
The Interplay Between Pulmonary Hypertension and Atrial Fibrillation: A Comprehensive Overview



Abstract
1. Introduction
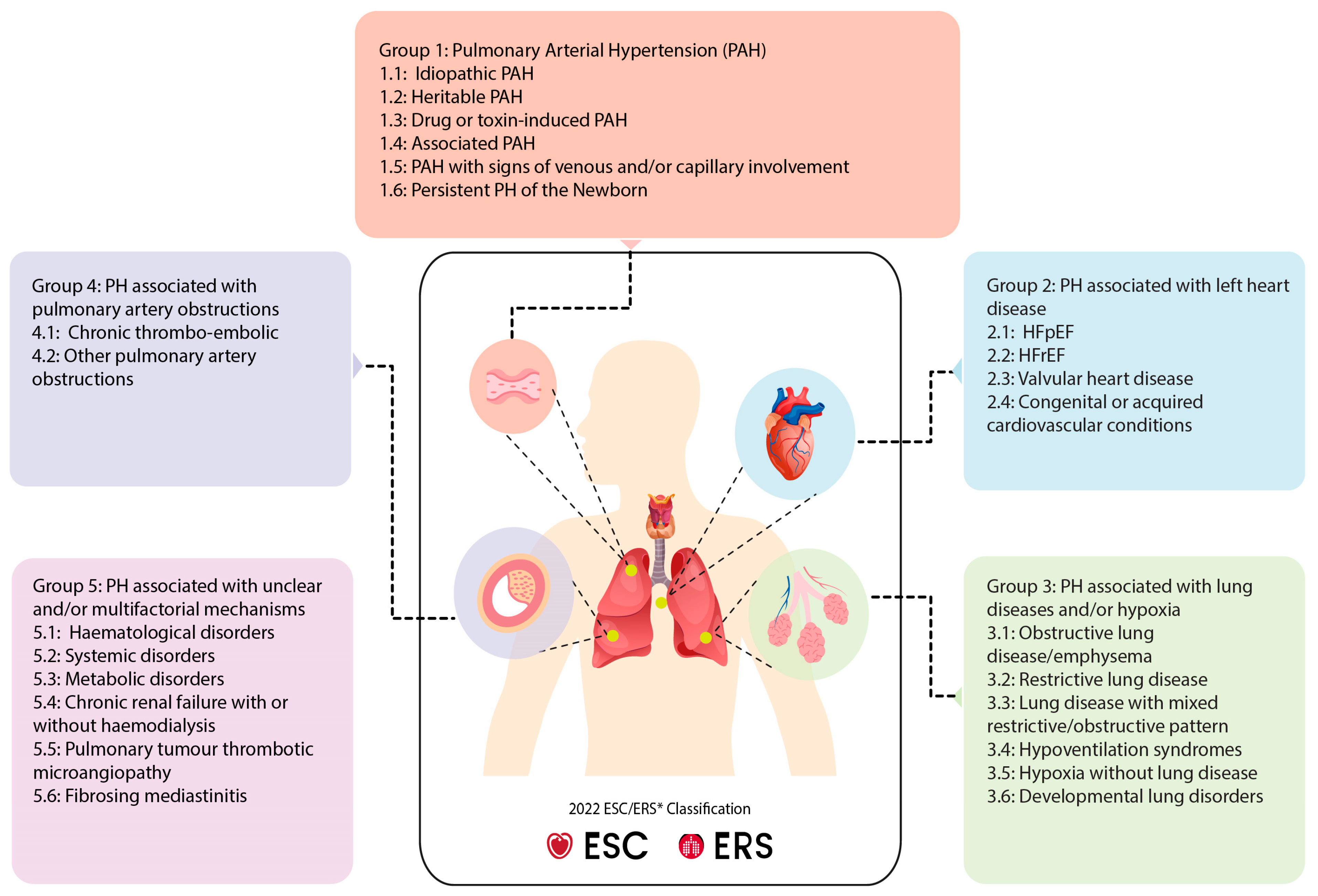
2. Clinical Classification of PH
3. Epidemiology and Prevalence of AF in PH
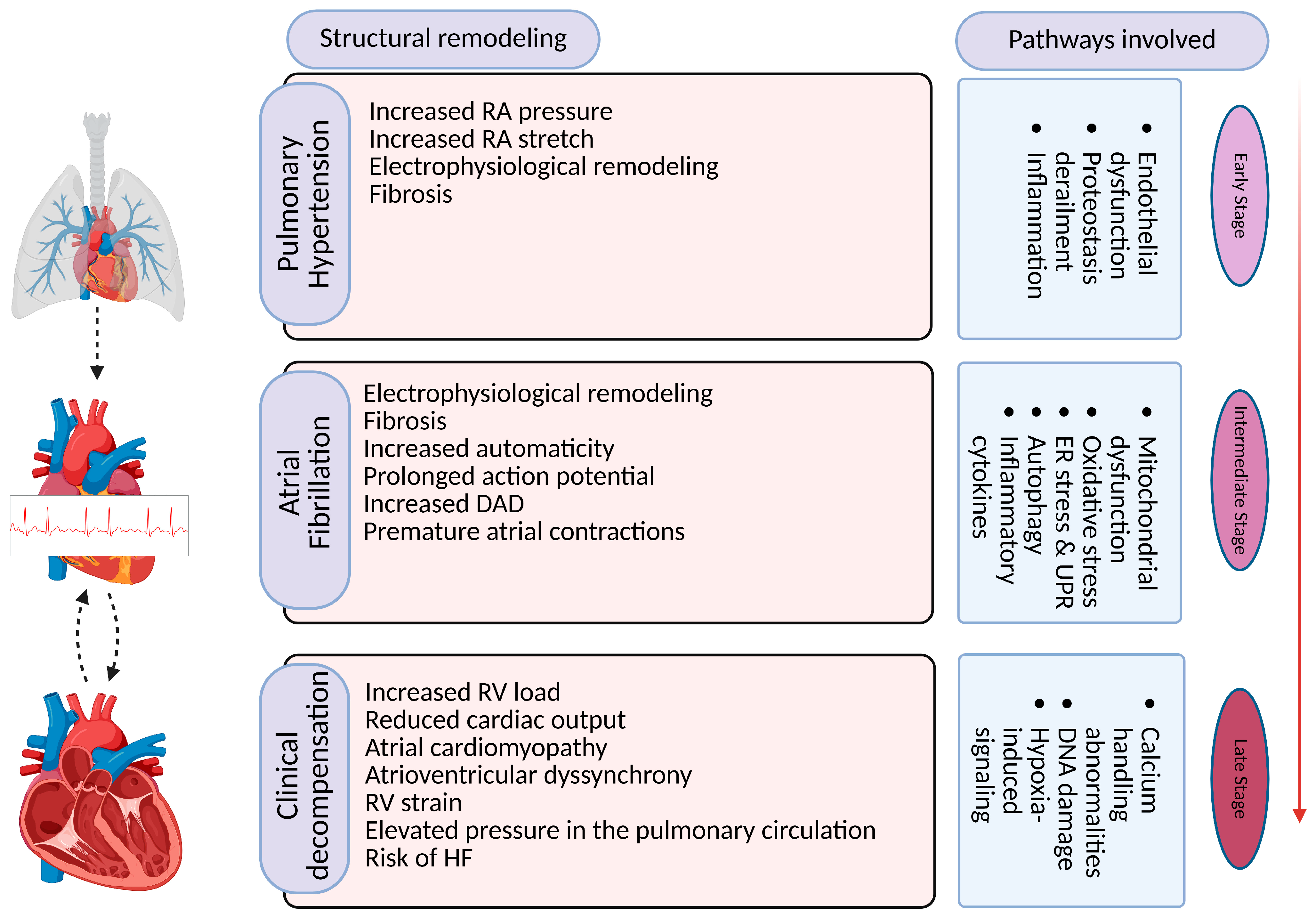
4. Molecular Mechanisms Underlying AF Development in PH
4.1. Role of Heat Shock Proteins
4.2. Oxidative Stress and DNA Damage in PH and AF
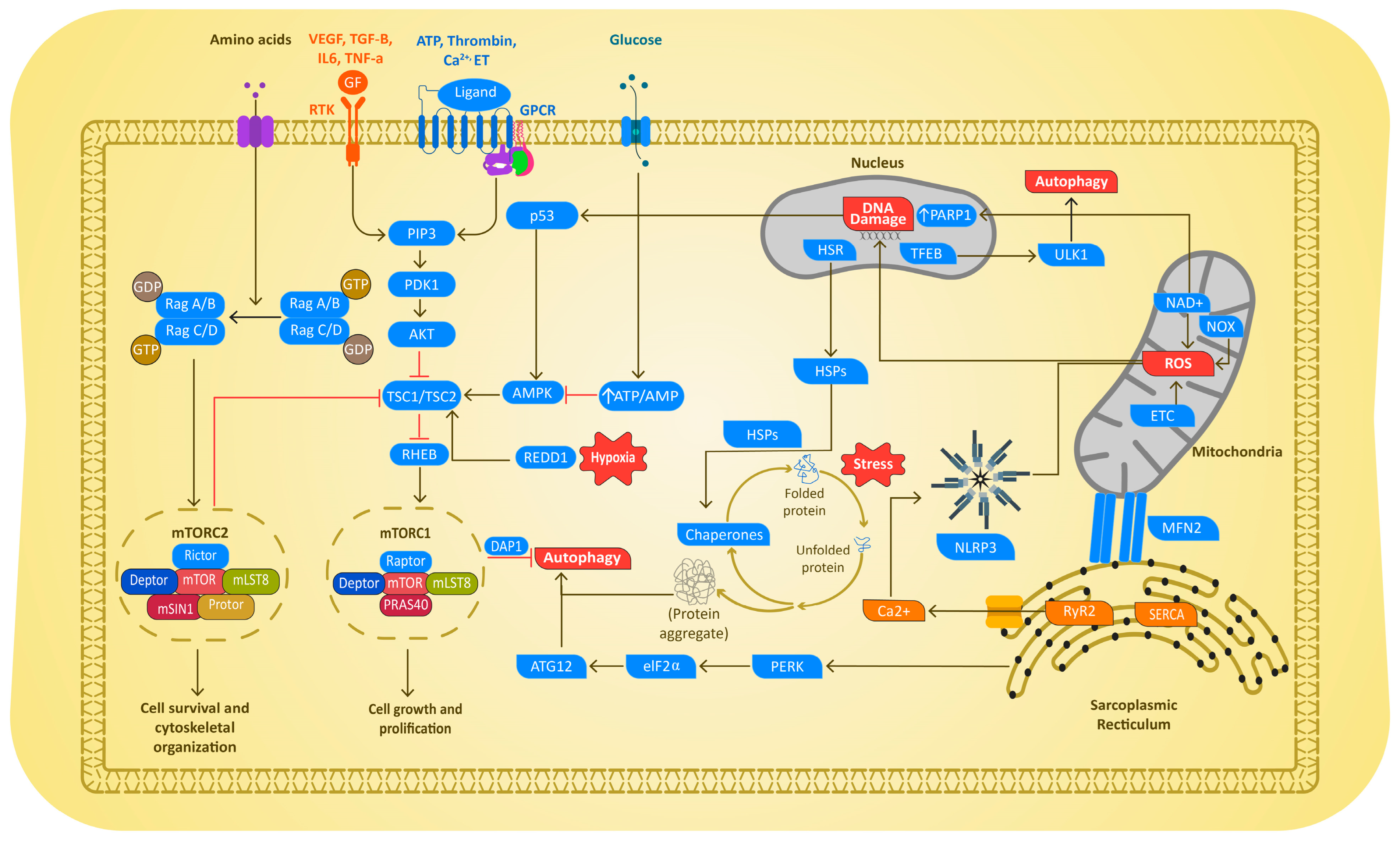
4.3. Autophagic Protein Degradation
4.4. Thromboembolism and Endothelial Dysfunction
4.5. Inflammation
5. Novel Druggable Targets
6. Knowledge Gaps
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kurakula, K.; Smolders, V.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Corris, P.A.; Seeger, W. Call it by the correct name-pulmonary hypertension not pulmonary arterial hypertension: Growing recognition of the global health impact for a well-recognized condition and the role of the Pulmonary Vascular Research Institute. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L992–L994. [Google Scholar] [CrossRef]
- Wanamaker, B.; Cascino, T.; McLaughlin, V.; Oral, H.; Latchamsetty, R.; Siontis, K.C. Atrial Arrhythmias in Pulmonary Hypertension: Pathogenesis, Prognosis and Management. Arrhythm. Electrophysiol. Rev. 2018, 7, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Rottlaender, D.; Motloch, L.J.; Schmidt, D.; Reda, S.; Larbig, R.; Wolny, M.; Dumitrescu, D.; Rosenkranz, S.; Erdmann, E.; Hoppe, U.C. Clinical impact of atrial fibrillation in patients with pulmonary hypertension. PLoS ONE 2012, 7, e33902. [Google Scholar] [CrossRef]
- Sammut, M.A.; Condliffe, R.; Elliot, C.; Hameed, A.; Lewis, R.; Kiely, D.G.; Kyriacou, A.; Middleton, J.T.; Raithatha, A.; Rothman, A.; et al. Atrial flutter and fibrillation in patients with pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension in the ASPIRE registry: Comparison of rate versus rhythm control approaches. Int. J. Cardiol. 2023, 371, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Tongers, J.; Schwerdtfeger, B.; Klein, G.; Kempf, T.; Schaefer, A.; Knapp, J.M.; Niehaus, M.; Korte, T.; Hoeper, M.M. Incidence and clinical relevance of supraventricular tachyarrhythmias in pulmonary hypertension. Am. Heart J. 2007, 153, 127–132. [Google Scholar] [CrossRef]
- Ruiz-Cano, M.J.; Gonzalez-Mansilla, A.; Escribano, P.; Delgado, J.; Arribas, F.; Torres, J.; Flox, A.; Riva, M.; Gomez, M.A.; Saenz, C. Clinical implications of supraventricular arrhythmias in patients with severe pulmonary arterial hypertension. Int. J. Cardiol. 2011, 146, 105–106. [Google Scholar] [CrossRef]
- Olsson, K.M.; Nickel, N.P.; Tongers, J.; Hoeper, M.M. Atrial flutter and fibrillation in patients with pulmonary hypertension. Int. J. Cardiol. 2013, 167, 2300–2305. [Google Scholar] [CrossRef]
- Cannillo, M.; Grosso Marra, W.; Gili, S.; D’Ascenzo, F.; Morello, M.; Mercante, L.; Mistretta, E.; Salera, D.; Zema, D.; Bissolino, A.; et al. Supraventricular Arrhythmias in Patients With Pulmonary Arterial Hypertension. Am. J. Cardiol. 2015, 116, 1883–1889. [Google Scholar] [CrossRef]
- Wen, L.; Sun, M.L.; An, P.; Jiang, X.; Sun, K.; Zheng, L.; Liu, Q.Q.; Wang, L.; Zhao, Q.H.; He, J.; et al. Frequency of supraventricular arrhythmias in patients with idiopathic pulmonary arterial hypertension. Am. J. Cardiol. 2014, 114, 1420–1425. [Google Scholar] [CrossRef]
- Witte, C.; Meyer Zur Heide Genannt Meyer-Arend, J.U.; Andrié, R.; Schrickel, J.W.; Hammerstingl, C.; Schwab, J.O.; Nickenig, G.; Skowasch, D.; Pizarro, C. Heart Rate Variability and Arrhythmic Burden in Pulmonary Hypertension. Adv. Exp. Med. Biol. 2016, 934, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Genuardi, M.V.; Koczo, A.; Zou, R.H.; Thoma, F.W.; Handen, A.; Craig, E.; Hogan, C.M.; Girard, T.; Althouse, A.D.; et al. Atrial arrhythmias are associated with increased mortality in pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045894018790316. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, V.; Peloquin, G.; Bourji, K.I.; Diab, N.; Sato, T.; Enobun, B.; Housten-Harris, T.; Damico, R.; Kolb, T.M.; Mathai, S.C.; et al. Pulmonary arterial hypertension and atrial arrhythmias: Incidence, risk factors, and clinical impact. Pulm. Circ. 2018, 8, 2045894018769874. [Google Scholar] [CrossRef] [PubMed]
- Fingrova, Z.; Ambroz, D.; Jansa, P.; Kuchar, J.; Lindner, J.; Kunstyr, J.; Aschermann, M.; Linhart, A.; Havranek, S. The prevalence and clinical outcome of supraventricular tachycardia in different etiologies of pulmonary hypertension. PLoS ONE 2021, 16, e0245752. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Pausch, C.; Grünig, E.; Klose, H.; Staehler, G.; Huscher, D.; Pittrow, D.; Olsson, K.M.; Vizza, C.D.; Gall, H.; et al. Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. J. Heart Lung Transplant. 2020, 39, 1435–1444. [Google Scholar] [CrossRef]
- Hansdottir, S.; Groskreutz, D.J.; Gehlbach, B.K. WHO’s in second?: A practical review of World Health Organization group 2 pulmonary hypertension. Chest 2013, 144, 638–650. [Google Scholar] [CrossRef]
- Al-Omary, M.S.; Sugito, S.; Boyle, A.J.; Sverdlov, A.L.; Collins, N.J. Pulmonary Hypertension Due to Left Heart Disease: Diagnosis, Pathophysiology, and Therapy. Hypertension 2020, 75, 1397–1408. [Google Scholar] [CrossRef]
- Gopinathannair, R.; Chen, L.Y.; Chung, M.K.; Cornwell, W.K.; Furie, K.L.; Lakkireddy, D.R.; Marrouche, N.F.; Natale, A.; Olshansky, B.; Joglar, J.A. Managing Atrial Fibrillation in Patients With Heart Failure and Reduced Ejection Fraction: A Scientific Statement From the American Heart Association. Circ. Arrhythm. Electrophysiol. 2021, 14, e000078. [Google Scholar] [CrossRef]
- Rådegran, G.; Kjellström, B.; Ekmehag, B.; Larsen, F.; Rundqvist, B.; Blomquist, S.B.; Gustafsson, C.; Hesselstrand, R.; Karlsson, M.; Kornhall, B.; et al. Characteristics and survival of adult Swedish PAH and CTEPH patients 2000–2014. Scand. Cardiovasc. J. 2016, 50, 243–250. [Google Scholar] [CrossRef]
- Brundel, B.; Ai, X.; Hills, M.T.; Kuipers, M.F.; Lip, G.Y.H.; de Groot, N.M.S. Atrial fibrillation. Nat. Rev. Dis. Primers 2022, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Heidbuchel, H.; Dagres, N.; Antz, M.; Kuck, K.H.; Lazure, P.; Murray, S.; Carrera, C.; Hindricks, G.; Vahanian, A. Major knowledge gaps and system barriers to guideline implementation among European physicians treating patients with atrial fibrillation: A European Society of Cardiology international educational needs assessment. Europace 2018, 20, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.G.; Deyell, M.W.; Bennett, R.; Macle, L. Assessment and management of asymptomatic atrial fibrillation. Heart 2024, 110, 675–682. [Google Scholar] [CrossRef]
- Kotecha, D.; Piccini, J.P. Atrial fibrillation in heart failure: What should we do? Eur. Heart J. 2015, 36, 3250–3257. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Borlaug, B.A. Pulmonary hypertension due to left heart disease. Circulation 2012, 126, 975–990. [Google Scholar] [CrossRef]
- Li, N.; Brundel, B. Inflammasomes and Proteostasis Novel Molecular Mechanisms Associated With Atrial Fibrillation. Circ. Res. 2020, 127, 73–90. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Li, J.; Liu, J.; Baks-Te Bulte, L.; Wiersma, M.; Malik, N.U.; van Marion, D.M.S.; Tolouee, M.; Hoogstra-Berends, F.; et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD+ depletion in experimental atrial fibrillation. Nat. Commun. 2019, 10, 1307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, C.T.; Qi, X.; Meijering, R.A.; Hoogstra-Berends, F.; Tadevosyan, A.; Cubukcuoglu Deniz, G.; Durdu, S.; Akar, A.R.; Sibon, O.C.; et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef]
- van Wijk, S.W.; Ramos, K.S.; Brundel, B. Cardioprotective Role of Heat Shock Proteins in Atrial Fibrillation: From Mechanism of Action to Therapeutic and Diagnostic Target. Int. J. Mol. Sci. 2021, 22, 442. [Google Scholar] [CrossRef]
- Liu, D.; Li, Y.; Zhao, Q. Effects of Inflammatory Cell Death Caused by Catheter Ablation on Atrial Fibrillation. J. Inflamm. Res. 2023, 16, 3491–3508. [Google Scholar] [CrossRef]
- Gomez-Puerto, M.C.; van Zuijen, I.; Huang, C.J.; Szulcek, R.; Pan, X.; van Dinther, M.A.; Kurakula, K.; Wiesmeijer, C.C.; Goumans, M.J.; Bogaard, H.J.; et al. Autophagy contributes to BMP type 2 receptor degradation and development of pulmonary arterial hypertension. J. Pathol. 2019, 249, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.L.; Szulcek, R.; Bourgeois, A.; Lampron, M.C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Aldred, M.A. DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes. 2020, 11, 1224. [Google Scholar] [CrossRef]
- Kurakula, K.; Hagdorn, Q.A.J.; van der Feen, D.E.; Vonk Noordegraaf, A.; Ten Dijke, P.; de Boer, R.A.; Bogaard, H.J.; Goumans, M.J.; Berger, R.M.F. Inhibition of the prolyl isomerase Pin1 improves endothelial function and attenuates vascular remodelling in pulmonary hypertension by inhibiting TGF-beta signalling. Angiogenesis 2022, 25, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26, 170094. [Google Scholar] [CrossRef]
- Zakynthinos, G.E.; Tsolaki, V.; Oikonomou, E.; Vavouranakis, M.; Siasos, G.; Zakynthinos, E. Metabolic Syndrome and Atrial Fibrillation: Different Entities or Combined Disorders. J. Pers. Med. 2023, 13, 1323. [Google Scholar] [CrossRef]
- Ranchoux, B.; Nadeau, V.; Bourgeois, A.; Provencher, S.; Tremblay, É.; Omura, J.; Coté, N.; Abu-Alhayja’a, R.; Dumais, V.; Nachbar, R.T.; et al. Metabolic Syndrome Exacerbates Pulmonary Hypertension due to Left Heart Disease. Circ. Res. 2019, 125, 449–466. [Google Scholar] [CrossRef]
- Havranek, S.; Fingrova, Z.; Skala, T.; Reichenbach, A.; Dusik, M.; Jansa, P.; Ambroz, D.; Dytrych, V.; Klimes, D.; Hutyra, M.; et al. Catheter ablation of atrial fibrillation and atrial tachycardia in patients with pulmonary hypertension: A randomized study. Europace 2023, 25, euad131. [Google Scholar] [CrossRef]
- Medi, C.; Kalman, J.M.; Ling, L.H.; Teh, A.W.; Lee, G.; Lee, G.; Spence, S.J.; Kaye, D.M.; Kistler, P.M. Atrial electrical and structural remodeling associated with longstanding pulmonary hypertension and right ventricular hypertrophy in humans. J. Cardiovasc. Electrophysiol. 2012, 23, 614–620. [Google Scholar] [CrossRef]
- Sanders, P.; Morton, J.B.; Davidson, N.C.; Spence, S.J.; Vohra, J.K.; Sparks, P.B.; Kalman, J.M. Electrical remodeling of the atria in congestive heart failure: Electrophysiological and electroanatomic mapping in humans. Circulation 2003, 108, 1461–1468. [Google Scholar] [CrossRef]
- Hiram, R.; Provencher, S. Pulmonary Disease, Pulmonary Hypertension and Atrial Fibrillation. Card. Electrophysiol. Clin. 2021, 13, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Archer, S.L.; Dorfmüller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [PubMed]
- Sakao, S.; Tatsumi, K. Vascular remodeling in pulmonary arterial hypertension: Multiple cancer-like pathways and possible treatment modalities. Int. J. Cardiol. 2011, 147, 4–12. [Google Scholar] [CrossRef]
- Wang, G.K.; Li, S.H.; Zhao, Z.M.; Liu, S.X.; Zhang, G.X.; Yang, F.; Wang, Y.; Wu, F.; Zhao, X.X.; Xu, Z.Y. Inhibition of heat shock protein 90 improves pulmonary arteriole remodeling in pulmonary arterial hypertension. Oncotarget 2016, 7, 54263–54273. [Google Scholar] [CrossRef]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 Accumulation Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103. [Google Scholar] [CrossRef]
- Rodriguez-Iturbe, B.; Johnson, R.J.; Sanchez-Lozada, L.G.; Pons, H. HSP70 and Primary Arterial Hypertension. Biomolecules 2023, 13, 272. [Google Scholar] [CrossRef]
- Salibe-Filho, W.; Araujo, T.L.; GMelo, E.; BCTCoimbra, L.; Lapa, M.S.; Acencio, M.M.; Freitas-Filho, O.; Capelozzi, V.L.; Teixeira, L.R.; Fernandes, C.J.; et al. Shear stress-exposed pulmonary artery endothelial cells fail to upregulate HSP70 in chronic thromboembolic pulmonary hypertension. PLoS ONE 2020, 15, e0242960. [Google Scholar] [CrossRef]
- Cao, J.; Yang, L.; Wang, L.; Zhao, Q.; Wu, D.; Li, M.; Mu, Y. Heat shock protein 70 attenuates hypoxia-induced apoptosis of pulmonary microvascular endothelial cells isolated from neonatal rats. Mol. Med. Rep. 2021, 24, 690. [Google Scholar] [CrossRef]
- Brundel, B.J.; Shiroshita-Takeshita, A.; Qi, X.; Yeh, Y.H.; Chartier, D.; van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Nattel, S. Induction of heat shock response protects the heart against atrial fibrillation. Circ. Res. 2006, 99, 1394–1402. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; van Marion, D.M.S.; Zhang, D.; Brundel, B. Heat shock protein inducer GGA*-59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 134, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.; Henning, R.H.; Ke, L.; van Gelder, I.C.; Crijns, H.J.; Kampinga, H.H. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 2006, 41, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Yeh, H.I.; Tsao, H.M.; Tai, C.T.; Lin, Y.J.; Chang, S.L.; Lo, L.W.; Tuan, T.C.; Suenari, K.; Li, C.H.; et al. Electrophysiological correlation and prognostic impact of heat shock protein 27 in atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2012, 5, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Mandal, K.; Torsney, E.; Poloniecki, J.; Camm, A.J.; Xu, Q.; Jahangiri, M. Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann. Thorac. Surg. 2005, 79, 865–871; discussion 871. [Google Scholar] [CrossRef]
- St Rammos, K.; Koullias, G.J.; Hassan, M.O.; Argyrakis, N.P.; Voucharas, C.G.; Scarupa, S.J.; Cowte, T.G. Low preoperative HSP70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovasc. Surg. 2002, 10, 228–232. [Google Scholar] [CrossRef]
- Cao, H.; Xue, L.; Xu, X.; Wu, Y.; Zhu, J.; Chen, L.; Chen, D.; Chen, Y. Heat shock proteins in stabilization of spontaneously restored sinus rhythm in permanent atrial fibrillation patients after mitral valve surgery. Cell Stress Chaperones 2011, 16, 517–528. [Google Scholar] [CrossRef]
- Federici, C.; Drake, K.M.; Rigelsky, C.M.; McNelly, L.N.; Meade, S.L.; Comhair, S.A.; Erzurum, S.C.; Aldred, M.A. Increased Mutagen Sensitivity and DNA Damage in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 219–228. [Google Scholar] [CrossRef]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef]
- Stam, K.; Cai, Z.; van der Velde, N.; van Duin, R.; Lam, E.; van der Velden, J.; Hirsch, A.; Duncker, D.J.; Merkus, D. Cardiac remodelling in a swine model of chronic thromboembolic pulmonary hypertension: Comparison of right vs. left ventricle. J. Physiol. 2019, 597, 4465–4480. [Google Scholar] [CrossRef]
- Wang, E.L.; Jia, M.M.; Luo, F.M.; Li, T.; Peng, J.J.; Luo, X.J.; Song, F.L.; Yang, J.F.; Peng, J.; Liu, B. Coordination between NADPH oxidase and vascular peroxidase 1 promotes dysfunctions of endothelial progenitor cells in hypoxia-induced pulmonary hypertensive rats. Eur. J. Pharmacol. 2019, 857, 172459. [Google Scholar] [CrossRef]
- Agarwal, S.; Sharma, H.; Chen, L.; Dhillon, N.K. NADPH oxidase-mediated endothelial injury in HIV- and opioid-induced pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1097–L1108. [Google Scholar] [CrossRef]
- Xu, D.; Hu, Y.H.; Gou, X.; Li, F.Y.; Yang, X.Y.; Li, Y.M.; Chen, F. Oxidative Stress and Antioxidative Therapy in Pulmonary Arterial Hypertension. Molecules 2022, 27, 3724. [Google Scholar] [CrossRef]
- Wu, J.; Pan, W.; Wang, C.; Dong, H.; Xing, L.; Hou, J.; Fang, S.; Li, H.; Yang, F.; Yu, B. H(2)S attenuates endoplasmic reticulum stress in hypoxia-induced pulmonary artery hypertension. Biosci. Rep. 2019, 39, 304. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Senchanthisai, S.; Sowden, M.; Pang, J.; White, R.J.; Berk, B.C. Endothelial-to-Mesenchymal Transition and Inflammation Play Key Roles in Cyclophilin A-Induced Pulmonary Arterial Hypertension. Hypertension 2020, 76, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R. Signaling Pathways Involved in the Development of Bronchopulmonary Dysplasia and Pulmonary Hypertension. Children 2020, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Poyatos, P.; Gratacós, M.; Samuel, K.; Orriols, R.; Tura-Ceide, O. Oxidative Stress and Antioxidant Therapy in Pulmonary Hypertension. Antioxidants 2023, 12, 1006. [Google Scholar] [CrossRef]
- Huetsch, J.C.; Suresh, K.; Shimoda, L.A. Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension. Antioxidants 2019, 8, 56. [Google Scholar] [CrossRef]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A.; Honeycutt, C.; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar] [CrossRef]
- Nabeebaccus, A.A.; Reumiller, C.M.; Shen, J.; Zoccarato, A.; Santos, C.X.C.; Shah, A.M. The regulation of cardiac intermediary metabolism by NADPH oxidases. Cardiovasc. Res. 2023, 118, 3305–3319. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. Association of atrial nicotinamide adenine dinucleotide phosphate oxidase activity with the development of atrial fibrillation after cardiac surgery. J. Am. Coll. Cardiol. 2008, 51, 68–74. [Google Scholar] [CrossRef]
- Chen, W.J.; Chang, S.H.; Chan, Y.H.; Lee, J.L.; Lai, Y.J.; Chang, G.J.; Tsai, F.C.; Yeh, Y.H. Tachycardia-induced CD44/NOX4 signaling is involved in the development of atrial remodeling. J. Mol. Cell. Cardiol. 2019, 135, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Youn, J.Y.; Kim, A.Y.; Ramirez, R.J.; Gao, L.; Ngo, D.; Chen, P.; Scovotti, J.; Mahajan, A.; Cai, H. NOX4-Dependent Hydrogen Peroxide Overproduction in Human Atrial Fibrillation and HL-1 Atrial Cells: Relationship to Hypertension. Front. Physiol. 2012, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Mondragón, R.; Lozhkin, A.; Vendrov, A.E.; Runge, M.S.; Isom, L.L.; Madamanchi, N.R. NADPH Oxidases and Oxidative Stress in the Pathogenesis of Atrial Fibrillation. Antioxidants 2023, 12, 1833. [Google Scholar] [CrossRef]
- Ramos, K.S.; Brundel, B. DNA Damage, an Innocent Bystander in Atrial Fibrillation and Other Cardiovascular Diseases? Front. Cardiovasc. Med. 2020, 7, 67. [Google Scholar] [CrossRef]
- Newman, J.H.; Wheeler, L.; Lane, K.B.; Loyd, E.; Gaddipati, R.; Phillips, J.A., 3rd; Loyd, J.E. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N. Engl. J. Med. 2001, 345, 319–324. [Google Scholar] [CrossRef]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Trégouët, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef]
- Drake, K.M.; Comhair, S.A.; Erzurum, S.C.; Tuder, R.M.; Aldred, M.A. Endothelial chromosome 13 deletion in congenital heart disease-associated pulmonary arterial hypertension dysregulates SMAD9 signaling. Am. J. Respir. Crit. Care Med. 2015, 191, 850–854. [Google Scholar] [CrossRef]
- Caron, M.C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.P.; Langelier, M.F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Lampron, M.C.; Vitry, G.; Nadeau, V.; Grobs, Y.; Paradis, R.; Samson, N.; Tremblay, È.; Boucherat, O.; Meloche, J.; Bonnet, S.; et al. PIM1 (Moloney Murine Leukemia Provirus Integration Site) Inhibition Decreases the Nonhomologous End-Joining DNA Damage Repair Signaling Pathway in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Fessel, J.P.; West, J.D. Redox biology in pulmonary arterial hypertension (2013 Grover Conference Series). Pulm. Circ. 2015, 5, 599–609. [Google Scholar] [CrossRef]
- Pool, L.; Wijdeveld, L.; de Groot, N.M.S.; Brundel, B. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. Int. J. Mol. Sci. 2021, 22, 8463. [Google Scholar] [CrossRef]
- Courboulin, A.; Paulin, R.; Giguère, N.J.; Saksouk, N.; Perreault, T.; Meloche, J.; Paquet, E.R.; Biardel, S.; Provencher, S.; Côté, J.; et al. Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 2011, 208, 535–548. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Ramos, K.S.; Baks, L.; Wiersma, M.; Lanters, E.A.H.; Bogers, A.; de Groot, N.M.S.; Brundel, B. Blood-based 8-hydroxy-2′-deoxyguanosine level: A potential diagnostic biomarker for atrial fibrillation. Heart Rhythm 2021, 18, 271–277. [Google Scholar] [CrossRef]
- Wiersma, M.; van Marion, D.M.S.; Bouman, E.J.; Li, J.; Zhang, D.; Ramos, K.S.; Lanters, E.A.H.; de Groot, N.M.S.; Brundel, B. Cell-Free Circulating Mitochondrial DNA: A Potential Blood-Based Marker for Atrial Fibrillation. Cells 2020, 9, 1159. [Google Scholar] [CrossRef]
- Pool, L.; van Wijk, S.W.; van Schie, M.S.; Taverne, Y.; de Groot, N.M.S.; Brundel, B. Quantifying DNA Lesions and Circulating Free DNA: Diagnostic Marker for Electropathology and Clinical Stage of AF. JACC Clin. Electrophysiol. 2024, 11, 321–332. [Google Scholar] [CrossRef]
- Zhang, C.F.; Zhao, F.Y.; Xu, S.L.; Liu, J.; Xing, X.Q.; Yang, J. Autophagy in pulmonary hypertension: Emerging roles and therapeutic implications. J. Cell. Physiol. 2019, 234, 16755–16767. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Smith, A.; Guo, L.; Alastalo, T.P.; Li, M.; Sawada, H.; Liu, X.; Chen, Z.H.; Ifedigbo, E.; Jin, Y.; et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2011, 183, 649–658. [Google Scholar] [CrossRef]
- Li, L.; Wang, X.; Wang, L.; Qu, L.; Zhu, X.; Li, M.; Dang, X.; Li, P.; Gao, Y.; Peng, Z.; et al. Mammalian target of rapamycin overexpression antagonizes chronic hypoxia-triggered pulmonary arterial hypertension via the autophagic pathway. Int. J. Mol. Med. 2015, 36, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.J.; Du, J.; Welak, S.; Guan, T.; Eis, A.; Shi, Y.; Konduri, G.G. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L651–L663. [Google Scholar] [CrossRef]
- Dalvi, P.; Sharma, H.; Chinnappan, M.; Sanderson, M.; Allen, J.; Zeng, R.; Choi, A.; O’Brien-Ladner, A.; Dhillon, N.K. Enhanced autophagy in pulmonary endothelial cells on exposure to HIV-Tat and morphine: Role in HIV-related pulmonary arterial hypertension. Autophagy 2016, 12, 2420–2438. [Google Scholar] [CrossRef]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.J.; Dunmore, B.J.; Morrell, N.W. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef]
- Kato, F.; Sakao, S.; Takeuchi, T.; Suzuki, T.; Nishimura, R.; Yasuda, T.; Tanabe, N.; Tatsumi, K. Endothelial cell-related autophagic pathways in Sugen/hypoxia-exposed pulmonary arterial hypertensive rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L899–L915. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Manhas, A.; Kaur, G.; Jagavelu, K.; Hanif, K. Inhibition of fatty acid synthase is protective in pulmonary hypertension. Br. J. Pharmacol. 2016, 173, 2030–2045. [Google Scholar] [CrossRef]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef]
- He, Y.; Zuo, C.; Jia, D.; Bai, P.; Kong, D.; Chen, D.; Liu, G.; Li, J.; Wang, Y.; Chen, G.; et al. Loss of DP1 Aggravates Vascular Remodeling in Pulmonary Arterial Hypertension via mTORC1 Signaling. Am. J. Respir. Crit. Care Med. 2020, 201, 1263–1276. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, J.; Wang, C.; Jain, P.P.; Xiong, M.; Shi, X.; Lei, Y.; Chen, S.; Yin, Q.; Thistlethwaite, P.A.; et al. MDM2-Mediated Ubiquitination of Angiotensin-Converting Enzyme 2 Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2020, 142, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Al-Qazazi, R.; Lima, P.D.A.; Prisco, S.Z.; Potus, F.; Dasgupta, A.; Chen, K.H.; Tian, L.; Bentley, R.E.T.; Mewburn, J.; Martin, A.Y.; et al. Macrophage-NLRP3 Activation Promotes Right Ventricle Failure in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2022, 206, 608–624. [Google Scholar] [CrossRef]
- Ren, X.; Johns, R.A.; Gao, W.D. EXPRESS: Right Heart in Pulmonary Hypertension: From Adaptation to Failure. Pulm. Circ. 2019, 9, 2045894019845611. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaço, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Shen, C.; Tan, J.; Wu, Z.; Wang, W.; Chen, Y.; Dai, Y.; Yang, X.; Ye, S.; Chen, J.; et al. Periostin: A Potential Therapeutic Target For Pulmonary Hypertension? Circ. Res. 2020, 127, 1138–1152. [Google Scholar] [CrossRef]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6, e006458. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Brundel, B.; Wiersma, M. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells 2019, 8, 1617. [Google Scholar] [CrossRef]
- Yan, J.; Thomson, J.K.; Zhao, W.; Wu, X.; Gao, X.; DeMarco, D.; Kong, W.; Tong, M.; Sun, J.; Bakhos, M.; et al. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J. Mol. Cell. Cardiol. 2018, 114, 105–115. [Google Scholar] [CrossRef]
- Yan, J.; Bare, D.J.; DeSantiago, J.; Zhao, W.; Mei, Y.; Chen, Z.; Ginsburg, K.; Solaro, R.J.; Wolska, B.M.; Bers, D.M.; et al. JNK2, a Newly-Identified SERCA2 Enhancer, Augments an Arrhythmic [Ca2+](SR) Leak-Load Relationship. Circ. Res. 2021, 128, 455–470. [Google Scholar] [CrossRef]
- Yan, J.; Kong, W.; Zhang, Q.; Beyer, E.C.; Walcott, G.; Fast, V.G.; Ai, X. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc. Res. 2013, 97, 589–597. [Google Scholar] [CrossRef]
- Wei, Z.; Bing, Z.; Shaohuan, Q.; Yanran, W.; Shuo, S.; Bi, T.; Feiyu, Z.; Heng, Z.; Qin, G.; Pinfang, K. Expression of miRNAs in plasma exosomes derived from patients with atrial fibrillation. Clin. Cardiol. 2020, 43, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Su, K.N.; Ma, Y.; Cacheux, M.; Ilkan, Z.; Raad, N.; Muller, G.K.; Wu, X.; Guerrera, N.; Thorn, S.L.; Sinusas, A.J.; et al. Atrial AMP-activated protein kinase is critical for prevention of dysregulation of electrical excitability and atrial fibrillation. JCI Insight 2022, 7, e141213. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Daou, D.; Luo, Y.; Link, M.S.; Lavandero, S.; Gillette, T.G.; Hill, J.A. Impaired AMP-Activated Protein Kinase Signaling in Heart Failure With Preserved Ejection Fraction-Associated Atrial Fibrillation. Circulation 2022, 146, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Ebana, Y.; Sun, Y.; Yang, X.; Watanabe, T.; Makita, S.; Ozaki, K.; Tanaka, T.; Arai, H.; Furukawa, T. Pathway analysis with genome-wide association study (GWAS) data detected the association of atrial fibrillation with the mTOR signaling pathway. Int. J. Cardiol. Heart Vasc. 2019, 24, 100383. [Google Scholar] [CrossRef]
- Bullón, P.; Cano-García, F.J.; Alcocer-Gómez, E.; Varela-López, A.; Roman-Malo, L.; Ruiz-Salmerón, R.J.; Quiles, J.L.; Navarro-Pando, J.M.; Battino, M.; Ruiz-Cabello, J.; et al. Could NLRP3-Inflammasome Be a Cardiovascular Risk Biomarker in Acute Myocardial Infarction Patients? Antioxid. Redox Signal 2017, 27, 269–275. [Google Scholar] [CrossRef]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.D.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef]
- Babapoor-Farrokhran, S.; Gill, D.; Alzubi, J.; Mainigi, S.K. Atrial fibrillation: The role of hypoxia-inducible factor-1-regulated cytokines. Mol. Cell. Biochem. 2021, 476, 2283–2293. [Google Scholar] [CrossRef]
- Spengler, K.; Kryeziu, N.; Große, S.; Mosig, A.S.; Heller, R. VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKα1. Cells 2020, 9, 687. [Google Scholar] [CrossRef]
- Gramley, F.; Lorenzen, J.; Jedamzik, B.; Gatter, K.; Koellensperger, E.; Munzel, T.; Pezzella, F. Atrial fibrillation is associated with cardiac hypoxia. Cardiovasc. Pathol. 2010, 19, 102–111. [Google Scholar] [CrossRef]
- Coons, J.C.; Pogue, K.; Kolodziej, A.R.; Hirsch, G.A.; George, M.P. Pulmonary Arterial Hypertension: A Pharmacotherapeutic Update. Curr. Cardiol. Rep. 2019, 21, 141. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Farber, H.W.; Ghofrani, H.A.; Benza, R.L.; Busse, D.; Meier, C.; Hoeper, M.M. Risk assessment in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53, 1802004. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczyk, R.; Błaszczak, P.; Kowal, K.; Jasiewicz, M.; Knapp, M.; Szpakowicz, A.; Ptaszyńska-Kopczyńska, K.; Sobkowicz, B.; Waszkiewicz, E.; Grzywna, R.; et al. The significance of diminished sTWEAK and P-selectin content in platelets of patients with pulmonary arterial hypertension. Cytokine 2018, 107, 52–58. [Google Scholar] [CrossRef]
- Szulcek, R.; Happé, C.M.; Rol, N.; Fontijn, R.D.; Dickhoff, C.; Hartemink, K.J.; Grünberg, K.; Tu, L.; Timens, W.; Nossent, G.D.; et al. Delayed Microvascular Shear Adaptation in Pulmonary Arterial Hypertension. Role of Platelet Endothelial Cell Adhesion Molecule-1 Cleavage. Am. J. Respir. Crit. Care Med. 2016, 193, 1410–1420. [Google Scholar] [CrossRef]
- Ogawa, A.; Matsubara, H. Increased levels of platelet-derived microparticles in pulmonary hypertension. Thromb. Res. 2020, 195, 120–124. [Google Scholar] [CrossRef]
- Hickey, P.M.; Lawrie, A.; Condliffe, R. Circulating Protein Biomarkers in Systemic Sclerosis Related Pulmonary Arterial Hypertension: A Review of Published Data. Front. Med. 2018, 5, 175. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, L.; Jia, Y.; Balistrieri, A.; Fraidenburg, D.R.; Wang, J.; Tang, H.; Yuan, J.X. Pathogenic Mechanisms of Pulmonary Arterial Hypertension: Homeostasis Imbalance of Endothelium-Derived Relaxing and Contracting Factors. JACC Asia 2022, 2, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Handler, S.S.; Jin, J.; Ogawa, M.T.; Feinstein, J.A.; Lo, C. Abnormal platelet aggregation in pediatric pulmonary hypertension. Pulm. Circ. 2022, 12, e12104. [Google Scholar] [CrossRef]
- Aytekin, M.; Aulak, K.S.; Haserodt, S.; Chakravarti, R.; Cody, J.; Minai, O.A.; Dweik, R.A. Abnormal platelet aggregation in idiopathic pulmonary arterial hypertension: Role of nitric oxide. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L512–L520. [Google Scholar] [CrossRef]
- Myllylahti, L.; Ropponen, J.; Lax, M.; Lassila, R.; Nykänen, A.I. Upregulation of Coagulation Factor VIII and Fibrinogen After Pulmonary Endarterectomy in Patients with Chronic Thromboembolic Pulmonary Hypertension. Clin. Appl. Thromb. Hemost. 2023, 29, 10760296231158369. [Google Scholar] [CrossRef]
- Lu, M.; Blaine, K.P.; Cullinane, A.; Hall, C.; Dulau-Florea, A.; Sun, J.; Chenwi, H.F.; Graninger, G.M.; Harper, B.; Thompson, K.; et al. Pulmonary arterial hypertension patients display normal kinetics of clot formation using thrombelastography. Pulm. Circ. 2021, 11, 20458940211022204. [Google Scholar] [CrossRef] [PubMed]
- Katta, S.; Vadapalli, S.; Sastry, B.K.; Nallari, P. t-plasminogen activator inhibitor-1 polymorphism in idiopathic pulmonary arterial hypertension. Indian J. Hum. Genet. 2008, 14, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Troncoso, M.F.; Ortiz-Quintero, J.; Garrido-Moreno, V.; Sanhueza-Olivares, F.; Guerrero-Moncayo, A.; Chiong, M.; Castro, P.F.; García, L.; Gabrielli, L.; Corbalán, R.; et al. VCAM-1 as a predictor biomarker in cardiovascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166170. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Pignatelli, P.; Perticone, F.; Sciacqua, A.; Carnevale, R.; Farcomeni, A.; Basili, S.; Corazza, G.R.; Davì, G.; Lip, G.Y.H.; et al. Aspirin and renal insufficiency progression in patients with atrial fibrillation and chronic kidney disease. Int. J. Cardiol. 2016, 223, 619–624. [Google Scholar] [CrossRef]
- Semczuk-Kaczmarek, K.; Płatek, A.E.; Ryś, A.; Adamowicz, J.; Legosz, P.; Kotkowski, M.; Dudzik-Płocica, A.; Gorko, D.; Szymański, F.M.; Filipiak, K.J. CHA2DS2-VASc score and fibrinogen concentration in patients with atrial fibrillation. Adv. Clin. Exp. Med. 2019, 28, 1451–1457. [Google Scholar] [CrossRef]
- Goette, A.; Ittenson, A.; Hoffmanns, P.; Reek, S.; Hartung, W.; Klein, H.; Ansorge, S.; Geller, J.C. Increased expression of P-selectin in patients with chronic atrial fibrillation. Pacing Clin. Electrophysiol. 2000, 23, 1872–1875. [Google Scholar] [CrossRef]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef]
- Guignabert, C.; Humbert, M. Targeting transforming growth factor-β receptors in pulmonary hypertension. Eur. Respir. J. 2021, 57, 2002341. [Google Scholar] [CrossRef]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear factor κ-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS ONE 2013, 8, e75415. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Dernellis, J.; Panaretou, M. C-reactive protein and paroxysmal atrial fibrillation: Evidence of the implication of an inflammatory process in paroxysmal atrial fibrillation. Acta Cardiol. 2001, 56, 375–380. [Google Scholar] [CrossRef]
- Li, J.; Solus, J.; Chen, Q.; Rho, Y.H.; Milne, G.; Stein, C.M.; Darbar, D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm 2010, 7, 438–444. [Google Scholar] [CrossRef]
- Nortamo, S.; Ukkola, O.; Lepojärvi, S.; Kenttä, T.; Kiviniemi, A.; Junttila, J.; Huikuri, H.; Perkiömäki, J. Association of sST2 and hs-CRP levels with new-onset atrial fibrillation in coronary artery disease. Int. J. Cardiol. 2017, 248, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, A.E.; Vatasescu, R.G.; Stanciu, M.M.; Serdarevic, N.; Dorobantu, M. The role of pro-fibrotic biomarkers in paroxysmal and persistent atrial fibrillation. Cytokine 2018, 103, 63–68. [Google Scholar] [CrossRef]
- Gaudino, M.; Andreotti, F.; Zamparelli, R.; Di Castelnuovo, A.; Nasso, G.; Burzotta, F.; Iacoviello, L.; Donati, M.B.; Schiavello, R.; Maseri, A.; et al. The -174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation 2003, 108 (Suppl. S1), Ii195–Ii199. [Google Scholar] [CrossRef]
- Wu, N.; Xu, B.; Liu, Y.; Chen, X.; Tang, H.; Wu, L.; Xiang, Y.; Zhang, M.; Shu, M.; Song, Z.; et al. Elevated plasma levels of Th17-related cytokines are associated with increased risk of atrial fibrillation. Sci. Rep. 2016, 6, 26543. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Guo, Y.; Li, S.; Yu, B.; Zhu, S.; Li, S.; Li, N.; Tian, Z.; Peng, C.; Cheng, J.; et al. Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace 2010, 12, 1713–1718. [Google Scholar] [CrossRef]
- Dwyer, N.; Kilpatrick, D. Bosentan for the treatment of adult pulmonary hypertension. Future Cardiol. 2011, 7, 19–37. [Google Scholar] [CrossRef]
- Wu, Y.; Adi, D.; Long, M.; Wang, J.; Liu, F.; Gai, M.T.; Aierken, A.; Li, M.Y.; Li, Q.; Wu, L.Q.; et al. 4-Phenylbutyric Acid Induces Protection against Pulmonary Arterial Hypertension in Rats. PLoS ONE 2016, 11, e0157538. [Google Scholar] [CrossRef]
- Boucherat, O.; Chabot, S.; Paulin, R.; Trinh, I.; Bourgeois, A.; Potus, F.; Lampron, M.C.; Lambert, C.; Breuils-Bonnet, S.; Nadeau, V.; et al. HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 4546. [Google Scholar] [CrossRef] [PubMed]
- Shimauchi, T.; Boucherat, O.; Yokokawa, T.; Grobs, Y.; Wu, W.; Orcholski, M.; Martineau, S.; Omura, J.; Tremblay, E.; Shimauchi, K.; et al. PARP1-PKM2 Axis Mediates Right Ventricular Failure Associated With Pulmonary Arterial Hypertension. JACC Basic Transl. Sci. 2022, 7, 384–403. [Google Scholar] [CrossRef] [PubMed]
- Sztormowska-Achranowicz, K.; Jankowski, Z.; Kocić, I. Protective effect of nicotinamide and L-arginine against monocrotaline-induced pulmonary hypertension in rats: Gender dependence. Pharmacol. Rep. 2020, 72, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Yared, J.P.; Bakri, M.H.; Erzurum, S.C.; Moravec, C.S.; Laskowski, D.M.; Van Wagoner, D.R.; Mascha, E.; Thornton, J. Effect of dexamethasone on atrial fibrillation after cardiac surgery: Prospective, randomized, double-blind, placebo-controlled trial. J. Cardiothorac. Vasc. Anesth. 2007, 21, 68–75. [Google Scholar] [CrossRef]
- Iskandar, S.; Reddy, M.; Afzal, M.R.; Rajasingh, J.; Atoui, M.; Lavu, M.; Atkins, D.; Bommana, S.; Umbarger, L.; Jaeger, M.; et al. Use of Oral Steroid and its Effects on Atrial Fibrillation Recurrence and Inflammatory Cytokines Post Ablation—The Steroid AF Study. J. Atr. Fibrillation 2017, 9, 1604. [Google Scholar] [CrossRef]
- Price, L.C.; Shao, D.; Meng, C.; Perros, F.; Garfield, B.E.; Zhu, J.; Montani, D.; Dorfmuller, P.; Humbert, M.; Adcock, I.M.; et al. Dexamethasone induces apoptosis in pulmonary arterial smooth muscle cells. Respir. Res. 2015, 16, 114. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 2017, 8, 99740–99756. [Google Scholar] [CrossRef]
- Starreveld, R.; Ramos, K.S.; Muskens, A.; Brundel, B.; de Groot, N.M.S. Daily Supplementation of L-Glutamine in Atrial Fibrillation Patients: The Effect on Heat Shock Proteins and Metabolites. Cells 2020, 9, 1729. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhao, J.; Gong, Y.; Wang, D.; Wang, X.; Yun, F.; Liu, Z.; Zhang, S.; Li, W.; Zhao, X.; et al. Autophagy exacerbates electrical remodeling in atrial fibrillation by ubiquitin-dependent degradation of L-type calcium channel. Cell Death Dis. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Europace 2016, 18, 1455–1490. [Google Scholar] [CrossRef] [PubMed]
- Harjola, V.P.; Mullens, W.; Banaszewski, M.; Bauersachs, J.; Brunner-La Rocca, H.P.; Chioncel, O.; Collins, S.P.; Doehner, W.; Filippatos, G.S.; Flammer, A.J.; et al. Organ dysfunction, injury and failure in acute heart failure: From pathophysiology to diagnosis and management. A review on behalf of the Acute Heart Failure Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2017, 19, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.H.; Ma, J.L.; Ding, D.; Ma, Y.J.; Wei, Y.P.; Jing, Z.C. Experimental animal models of pulmonary hypertension: Development and challenges. Animal Model. Exp. Med. 2022, 5, 207–216. [Google Scholar] [CrossRef]
- Bueno-Beti, C.; Sassi, Y.; Hajjar, R.J.; Hadri, L. Pulmonary Artery Hypertension Model in Rats by Monocrotaline Administration. Methods Mol. Biol. 2018, 1816, 233–241. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Registry | Sample Size | Patient Population * | Sex (%) * | Age (Years) | Incidence and Prevalence of Tachyarrhythmias | Ref. |
|---|---|---|---|---|---|---|
| Hannover Medical School, Hannover, Germany | 231 | iPAH = 70%, CTEPH = 12% | Female = 65% | 48 ± 14 ** | AFL = 6.49%, AF = 5.62%, AVNRT = 1.3% | [6] |
| PH Service of the Città della Salute e della Scienza of Turin. | 77 | PAH = 66%, CTEPH = 18% | Female = 53% | 63(48–70.7) *** | ParAF = 3.8%, PerAF = 10.38%, LSPerAF = 3.8% AVNRT = 1.2% | [9] |
| Hannover Medical School, Hannover, Germany | 239 | PAH = 66%, CTEPH = 34% | Female = 61% | 55 (49–66) *** | New onset of AF/AFL after 5 years = 25.1% | [8] |
| China | 280 | iPAH = 100% | Female = 68% | 39 ± 15 ** | AF = 5.71%, AFL = 4.64%, AT = 3.92% | [10] |
| University Hospital Bonn, Germany | 64 | PAH = 39%, CTEPH = 17% | Female = 47% | 66.4 ± 11.8 ** | AF = 32.1% | [11] |
| UPMC (Pittsburgh) hospitals | 297 | PAH = 90% CTEPH = 10% | Female = 67% | 57.6 ± 14.7 ** | AF = 15.48%, AFL = 8.41% | [12] |
| Johns Hopkins Pulmonary Hypertension Registry | 317 | iPAH = 37% SSc-PAH = 63% | Female = 84% | 56.7 ± 14.4 ** | AA (Total) = 13.2% AF = 5.99% AFL = 2.8% | [13] |
| General University Hospital, Prague | 755 | PAH = 44.23%, CTEPH = 17.21% | Female = 59% | 60 ± 15 ** | ParAF = 7.9% PerAF = 4.1% Permanent AF = 9% AF (total) = 21% | [14] |
| General Class | Drug(s) | Mechanism of Action | Condition | Trial Phase | Refs or Clinical Trial Identifier |
|---|---|---|---|---|---|
| DNA damage | ABT-888 | PARP1 inhibitor | AF | Preclinical | [27] |
| PH | Preclinical | [58,152] | |||
| Nicotinamide | Enhances NAD+ and NADH levels, reducing oxidative damage to proteins and DNA | AF | Preclinical | [27,28] | |
| PH | Preclinical | [153] | |||
| Oxidative stress | L-glutamine | Induce HSPs and normalizes metabolites | AF | II | [159] |
| PH | II | NCT01048905 | |||
| PQC system | 4-Phenyl-butyrate | ER stress inhibitor | AF | Preclinical | [106] |
| ER stress signaling inhibitor | PH | Preclinical | [150] | ||
| Tubastatin | HDAC6 inhibitor | AF | Preclinical | [28] | |
| PH | Preclinical | [151] | |||
| Autophagy | Chloroquine | Inhibition of autophagic flux [160] | AF | Preclinical | [161] |
| PH | NA | NCT04314817 | |||
| MCC950 | NLRP3-selective inhibitor | AF | Preclinical | [157] | |
| PF | Preclinical | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultan, D.; Brundel, B.J.J.M.; Kurakula, K. The Interplay Between Pulmonary Hypertension and Atrial Fibrillation: A Comprehensive Overview. Cells 2025, 14, 839. https://doi.org/10.3390/cells14110839
Sultan D, Brundel BJJM, Kurakula K. The Interplay Between Pulmonary Hypertension and Atrial Fibrillation: A Comprehensive Overview. Cells. 2025; 14(11):839. https://doi.org/10.3390/cells14110839
Chicago/Turabian StyleSultan, Danish, Bianca J. J. M. Brundel, and Kondababu Kurakula. 2025. "The Interplay Between Pulmonary Hypertension and Atrial Fibrillation: A Comprehensive Overview" Cells 14, no. 11: 839. https://doi.org/10.3390/cells14110839
APA StyleSultan, D., Brundel, B. J. J. M., & Kurakula, K. (2025). The Interplay Between Pulmonary Hypertension and Atrial Fibrillation: A Comprehensive Overview. Cells, 14(11), 839. https://doi.org/10.3390/cells14110839