, USA). The samples were washed with a staining buffer (2% FBS in PBS), resuspended in the staining buffer, and analyzed using the CytoFLEX 13-color cytometry (Beckman Coulter, Brea, CA, USA). The expression levels of immune cells were quantified using the CytExpert analysis software version 2.3.0.84 (Beckman Coulter, Brea, CA, USA).
2.12. Immunohistochemistry Staining
Tumor specimens were formalin-fixed, paraffin-embedded, and cut into 3 μm thick slides. Samples were deparaffinized with xylene for 15 min and sequentially rehydrated with different concentrations of ethanol. The samples were heat-inactivated in 10 mM sodium citrate buffer (0.05% Tween-20, pH 6.0) for 40 min at 100 °C for antigen retrieval, and then blocked using the BlockPRO blocking buffer (Visual Protein, Taipei, Taiwan) for 30 min at room temperature. Samples were then incubated with primary antibodies against STING (1:100; Cell Signaling Technology, Danvers, MA, USA) for 2 h at room temperature. To exclude non-specific signals, a negative control without the STING antibody was prepared for use. Endogenous peroxidase activity was inactivated by incubation with 3% hydrogen peroxide at room temperature for 10 min. The secondary antibody SignalStain® Boost IHC Detection Reagent (HRP, Rabbit) (Cell Signaling Technology) was used and incubated for 30 min at room temperature. Subsequently, 3,3-diaminobenzidine (DAB) was used as a chromogen for 10 min using the DAB Substrate–Chromogen system (Dako Liquid DAB + Substrate Chromogen System; Agilent Dako, Santa Clara, CA, USA). The slides were washed with PBS and then counterstained with hematoxylin (Merck, Darmstadt, Germany), dehydrated with ethanol, stabilized with xylene, and mounted using a mounting solution (Merck). Cells expressing STING were visualized, and images were captured using a BX51 light microscope. Cells expressing STING were obtained from five different areas and scored as follows: 0, no expression; 1, mild expression; 2, moderate expression; and 3, severe expression, to indicate the different protein expression levels.
2.13. Statistical Analysis
The data are expressed as mean ± standard deviation, and one-way analysis of variance (ANOVA) with post hoc multiple comparison analysis was performed with an LSD test. Statistical analysis shows the significant differences between the control and ciprofloxacin-treated groups: * p < 0.05, ** p < 0.01, and *** p < 0.001. Significant differences between the ciprofloxacin and STING siRNA or STING siRNA plus ciprofloxacin-treated groups are indicated by # p < 0.05. Significant differences between ciprofloxacin plus anti-PD1 and anti-PD1 or ciprofloxacin-treated groups are indicated by # p < 0.05.
4. Discussion
In this study, we validated the ciprofloxacin-induced cGAS-STING signaling pathway activation in CRC. The activation of this DNA-sensing pathway may be associated with enhanced anti-PD1 activity in colon cancer treatment.
The putative mechanism of action of ciprofloxacin involves the disruption of bacterial DNA topoisomerase II and gyrase activity to inhibit DNA replication. This function allows ciprofloxacin to treat Gram-positive and -negative bacterial infections [
17]. However, we found a novel effect of ciprofloxacin in promoting cytosolic DNA formation and the activation of the cGAS-STING signaling pathway. This novel effect may account for the enhanced anti-PD1 effect. Previous studies have reported that ciprofloxacin also inhibits anti-tumor growth in several cancer cell lines, including colorectal carcinoma cells, triple-negative breast cancer, bladder cancer, and prostate cancer [
21,
22,
23,
24,
25]. Ciprofloxacin suppresses DNA synthesis; disrupts mitochondrial membrane potential; upregulates Bax and caspase-3, -8, and -9; and enhances apoptosis in rat and human CRC cells [
21]. Ciprofloxacin induces cell cycle arrest at the S/G2-M phase and the expression of cell cycle-related proteins TP53 and CDKN1 and pro-apoptotic proteins in bladder and prostate cancers [
22,
23,
24]. Similar mechanisms have been demonstrated to elevate p53 expression and trigger apoptosis via the activation of the p53/Bax/Bcl-2 signaling pathway in MDA-MB-231 breast cancer cells after ciprofloxacin treatment [
25]. In our study, we found that cell viability, morphology, and cell cycle were not altered by ciprofloxacin; however, cell proliferation was only slightly inhibited. This discrepancy may be due to the different cell lines and ciprofloxacin concentrations used in the experiments. The concentration of ciprofloxacin in these studies ranged from 50 to 500 μg/mL (approximately 150–1500 μM), which was much higher than that used in our study. Another issue is that ciprofloxacin does not impact the cell cycle but induces cytosolic DNA elevation. This may be the putative mechanism of ciprofloxacin shown to inhibit topoisomerase II activity and cause double-stranded DNA break and DNA damage. The use of higher concentrations of ciprofloxacin to verify tumor growth inhibition, cell cycle arrest, and apoptosis in mouse colorectal CT26 cells should be further investigated.
Antimicrobial fluoroquinolones are widely used in clinical practice. Fluoroquinolones inhibit enzymatic activity to prevent DNA ligation, resulting in single- and double-stranded DNA breaks [
26]. Ciprofloxacin inhibits bacterial DNA gyrase and topoisomerase IV, leading to double-stranded break formation. dsDNA is further processed into single-stranded DNA for binding to the RecA protein and for DNA repair. In mammalian cells, ciprofloxacin interacts with topoisomerase II, leading to double-stranded breaks and ultimately cell death. Some ciprofloxacin derivatives have been reported that target topoisomerase I/II to form single-stranded and double-stranded breaks, thereby inducing tumor cell apoptosis [
27]. The mechanisms of ciprofloxacin increase the amount of cytosolic double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA). In human cells, two isozymes of topoisomerase II, IIα and IIβ, are encoded and involved in cancer development [
28]. Therefore, exploring fluoroquinolones as anticancer drugs that target human topoisomerase II is a reasonable option. Hangas et al. reported that ciprofloxacin inhibited mitochondrial topoisomerase II activity, followed by the silencing of mitochondrial transcription, replication, and aberrant mitochondria, resulting in the disturbed proliferation and differentiation of C2C12 cells [
29]. Ciprofloxacin causes a reduction in mitochondrial DNA (mtDNA) content, energy formation, and calcium homeostasis in leukemia Jurkat cells [
30]. The cytotoxicity of ciprofloxacin is derived from site-specific, double-stranded mtDNA breaks and the loss of mtDNA in tumor cells [
31]. These observations indicate that ciprofloxacin can target mitochondrial DNA and release dsDNA in the cytosol. Another study demonstrated that using plasmid DNA and treating with high concentrations of ciprofloxacin (200–300 μM) inhibited human topoisomerase IIα and IIβ activity and led to DNA breaks but not at low concentrations (3–30 μM [approximately 1–10 μg/mL]), which is expected to be similar to the serum ciprofloxacin levels in patients [
32]. Ciprofloxacin specifically targets bacterial topoisomerases but does not have the same effect on human topoisomerases, which is crucial for its antibacterial activity while minimizing harm to human cells [
32]. Mitochondrial damage by cisplatin induces mitochondrial DNA release into the cytosol, with the subsequent activation of the cGAS-STING signaling pathway [
33]. In our study, ciprofloxacin induced ssDNA and dsDNA formation in the cytosol and elevated the levels of component molecules of the cGAS-STING pathway. However, whether ssDNA and dsDNA originate from mitochondrial or chromatin DNA remains unknown. Mitochondria originated from endosymbiotic Alphaproteobacteria and integrated into eukaryotic cells [
34]. We speculate that ciprofloxacin may cause mitochondrial DNA breaks in CT26 cells by inhibiting topoisomerase II activity by subsequently forming ssDNA and dsDNA, and releasing them into the cytosol, thus further activating the cGAS-STING pathway. This suggests that mitochondrial DNA could act as a damage-associated molecular pattern signal for cGAS recognition and cause an innate immune system response [
35]. However, we cannot rule out the possibility that ciprofloxacin damages the nucleus and releases DNA into the cytosol after treatment with high concentrations of ciprofloxacin.
Topoisomerases relax supercoiled DNA and regulate its topological structure. Topoisomerases are responsible for DNA replication, transcription, genome integrity, and regulation of cell cycle progression. Topoisomerases have been used as targets for the design and development of anticancer drugs that disrupt their activity. Topoisomerase II inhibitors are subdivided into two categories: Topo II catalytic inhibitors and Topo II poisons [
36]. Ciprofloxacin is a Topo II poison inhibitor that increases the covalent complexes of DNA and topoisomerase II, forming double-stranded breaks, and is toxic to bacteria. Other Topo II poisons that have been developed as clinical chemotherapy drugs include doxorubicin, etoposide, and teniposide [
36]. Teniposide has been reported to induce the expression of the MC38 and CT26 tumor cell immunogenic cell death marker HMGB1, T cell activation, and DC maturation. Teniposide induces tumor immunogenicity by activating NF-κB and STING-dependent type I IFN production [
19]. Ultrasound-responsive low-dose doxorubicin liposomes enhance doxorubicin release into the tumor cell nucleus and induce tumor mitochondrial oxidation. The released tumor mitochondria DNA triggers the cGAS-STING pathway activation and enhances T cell immunity [
37]. Another report demonstrated that ciprofloxacin causes nuclear and mitochondrial DNA release into the cytosol, mitochondrial damage, the formation of reactive oxygen species, and STING protein activation in the mouse aorta [
38]. These results imply that topoisomerase II inhibitors, including ciprofloxacin, can cause harmful nuclear or mitochondrial DNA release into the cytosol for DNA-sensing signaling and cGAS-STING pathway activation.
However, the safety of using ciprofloxacin is a concern. The most common adverse effects of fluoroquinolones, including ciprofloxacin, are gastrointestinal disorders. Other severe side effects include tendinopathy, peripheral neuropathy, QTc prolongation, cardiac arrhythmias, neuropsychiatric disorders, aortic aneurysms, and aortic dissection [
17,
39]. Clinically relevant studies have shown that adverse effects are mild after oral administration. In our in vivo study, ciprofloxacin did not show any biological toxicity, including effects on body weight, liver and renal function, or hematological profiles. The results imply that, in our system, low-dose ciprofloxacin is harmless compared to orally administered ciprofloxacin twice a day for 1–2 weeks at doses of 250–750 mg, according to the severity of infections in the clinic. Furthermore, a previous report showed that patients with cancer who were orally administered 750 mg of ciprofloxacin every 8 h experienced mild side effects, mostly gastrointestinal disorders [
40]. Ciprofloxacin is effective and safe for treating infections in patients with cancer [
40]. However, toxicities and side effects should be monitored after a clinical dose of ciprofloxacin is administered in in vivo anti-tumor studies in the future.
The topoisomerase II inhibitor, teniposide, has been reported to enhance anti-PD1 anti-tumor efficacy in mouse CRC. Teniposide-potentiated immune checkpoint blocker anti-tumor immunity is associated with intratumor STING activation [
19]. These results are consistent with our results showing that ciprofloxacin potentiates the anti-PD1 anti-tumor effect in a CT26 syngeneic animal model. Tumor specimens also showed the highest expression of STING protein with the combination therapy. Ciprofloxacin exerts a synergistic effect on tumor growth suppression in combination with immunotherapy. Another study showed that the topoisomerase II inhibitor idarubicin could sensitize anti-PD1 immunotherapy by CD8
+ T cells and activate the TME immune status in hepatocellular carcinoma [
41]. However, a retrospective cohort study showed that concurrent treatment with antibiotics and immune checkpoint inhibitors was associated with higher mortality in patients with hepatocellular carcinoma [
42]. Broad-spectrum antibiotics have demonstrated negative outcomes with immune checkpoint inhibitor treatment in patients with cancer in clinical settings. These results shed light on how antibiotics modulate the gut microbiota and induce dysbiosis to influence immunotherapy efficacy [
43]. The gut microbiome plays an important role in immune function and is a potential target of immunotherapy. Antibiotics disrupt the microbiota balance and hinder the efficacy of immunotherapy. Retrospect studies demonstrated that antibiotic administration is inconsistent. However, whether antibiotics are beneficial for immunotherapy remains unclear. The results remain controversial because the dosage, duration, and types of antibiotic treatment should be considered when patients with cancer receive immunotherapy. The favorable outcomes of the combination of ciprofloxacin and immunotherapy should be investigated in more detail. More research is needed to understand the specific mechanisms by which ciprofloxacin modulates the immunotherapy response and develop a new regimen for patients to enhance immunotherapy efficacy after ciprofloxacin use. Next-generation sequencing technology such as 16S rRNA sequencing and metagenomics, dietary interventions, microbiota depletion models, or fecal microbiota transplantation methods could be used to analyze or modulate the gut microbiota and to verify the mechanisms of concurrent ciprofloxacin and immunotherapy treatment that influence the gut microbiota in the future.
Many studies have shown that ciprofloxacin exerts chemosensitizing effects. The combination of ciprofloxacin with chemotherapeutic drugs such as doxorubicin and docetaxel enhance hormone-refractory prostate cancer cell growth inhibition [
44]. Ciprofloxacin reverses multidrug resistance and enhances tumor cell-sensitized ABCB1 substrates to maintain chemo-drug concentration in cells [
45]. Geller et al. found that ciprofloxacin abrogates the bacteria-induced conversion of the chemo-drug gemcitabine into its inactive form to resist tumor effects [
46]. This indicates that ciprofloxacin has the potential to be developed as a combination therapy with other clinical chemotherapeutic drugs to improve cancer treatment.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}