
Innate Lymphoid Cells in Inflammatory Bowel Disease


Abstract
1. Overview of IBD
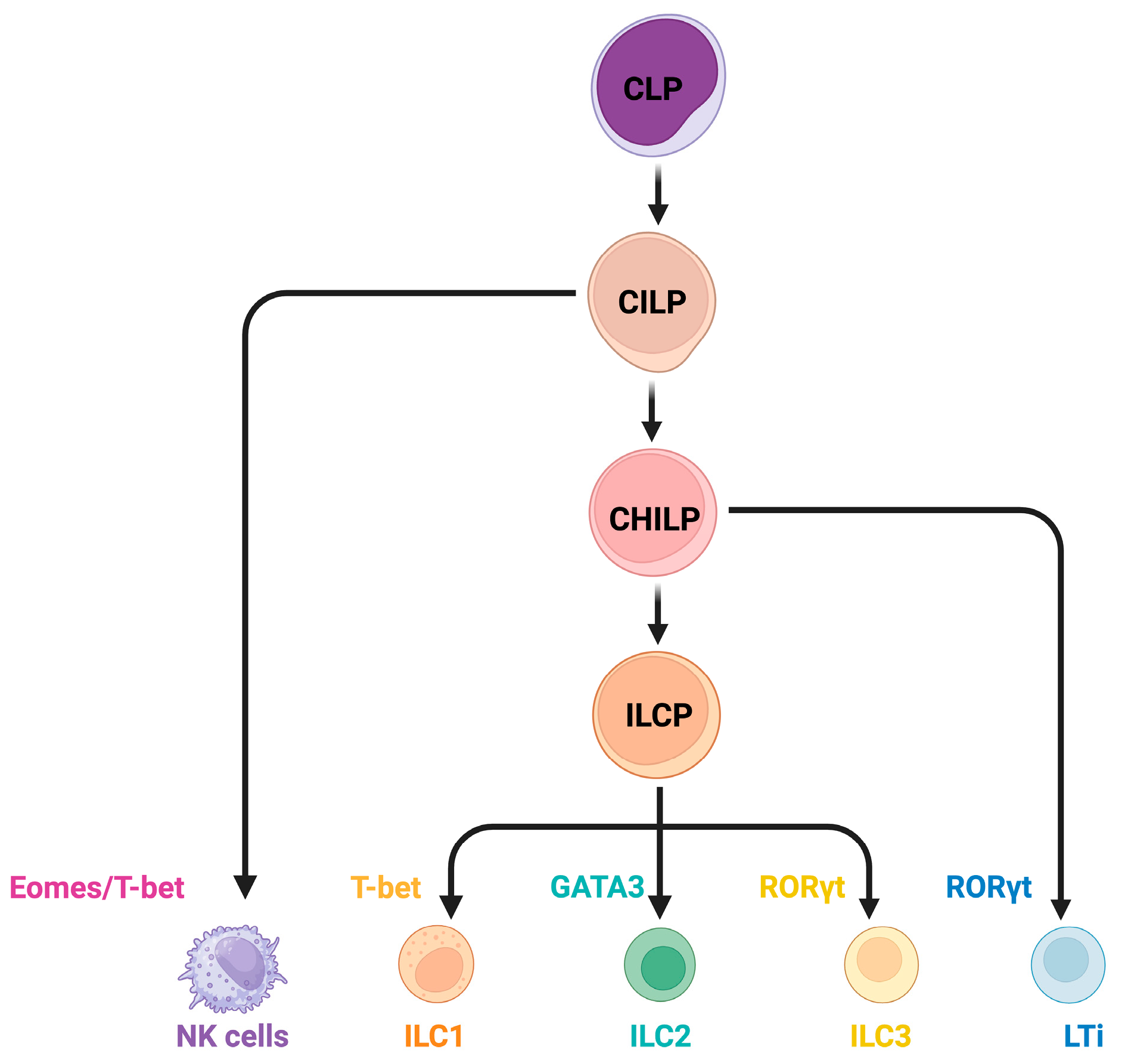
2. Overview of ILCs
3. ILCs in Homeostasis and IBD
3.1. ILC1
3.1.1. ILC1 in Homeostasis
3.1.2. ILC1 in IBD
3.2. ILC2
3.2.1. ILC2 in Homeostasis
3.2.2. ILC2 in IBD
3.3. ILC3
3.3.1. ILC3 in Homeostasis
3.3.2. ILC3s in IBD
3.4. NK Cells
3.4.1. NK Cells in Homeostasis
3.4.2. NK Cells in IBD
4. ILCs in IBD Therapies
5. Challenges and Future Directions in ILCs in IBD
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dolinger, M.; Torres, J.; Vermeire, S. Crohn’s disease. Lancet 2024, 403, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Berre, C.L.; Honap, S.; Peyrin-Biroulet, L. Ulcerative colitis. Lancet 2023, 402, 571–584. [Google Scholar] [CrossRef]
- Olén, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Colorectal cancer in Crohn’s disease: A Scandinavian population-based cohort study. Lancet Gastroenterol. Hepatol. 2020, 5, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Lewis, J.D.; Parlett, L.E.; Jonsson Funk, M.L.; Brensinger, C.; Pate, V.; Wu, Q.; Dawwas, G.K.; Weiss, A.; Constant, B.D.; McCauley, M.; et al. Incidence, Prevalence, and Racial and Ethnic Distribution of Inflammatory Bowel Disease in the United States. Gastroenterology 2023, 165, 1197–1205.e1192. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Zhou, J.; Wang, Z.; Liu, D.; Zhang, H.; Xie, S.; Wu, K. Epidemiology, pathogenesis, diagnosis, and treatment of inflammatory bowel disease: Insights from the past two years. Chin. Med. J. 2025, 138, 763–776. [Google Scholar] [CrossRef]
- Buie, M.J.; Quan, J.; Windsor, J.W.; Coward, S.; Hansen, T.M.; King, J.A.; Kotze, P.G.; Gearry, R.B.; Ng, S.C.; Mak, J.W.Y.; et al. Global Hospitalization Trends for Crohn’s Disease and Ulcerative Colitis in the 21st Century: A Systematic Review With Temporal Analyses. Clin. Gastroenterol. Hepatol. 2023, 21, 2211–2221. [Google Scholar] [CrossRef]
- Xu, L.; He, B.; Sun, Y.; Li, J.; Shen, P.; Hu, L.; Liu, G.; Wang, J.; Duan, L.; Zhan, S.; et al. Incidence of Inflammatory Bowel Disease in Urban China: A Nationwide Population-based Study. Clin. Gastroenterol. Hepatol. 2023, 21, 3379–3386.e3329. [Google Scholar] [CrossRef]
- Wang, S.; Dong, Z.; Wan, X. Global, regional, and national burden of inflammatory bowel disease and its associated anemia, 1990 to 2019 and predictions to 2050: An analysis of the global burden of disease study 2019. Autoimmun. Rev. 2024, 23, 103498. [Google Scholar] [CrossRef]
- Burisch, J.; Zhao, M.; Odes, S.; Cruz, P.D.; Vermeire, S.; Bernstein, C.N.; Kaplan, G.G.; Duricova, D.; Greenberg, D.; Melberg, H.O.; et al. The cost of inflammatory bowel disease in high-income settings: A Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 2023, 8, 458–492. [Google Scholar] [CrossRef]
- Colombel, J.-F.; Narula, N.; Peyrin-Biroulet, L. Management Strategies to Improve Outcomes of Patients With Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 351–361.e355. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, N.A.; Heap, G.A.; Green, H.D.; Hamilton, B.; Bewshea, C.; Walker, G.J.; Thomas, A.; Nice, R.; Perry, M.H.; Bouri, S. Predictors of anti-TNF treatment failure in anti-TNF-naive patients with active luminal Crohn’s disease: A prospective, multicentre, cohort study. Lancet Gastroenterol. Hepatol. 2019, 4, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Temido, M.J.; Honap, S.; Jairath, V.; Vermeire, S.; Danese, S.; Portela, F.; Peyrin-Biroulet, L. Overcoming the challenges of overtreating and undertreating inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Ciorba, M.A.; Konnikova, L.; Hirota, S.A.; Lucchetta, E.M.; Turner, J.R.; Slavin, A.; Johnson, K.; Condray, C.D.; Hong, S.; Cressall, B.K.; et al. Challenges in IBD Research 2024: Preclinical Human IBD Mechanisms. Inflamm. Bowel Dis. 2024, 30, S5–S18. [Google Scholar] [CrossRef]
- Gibson, G.; Rioux, J.D.; Cho, J.H.; Haritunians, T.; Thoutam, A.; Abreu, M.T.; Brant, S.R.; Kugathasan, S.; McCauley, J.L.; Silverberg, M.; et al. Eleven Grand Challenges for Inflammatory Bowel Disease Genetics and Genomics. Inflamm. Bowel Dis. 2025, 31, 272–284. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, R.; Gao, H.; Jung, S.; Gao, X.; Sun, R.; Liu, X.; Kim, Y.; Lee, H.-S.; Kawai, Y.; et al. Genetic architecture of the inflammatory bowel diseases across East Asian and European ancestries. Nat. Genet. 2023, 55, 796–806. [Google Scholar] [CrossRef]
- Hugot, J.-P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.-P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Colonna, M. Innate Lymphoid Cells in Mucosal Immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Araujo, L.P.; Edwards, M.; Irie, K.; Huang, Y.; Kawano, Y.; Tran, A.; De Michele, S.; Bhagat, G.; Wang, H.H.; Ivanov, I.I. Context-dependent role of group 3 innate lymphoid cells in mucosal protection. Sci. Immunol. 2024, 9, eade7530. [Google Scholar] [CrossRef]
- Peng, V.; Jaeger, N.; Colonna, M. Innate Lymphoid Cells and Inflammatory Bowel Disease. In Innate Lymphoid Cells; Sun, X.-H., Ed.; Springer Nature: Singapore, 2022; pp. 97–112. [Google Scholar]
- Zhang, J.; Marotel, M.; Fauteux-Daniel, S.; Mathieu, A.-L.; Viel, S.; Marçais, A.; Walzer, T. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur. J. Immunol. 2018, 48, 738–750. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue–inducer cells are interleukin 17–producing precursors to RORC+ CD127+ natural killer–like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef]
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing Lymph Nodes Collect CD4+CD3− LTβ+ Cells That Can Differentiate to APC, NK Cells, and Follicular Cells but Not T or B Cells. Immunity 1997, 7, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Liu, C.; Gong, Y.; Zhang, H.; Yang, H.; Zeng, Y.; Bian, Z.; Xin, Q.; Bai, Z.; Zhang, M.; He, J.; et al. Delineating spatiotemporal and hierarchical development of human fetal innate lymphoid cells. Cell Res. 2021, 31, 1106–1122. [Google Scholar] [CrossRef]
- Cherrier, D.E.; Serafini, N.; Di Santo, J.P. Innate Lymphoid Cell Development: A T Cell Perspective. Immunity 2018, 48, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 2018, 48, 1104–1117. [Google Scholar] [CrossRef]
- Hernández, D.C.; Juelke, K.; Müller, N.C.; Durek, P.; Ugursu, B.; Mashreghi, M.F.; Rückert, T.; Romagnani, C. An in vitro platform supports generation of human innate lymphoid cells from CD34(+) hematopoietic progenitors that recapitulate ex vivo identity. Immunity 2021, 54, 2417–2432.e2415. [Google Scholar] [CrossRef]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef]
- Bennstein, S.B.; Uhrberg, M. Circulating innate lymphoid cells (cILCs): Unconventional lymphocytes with hidden talents. J. Allergy Clin. Immunol. 2024, 154, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Hashimoto-Hill, S.; Kim, M. Migration and Tissue Tropism of Innate Lymphoid Cells. Trends Immunol. 2016, 37, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Abidi, A.; Laurent, T.; Bériou, G.; Bouchet-Delbos, L.; Fourgeux, C.; Louvet, C.; Triki-Marrakchi, R.; Poschmann, J.; Josien, R.; Martin, J. Characterization of Rat ILCs Reveals ILC2 as the Dominant Intestinal Subset. Front. Immunol. 2020, 11, 255. [Google Scholar] [CrossRef]
- Cortez, V.S.; Colonna, M. Diversity and function of group 1 innate lymphoid cells. Immunol. Lett. 2016, 179, 19–24. [Google Scholar] [CrossRef]
- Krämer, B.; Goeser, F.; Lutz, P.; Glässner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017, 13, e1006373. [Google Scholar] [CrossRef]
- Vély, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F.; et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef]
- Mazzurana, L.; Czarnewski, P.; Jonsson, V.; Wigge, L.; Ringnér, M.; Williams, T.C.; Ravindran, A.; Björklund, Å.K.; Säfholm, J.; Nilsson, G.; et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. 2021, 31, 554–568. [Google Scholar] [CrossRef]
- Kokkinou, E.; Pandey, R.V.; Mazzurana, L.; Gutierrez-Perez, I.; Tibbitt, C.A.; Weigel, W.; Soini, T.; Carrasco, A.; Rao, A.; Nagasawa, M.; et al. CD45RA(+)CD62L(-) ILCs in human tissues represent a quiescent local reservoir for the generation of differentiated ILCs. Sci. Immunol. 2022, 7, eabj8301. [Google Scholar] [CrossRef]
- Kokkinou, E.; Soini, T.; Pandey, R.V.; van Acker, A.; Theorell, J.; Czarnewski, P.; Kvedaraite, E.; Vandamme, N.; Lourda, M.; Sorini, C.; et al. The single-cell transcriptional landscape of innate and adaptive lymphocytes in pediatric-onset colitis. Cell Rep. Med. 2023, 4, 101038. [Google Scholar] [CrossRef] [PubMed]
- Weizman, O.-E.; Adams, N.M.; Schuster, I.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E. ILC1 confer early host protection at initial sites of viral infection. Cell 2017, 171, 795–808.e712. [Google Scholar] [CrossRef]
- Shannon, J.P.; Vrba, S.M.; Reynoso, G.V.; Wynne-Jones, E.; Kamenyeva, O.; Malo, C.S.; Cherry, C.R.; McManus, D.T.; Hickman, H.D. Group 1 innate lymphoid-cell-derived interferon-γ maintains anti-viral vigilance in the mucosal epithelium. Immunity 2021, 54, 276–290.e275. [Google Scholar] [CrossRef] [PubMed]
- Flommersfeld, S.; Böttcher, J.P.; Ersching, J.; Flossdorf, M.; Meiser, P.; Pachmayr, L.O.; Leube, J.; Hensel, I.; Jarosch, S.; Zhang, Q.; et al. Fate mapping of single NK cells identifies a type 1 innate lymphoid-like lineage that bridges innate and adaptive recognition of viral infection. Immunity 2021, 54, 2288–2304.e2287. [Google Scholar] [CrossRef]
- Coman, D.; Coales, I.; Roberts, L.B.; Neves, J.F. Helper-Like Type-1 Innate Lymphoid Cells in Inflammatory Bowel Disease. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Shui, J.W.; Larange, A.; Kim, G.; Vela, J.L.; Zahner, S.; Cheroutre, H.; Kronenberg, M. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature 2012, 488, 222–225. [Google Scholar] [CrossRef]
- Krabbendam, L.; Heesters, B.A.; Kradolfer, C.M.A.; Haverkate, N.J.E.; Becker, M.A.J.; Buskens, C.J.; Bemelman, W.A.; Bernink, J.H.; Spits, H. CD127+ CD94+ innate lymphoid cells expressing granulysin and perforin are expanded in patients with Crohn’s disease. Nat. Commun. 2021, 12, 5841. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127+ Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Yomogida, K.; Bigley, T.M.; Trsan, T.; Gilfillan, S.; Cella, M.; Yokoyama, W.M.; Egawa, T.; Colonna, M. Hobit confers tissue-dependent programs to type 1 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2117965118. [Google Scholar] [CrossRef]
- Friedrich, C.; Taggenbrock, R.L.R.E.; Doucet-Ladevèze, R.; Golda, G.; Moenius, R.; Arampatzi, P.; Kragten, N.A.M.; Kreymborg, K.; de Agüero, M.G.; Kastenmueller, W.; et al. Effector differentiation downstream of lineage commitment in ILC1 is driven by Hobit across tissues. Nat. Immunol. 2021, 22, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Vonarbourg, C. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt innate lymphocytes. Immunity 2010, 33, 736–751. [Google Scholar] [CrossRef]
- Fiancette, R.; Finlay, C.M.; Willis, C.; Bevington, S.L.; Soley, J.; Ng, S.T.H.; Baker, S.M.; Andrews, S.; Hepworth, M.R.; Withers, D.R. Reciprocal transcription factor networks govern tissue-resident ILC3 subset function and identity. Nat. Immunol. 2021, 22, 1245–1255. [Google Scholar] [CrossRef]
- McFarland, A.P.; Yalin, A.; Wang, S.-Y.; Cortez, V.S.; Landsberger, T.; Sudan, R.; Peng, V.; Miller, H.L.; Ricci, B.; David, E.; et al. Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity 2021, 54, 1320–1337.e1324. [Google Scholar] [CrossRef]
- Bai, L.; Vienne, M.; Tang, L.; Kerdiles, Y.; Etiennot, M.; Escalière, B.; Galluso, J.; Wei, H.; Sun, R.; Vivier, E.; et al. Liver type 1 innate lymphoid cells develop locally via an interferon-γ–dependent loop. Science 2021, 371, eaba4177. [Google Scholar] [CrossRef]
- Langer, V. IFN-γ drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption. J. Clin. Invest. 2019, 129, 4691–4707. [Google Scholar] [CrossRef]
- Creyns, B.; Jacobs, I.; Verstockt, B.; Cremer, J.; Ballet, V.; Vandecasteele, R.; Vanuytsel, T.; Ferrante, M.; Vermeire, S.; Van Assche, G.; et al. Biological Therapy in Inflammatory Bowel Disease Patients Partly Restores Intestinal Innate Lymphoid Cell Subtype Equilibrium. Front. Immunol. 2020, 11, 1847. [Google Scholar] [CrossRef]
- Forkel, M.; Van Tol, S.; Höög, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. J. Crohn’s Colitis 2019, 13, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Gamini, R.; Sécca, C.; Collins, P.L.; Zhao, S.; Peng, V.; Robinette, M.L.; Schettini, J.; Zaitsev, K.; Gordon, W.; et al. Subsets of ILC3−ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat. Immunol. 2019, 20, 980–991. [Google Scholar] [CrossRef]
- Jaeger, N.; Antonova, A.U.; Kreisel, D.; Roan, F.; Lantelme, E.; Ziegler, S.F.; Cella, M.; Colonna, M. Diversity of group 1 innate lymphoid cells in human tissues. Nat. Immunol. 2024, 25, 1460–1473. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, X.; Liu, Z.; Shao, F.; Yu, D.; Zhao, M.; Qin, X.; Wang, S. Microbiota regulates the TET1-mediated DNA hydroxymethylation program in innate lymphoid cell differentiation. Nat. Commun. 2024, 15, 4792. [Google Scholar] [CrossRef]
- Jowett, G.M.; Norman, M.D.A.; Yu, T.T.L.; Arévalo, P.R.; Hoogland, D.; Lust, S.T.; Read, E.; Hamrud, E.; Walters, N.J.; Niazi, U.; et al. ILC1 drive intestinal epithelial and matrix remodelling. Nat. Mater. 2021, 20, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Mori, R.; Ogino, T.; Murakami, M.; Kayama, H.; Okuzaki, D.; Ikeda, A.; Sekido, Y.; Hata, T.; Hamabe, A.; Takahashi, H.; et al. Group 1 innate lymphoid cells and inflammatory macrophages exacerbate fibrosis in creeping fat through IFN-γ secretion. J. Gastroenterol. 2025. [Google Scholar] [CrossRef]
- Frisbee, A.L.; Saleh, M.M.; Young, M.K.; Leslie, J.L.; Simpson, M.E.; Abhyankar, M.M.; Cowardin, C.A.; Ma, J.Z.; Pramoonjago, P.; Turner, S.D.; et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun. 2019, 10, 2712. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Reboldi, A.; Hall, J.A.; Berg, L.J. The Tec kinase ITK is essential for ILC2 survival and epithelial integrity in the intestine. Nat. Commun. 2019, 10, 784. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R.; Urban, J.F.; Paul, W.E. IL-25-responsive, lineage-negative KLRG1hi cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 2015, 16, 161–169. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin–EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sirohi, K.; Verma, M.; McKay, J.; Michalec, L.; Sripada, A.; Danhorn, T.; Rollins, D.; Good, J.; Gorska, M.M.; et al. Optimal identification of human conventional and nonconventional (CRTH2(-)IL7Rα(-)) ILC2s using additional surface markers. J. Allergy Clin. Immunol. 2020, 146, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, M.; Heesters, B.A.; Kradolfer, C.M.A.; Krabbendam, L.; Martinez-Gonzalez, I.; de Bruijn, M.J.W.; Golebski, K.; Hendriks, R.W.; Stadhouders, R.; Spits, H.; et al. KLRG1 and NKp46 discriminate subpopulations of human CD117(+)CRTH2(-) ILCs biased toward ILC2 or ILC3. J. Exp. Med. 2019, 216, 1762–1776. [Google Scholar] [CrossRef]
- Spits, H.; Mjösberg, J. Heterogeneity of type 2 innate lymphoid cells. Nat. Rev. Immunol. 2022, 22, 701–712. [Google Scholar] [CrossRef]
- Camelo, A.; Rosignoli, G.; Ohne, Y.; Stewart, R.A.; Overed-Sayer, C.; Sleeman, M.A.; May, R.D. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv. 2017, 1, 577–589. [Google Scholar] [CrossRef]
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial cell-derived cytokines: More than just signaling the alarm. J. Clin. Investig. 2019, 129, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, M.; Wang, Q.; Gao, L.; Jiang, H.; Shi, K.; Lin, Y.; Zhou, J.; Huang, J.; Qu, S.; et al. IL-33 released during challenge phase regulates allergic asthma in an age-dependent way. Cell. Mol. Immunol. 2024, 1–17. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.K.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.J. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging Functions of Amphiregulin in Orchestrating Immunity, Inflammation, and Tissue Repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef]
- Stockis, A.M.; Yano, H.; Parkhurst, C.N.; Mahlakõiv, T.; Chu, C.; Zhang, W.; He, Z.; Jarick, K.J.; Zhong, C.; Putzel, G.G.; et al. Neuropeptide regulation of non-redundant ILC2 responses at barrier surfaces. Nature 2022, 611, 787–793. [Google Scholar] [CrossRef]
- Cardoso, V.; Chesné, J.; Ribeiro, H.; García-Cassani, B.; Carvalho, T.; Bouchery, T.; Shah, K.; Barbosa-Morais, N.L.; Harris, N.; Veiga-Fernandes, H. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 2017, 549, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, S.; Zhou, K.; Wang, Y.; Chen, Y.; Hu, W.; Li, S.; Li, H.; Wang, Y.; Wang, Q.; et al. Neuromedin U programs eosinophils to promote mucosal immunity of the small intestine. Science 2023, 381, 1189–1196. [Google Scholar] [CrossRef]
- Szeto, A.C.H.; Clark, P.A.; Ferreira, A.C.F.; Heycock, M.; Griffiths, E.L.; Jou, E.; Mannion, J.; Luan, S.L.; Storrar, S.; Knolle, M.D.; et al. Mef2d potentiates type-2 immune responses and allergic lung inflammation. Science 2024, 384, eadl0370. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Luo, J.; Zeng, N.; Jiang, S.; Chen, W.; Hoyle, R.D.; Klenerman, P.; Pavord, I.D.; Xue, L. Neuromedin U promotes human type 2 immune responses. Mucosal Immunol. 2022, 15, 990–999. [Google Scholar] [CrossRef]
- Li, S.; Bostick, J.W.; Ye, J.; Qiu, J.; Zhang, B.; Urban, J.F., Jr.; Avram, D.; Zhou, L. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity 2018, 49, 915–928.e915. [Google Scholar] [CrossRef]
- Cella, M.; Colonna, M. Aryl hydrocarbon receptor: Linking environment to immunity. Semin. Immunol. 2015, 27, 310–314. [Google Scholar] [CrossRef]
- Bando, J.K.; Gilfillan, S.; Di Luccia, B.; Fachi, J.L.; Sécca, C.; Cella, M.; Colonna, M. ILC2s are the predominant source of intestinal ILC-derived IL-10. J. Exp. Med. 2019, 217, e20191520. [Google Scholar] [CrossRef]
- Golebski, K.; Layhadi, J.A.; Sahiner, U.; Steveling-Klein, E.H.; Lenormand, M.M.; Li, R.C.Y.; Bal, S.M.; Heesters, B.A.; Vilà-Nadal, G.; Hunewald, O.; et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity 2021, 54, 291–307.e297. [Google Scholar] [CrossRef]
- Ngo Thi Phuong, N.; Palmieri, V.; Adamczyk, A.; Klopfleisch, R.; Langhorst, J.; Hansen, W.; Westendorf, A.M.; Pastille, E. IL-33 Drives Expansion of Type 2 Innate Lymphoid Cells and Regulatory T Cells and Protects Mice From Severe, Acute Colitis. Front. Immunol. 2021, 12, 669787. [Google Scholar] [CrossRef]
- De Salvo, C.; Buela, K.-A.; Creyns, B.; Corridoni, D.; Rana, N.; Wargo, H.L.; Cominelli, C.L.; Delaney, P.G.; Rodriguez-Palacios, A.; Cominelli, F.; et al. NOD2 drives early IL-33–dependent expansion of group 2 innate lymphoid cells during Crohn’s disease–like ileitis. J. Clin. Investig. 2021, 131, e140624. [Google Scholar] [CrossRef] [PubMed]
- Mazzurana, L.; Bonfiglio, F.; Forkel, M.; D’Amato, M.; Halfvarson, J.; Mjösberg, J. Crohn’s Disease Is Associated With Activation of Circulating Innate Lymphoid Cells. Inflamm. Bowel Dis. 2021, 27, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Masterson, J.C.; Capocelli, K.E.; Hosford, L.; Biette, K.; McNamee, E.N.; de Zoeten, E.F.; Harris, R.; Fernando, S.D.; Jedlicka, P.; Protheroe, C.; et al. Eosinophils and IL-33 Perpetuate Chronic Inflammation and Fibrosis in a Pediatric Population with Stricturing Crohn’s Ileitis. Inflamm. Bowel Dis. 2015, 21, 2429–2440. [Google Scholar] [CrossRef]
- Bailey, J.R.; Bland, P.W.; Tarlton, J.F.; Peters, I.; Moorghen, M.; Sylvester, P.A.; Probert, C.S.J.; Whiting, C.V. IL-13 Promotes Collagen Accumulation in Crohn’s Disease Fibrosis by Down-Regulation of Fibroblast MMP Synthesis: A Role for Innate Lymphoid Cells? PLoS ONE 2012, 7, e52332. [Google Scholar] [CrossRef]
- Irie, E.; Ishihara, R.; Mizushima, I.; Hatai, S.; Hagihara, Y.; Takada, Y.; Tsunoda, J.; Iwata, K.; Matsubara, Y.; Yoshimatsu, Y.; et al. Enrichment of type I interferon signaling in colonic group 2 innate lymphoid cells in experimental colitis. Front. Immunol. 2022, 13, 982827. [Google Scholar] [CrossRef]
- Iliopoulou, L.; Tzaferis, C.; Prados, A.; Roumelioti, F.; Koliaraki, V.; Kollias, G. Different fibroblast subtypes propel spatially defined ileal inflammation through TNFR1 signalling in murine ileitis. Nat. Commun. 2025, 16, 3023. [Google Scholar] [CrossRef]
- Zhao, M.; Shao, F.; Yu, D.; Zhang, J.; Liu, Z.; Ma, J.; Xia, P.; Wang, S. Maturation and specialization of group 2 innate lymphoid cells through the lung-gut axis. Nat. Commun. 2022, 13, 7600. [Google Scholar] [CrossRef]
- Kim, J.; Ham, J.; Kang, H.R.; Bae, Y.-S.; Kim, T.; Kim, H.Y. JAK3 inhibitor suppresses multipotent ILC2s and attenuates steroid-resistant asthma. Sci. Adv. 2023, 9, eadi3770. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Whelan, K.; Allegretti, J.R.; Sokol, H. Diet and Microbiome-Directed Therapy 2.0 for IBD. Clin. Gastroenterol. Hepatol. 2025, 23, 406–418. [Google Scholar] [CrossRef]
- Adolph, T.E.; Zhang, J. Diet fuelling inflammatory bowel diseases: Preclinical and clinical concepts. Gut 2022, 71, 2574–2586. [Google Scholar] [CrossRef]
- Armstrong, H.K.; Bording-Jorgensen, M.; Santer, D.M.; Zhang, Z.; Valcheva, R.; Rieger, A.M.; Sung-Ho Kim, J.; Dijk, S.I.; Mahmood, R.; Ogungbola, O.; et al. Unfermented β-fructan Fibers Fuel Inflammation in Select Inflammatory Bowel Disease Patients. Gastroenterology 2023, 164, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Arifuzzaman, M.; Won, T.H.; Yano, H.; Uddin, J.; Emanuel, E.R.; Hu, E.; Zhang, W.; Li, T.-T.; Jin, W.-B.; Grier, A.; et al. Dietary fiber is a critical determinant of pathologic ILC2 responses and intestinal inflammation. J. Exp. Med. 2024, 221, e20232148. [Google Scholar] [CrossRef] [PubMed]
- Massironi, S.; Viganò, C.; Palermo, A.; Pirola, L.; Mulinacci, G.; Allocca, M.; Peyrin-Biroulet, L.; Danese, S. Inflammation and malnutrition in inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2023, 8, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Nagano, Y.; Morita, S.; Tanoue, T.; Yamane, H.; Ishikawa, K.; Sato, T.; Kubo, M.; Hori, S.; Taniguchi, T.; et al. Diet-mediated constitutive induction of novel IL-4+ ILC2 cells maintains intestinal homeostasis in mice. J. Exp. Med. 2023, 220, e20221773. [Google Scholar] [CrossRef]
- Gogoi, M.; Clark, P.A.; Ferreira, A.C.F.; Rodriguez Rodriguez, N.; Heycock, M.; Ko, M.; Murphy, J.E.; Chen, V.; Luan, S.-L.; Jolin, H.E.; et al. ILC2-derived LIF licences progress from tissue to systemic immunity. Nature 2024, 632, 885–892. [Google Scholar] [CrossRef]
- Rao, Z.; Liu, S.; Li, Z.; Wang, Q.; Gao, F.; Peng, H.; Ren, D.; Zang, Y.; Li, H.; Li, Y.; et al. Alarmin-loaded extracellular lipid droplets induce airway neutrophil infiltration during type 2 inflammation. Immunity 2024, 57, 2514–2529.e2517. [Google Scholar] [CrossRef]
- Stockis, J.; Yip, T.; Moreno-Vicente, J.; Burton, O.; Samarakoon, Y.; Schuijs, M.J.; Raghunathan, S.; Garcia, C.; Luo, W.; Whiteside, S.K.; et al. Cross-talk between ILC2 and Gata3 high Tregs locally constrains adaptive type 2 immunity. Sci. Immunol. 2024, 9, eadl1903. [Google Scholar] [CrossRef]
- Wang, Y.; Li, D.; Liu, Y.; Chen, S.; Dong, Z. Adaptive immune cells antagonize ILC2 homeostasis via SLAMF3 and SLAMF5. Sci. Adv. 2025, 11, eadp9894. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Domingo, P.; Romera-Hernandez, M.; Karrich, J.J.; Cornelissen, F.; Papazian, N.; Lindenbergh-Kortleve, D.J.; Butler, J.A.; Boon, L.; Coles, M.C.; Samsom, J.N.; et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J. Exp. Med. 2015, 212, 1783–1791. [Google Scholar] [CrossRef]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.M.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human NK cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef]
- Jarade, A.; Garcia, Z.; Marie, S.; Demera, A.; Prinz, I.; Bousso, P.; Di Santo, J.P.; Serafini, N. Inflammation triggers ILC3 patrolling of the intestinal barrier. Nat. Immunol. 2022, 23, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Fehlings, M.; Kløverpris, H.N.; McGovern, N.; Koo, S.-L.; Loh, C.Y.; Lim, S.; Kurioka, A.; Fergusson, J.R.; Tang, C.-L.; et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 2017, 46, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Takatori, H.; Kanno, Y.; Watford, W.T.; Tato, C.M.; Weiss, G.; Ivanov, I.I.; Littman, D.R.; O’Shea, J.J. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 2009, 206, 35–41. [Google Scholar] [CrossRef]
- Luci, C.; Reynders, A.; Ivanov, I.I.; Cognet, C.; Chiche, L.; Chasson, L.; Hardwigsen, J.; Anguiano, E.; Banchereau, J.; Chaussabel, D.; et al. Influence of the transcription factor RORγt on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 2009, 10, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Sawa, S.; Cherrier, M.; Lochner, M.; Satoh-Takayama, N.; Fehling, H.J.; Langa, F.; Di Santo, J.P.; Eberl, G. Lineage relationship analysis of RORγt+ innate lymphoid cells. Science 2010, 330, 665–669. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Vosshenrich, C.A.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef]
- Clark, P.A.; Gogoi, M.; Rodriguez-Rodriguez, N.; Ferreira, A.C.F.; Murphy, J.E.; Walker, J.A.; Crisp, A.; Jolin, H.E.; Shields, J.D.; McKenzie, A.N.J. Recipient tissue microenvironment determines developmental path of intestinal innate lymphoid progenitors. Nat. Commun. 2024, 15, 7809. [Google Scholar] [CrossRef]
- Das, A.; Martinez-Ruiz, G.U.; Bouladoux, N.; Stacy, A.; Moraly, J.; Vega-Sendino, M.; Zhao, Y.; Lavaert, M.; Ding, Y.; Morales-Sanchez, A.; et al. Transcription factor Tox2 is required for metabolic adaptation and tissue residency of ILC3 in the gut. Immunity 2024, 57, 1019–1036.e1019. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural Aryl Hydrocarbon Receptor Ligands Control Organogenesis of Intestinal Lymphoid Follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.-m.E.; Fish, K.; Fu, Y.-X.; Zhou, L. The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef]
- Chiang, H.Y.; Lu, H.H.; Sudhakar, J.N.; Chen, Y.W.; Shih, N.S.; Weng, Y.T.; Shui, J.W. IL-22 initiates an IL-18-dependent epithelial response circuit to enforce intestinal host defence. Nat. Commun. 2022, 13, 874. [Google Scholar] [CrossRef]
- He, G.W.; Lin, L.; DeMartino, J.; Zheng, X.; Staliarova, N.; Dayton, T.; Begthel, H.; van de Wetering, W.J.; Bodewes, E.; van Zon, J.; et al. Optimized human intestinal organoid model reveals interleukin-22-dependency of paneth cell formation. Cell Stem Cell 2022, 29, 1333–1345.e1336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhou, L.; Zhou, J.; Chu, C.; Zhang, C.; Sockolow, R.E.; Eberl, G.; Sonnenberg, G.F. ZBTB46 defines and regulates ILC3s that protect the intestine. Nature 2022, 609, 159–165. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, J.; Song, P.; Huang, J.; Yang, Z.; Han, J.; Wu, L.; Guo, X. p38α–eIF6–Nsun2 axis promotes ILC3’s rapid response to protect host from intestinal inflammation. J. Exp. Med. 2024, 222, e20240624. [Google Scholar] [CrossRef]
- Krzywinska, E.; Sobecki, M.; Nagarajan, S.; Zacharjasz, J.; Tambuwala, M.M.; Pelletier, A.; Cummins, E.; Gotthardt, D.; Fandrey, J.; Kerdiles, Y.M.; et al. The transcription factor HIF-1α mediates plasticity of NKp46+ innate lymphoid cells in the gut. J. Exp. Med. 2022, 219. [Google Scholar] [CrossRef] [PubMed]
- Pascal, M.; Kazakov, A.; Chevalier, G.; Dubrule, L.; Deyrat, J.; Dupin, A.; Saha, S.; Jagot, F.; Sailor, K.; Dulauroy, S.; et al. The neuropeptide VIP potentiates intestinal innate type 2 and type 3 immunity in response to feeding. Mucosal Immunol. 2022, 15, 629–641. [Google Scholar] [CrossRef]
- Seillet, C.; Luong, K.; Tellier, J.; Jacquelot, N.; Shen, R.D.; Hickey, P.; Wimmer, V.C.; Whitehead, L.; Rogers, K.; Smyth, G.K.; et al. The neuropeptide VIP confers anticipatory mucosal immunity by regulating ILC3 activity. Nat. Immunol. 2020, 21, 168–177. [Google Scholar] [CrossRef]
- Yu, H.B.; Yang, H.; Allaire, J.M.; Ma, C.; Graef, F.A.; Mortha, A.; Liang, Q.; Bosman, E.S.; Reid, G.S.; Waschek, J.A.; et al. Vasoactive intestinal peptide promotes host defense against enteric pathogens by modulating the recruitment of group 3 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2106634118. [Google Scholar] [CrossRef] [PubMed]
- Serafini, N.; Jarade, A.; Surace, L.; Goncalves, P.; Sismeiro, O.; Varet, H.; Legendre, R.; Coppee, J.-Y.; Disson, O.; Durum, S.K.; et al. Trained ILC3 responses promote intestinal defense. Science 2022, 375, 859–863. [Google Scholar] [CrossRef]
- Romera-Hernández, M.; Aparicio-Domingo, P.; Papazian, N.; Karrich, J.J.; Cornelissen, F.; Hoogenboezem, R.M.; Samsom, J.N.; Cupedo, T. Yap1-Driven Intestinal Repair Is Controlled by Group 3 Innate Lymphoid Cells. Cell Rep. 2020, 30, 37–45.e33. [Google Scholar] [CrossRef]
- Zhou, L.; Zhou, W.; Joseph, A.M.; Chu, C.; Putzel, G.G.; Fang, B.; Teng, F.; Lyu, M.; Yano, H.; Andreasson, K.I.; et al. Group 3 innate lymphoid cells produce the growth factor HB-EGF to protect the intestine from TNF-mediated inflammation. Nat. Immunol. 2022, 23, 251–261. [Google Scholar] [CrossRef]
- Constantinides, M.G.; McDonald, B.D.; Verhoef, P.A.; Bendelac, A. A committed precursor to innate lymphoid cells. Nature 2014, 508, 397–401. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, H.; Wu, S.; Liu, J.; Liu, H.; Wang, D.; Zhang, Y.; Niu, H.; Su, X.; Sun, J.; et al. PLZF restricts intestinal ILC3 function in gut defense. Cell. Mol. Immunol. 2023, 20, 379–388. [Google Scholar] [CrossRef]
- Liu, B.; Ye, B.; Zhu, X.; Yang, L.; Li, H.; Liu, N.; Zhu, P.; Lu, T.; He, L.; Tian, Y.; et al. An inducible circular RNA circKcnt2 inhibits ILC3 activation to facilitate colitis resolution. Nat. Commun. 2020, 11, 4076. [Google Scholar] [CrossRef] [PubMed]
- Cosovanu, C.; Neumann, C. The Many Functions of Foxp3(+) Regulatory T Cells in the Intestine. Front. Immunol. 2020, 11, 600973. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef]
- Griseri, T.; Asquith, M.; Thompson, C.; Powrie, F. OX40 is required for regulatory T cell–mediated control of colitis. J. Exp. Med. 2010, 207, 699–709. [Google Scholar] [CrossRef]
- Deng, T.; Suo, C.; Chang, J.; Yang, R.; Li, J.; Cai, T.; Qiu, J. ILC3-derived OX40L is essential for homeostasis of intestinal Tregs in immunodeficient mice. Cell. Mol. Immunol. 2020, 17, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Ulezko Antonova, A.; Lonardi, S.; Monti, M.; Missale, F.; Fan, C.; Coates, M.L.; Bugatti, M.; Jaeger, N.; Fernandes Rodrigues, P.; Brioschi, S.; et al. A distinct human cell type expressing MHCII and RORγt with dual characteristics of dendritic cells and type 3 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2023, 120, e2318710120. [Google Scholar] [CrossRef]
- Lyu, M.; Suzuki, H.; Kang, L.; Gaspal, F.; Zhou, W.; Goc, J.; Zhou, L.; Zhou, J.; Zhang, W.; Shen, Z.; et al. ILC3s select microbiota-specific regulatory T cells to establish tolerance in the gut. Nature 2022, 610, 744–751. [Google Scholar] [CrossRef]
- Kedmi, R.; Najar, T.A.; Mesa, K.R.; Grayson, A.; Kroehling, L.; Hao, Y.; Hao, S.; Pokrovskii, M.; Xu, M.; Talbot, J.; et al. A RORγt(+) cell instructs gut microbiota-specific T(reg) cell differentiation. Nature 2022, 610, 737–743. [Google Scholar] [CrossRef]
- Akagbosu, B.; Tayyebi, Z.; Shibu, G.; Paucar Iza, Y.A.; Deep, D.; Parisotto, Y.F.; Fisher, L.; Pasolli, H.A.; Thevin, V.; Elmentaite, R.; et al. Novel antigen-presenting cell imparts T(reg)-dependent tolerance to gut microbiota. Nature 2022, 610, 752–760. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria–specific CD4+ T cells. Science 2015. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Monticelli, L.A.; Fung, T.C.; Ziegler, C.G.K.; Grunberg, S.; Sinha, R.; Mantegazza, A.R.; Ma, H.-L.; Crawford, A.; Angelosanto, J.M.; et al. Innate lymphoid cells regulate CD4+ T cell responses to intestinal commensal bacteria. Nature 2013, 498, 113. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.F.; Wu, S.; Trsan, T.; Panda, S.K.; Fachi, J.L.; Liu, Y.; Du, S.; de Oliveira, S.; Antonova, A.U.; Khantakova, D.; et al. Rorγt-positive dendritic cells are required for the induction of peripheral regulatory T cells in response to oral antigens. Cell 2025. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Doty, A.L.; Tang, Y.; Berrie, D.; Iqbal, A.; Tan, S.A.; Clare-Salzler, M.J.; Wallet, S.M.; Glover, S.C. Enrichment of IL-17A(+) IFN-γ(+) and IL-22(+) IFN-γ(+) T cell subsets is associated with reduction of NKp44(+) ILC3s in the terminal ileum of Crohn’s disease patients. Clin. Exp. Immunol. 2017, 190, 143–153. [Google Scholar] [CrossRef]
- Elmentaite, R.; Kumasaka, N.; Roberts, K.; Fleming, A.; Dann, E.; King, H.W.; Kleshchevnikov, V.; Dabrowska, M.; Pritchard, S.; Bolt, L.; et al. Cells of the human intestinal tract mapped across space and time. Nature 2021, 597, 250–255. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e722. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liu, C.; Gu, Z.; Yang, X.; Lan, X.; Guo, X. Dysregulation of Wnt/β-catenin signaling contributes to intestinal inflammation through regulation of group 3 innate lymphoid cells. Nat. Commun. 2024, 15, 2820. [Google Scholar] [CrossRef]
- Ahmed, A.; Joseph, A.M.; Zhou, J.; Horn, V.; Uddin, J.; Lyu, M.; Goc, J.; Bank, J.R.I.L.C.; Sockolow, R.E.; Wing, J.B.; et al. CTLA-4-expressing ILC3s restrain interleukin-23-mediated inflammation. Nature 2024, 630, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.W.; Schroeder, J.-H.; Roberts, L.B.; Mohamed, R.; Cozzetto, D.; Beattie, G.; Omer, O.S.; Ross, E.M.; Heuts, F.; Jowett, G.M.; et al. CTLA-4 expressing innate lymphoid cells modulate mucosal homeostasis in a microbiota dependent manner. Nat. Commun. 2024, 15, 9520. [Google Scholar] [CrossRef]
- Ninnemann, J.; Winsauer, C.; Bondareva, M.; Kühl, A.A.; Lozza, L.; Durek, P.; Lissner, D.; Siegmund, B.; Kaufmann, S.H.E.; Mashreghi, M.-F.; et al. TNF hampers intestinal tissue repair in colitis by restricting IL-22 bioavailability. Mucosal Immunol. 2022, 15, 698–716. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Yang, J.; Zhai, Y.; Zhang, H.; Zhou, Y.; Hong, L.; Yuan, D.; Xia, R.; Liu, Y.; Pan, J.; et al. Nucleophosmin 1 promotes mucosal immunity by supporting mitochondrial oxidative phosphorylation and ILC3 activity. Nat. Immunol. 2024. [Google Scholar] [CrossRef]
- Horn, V.; Sonnenberg, G.F. Group 3 innate lymphoid cells in intestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 428–443. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Liu, G.; Yi, X.; Wu, J.; Cao, H.; Zhang, L.; Zhou, P.; Fan, Y.; Yu, Y.; et al. NRP1 instructs IL-17-producing ILC3s to drive colitis progression. Cell. Mol. Immunol. 2025, 1–15. [Google Scholar] [CrossRef]
- Wu, X.; Khatun, A.; Kasmani, M.Y.; Chen, Y.; Zheng, S.; Atkinson, S.; Nguyen, C.; Burns, R.; Taparowsky, E.J.; Salzman, N.H.; et al. Group 3 innate lymphoid cells require BATF to regulate gut homeostasis in mice. J. Exp. Med. 2022. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef]
- Siakavellas, S.I.; Bamias, G. Tumor Necrosis Factor-like Cytokine TL1A and Its Receptors DR3 and DcR3: Important New Factors in Mucosal Homeostasis and Inflammation. Inflamm. Bowel Dis. 2015, 21, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, J.G.; Woo, V.; Viladomiu, M.; Putzel, G.; Lima, S.; Diehl, G.E.; Marderstein, A.R.; Gandara, J.; Perez, A.R.; Withers, D.R.; et al. Microbiota-Induced TNF-like Ligand 1A Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation during Colitis. Immunity 2018, 49, 1077–1089.e1075. [Google Scholar] [CrossRef]
- Li, J.; Shi, W.; Sun, H.; Ji, Y.; Chen, Y.; Guo, X.; Sheng, H.; Shu, J.; Zhou, L.; Cai, T.; et al. Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nat. Commun. 2019, 10, 3371. [Google Scholar] [CrossRef]
- Powell, N.; Pantazi, E.; Pavlidis, P.; Tsakmaki, A.; Li, K.; Yang, F.; Parker, A.; Pin, C.; Cozzetto, D.; Minns, D.; et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut 2020, 69, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Tsakmaki, A.; Pantazi, E.; Li, K.; Cozzetto, D.; Digby- Bell, J.; Yang, F.; Lo, J.W.; Alberts, E.; Sa, A.C.C.; et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat. Commun. 2022, 13, 5820. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.S.; Gaifem, J.; Pereira, M.S.; Alves, M.F.; Silva, M.; Padrão, N.; Cavadas, B.; Moreira-Barbosa, C.; Alves, I.; Marcos-Pinto, R.; et al. Alterations in mucosa branched N-glycans lead to dysbiosis and downregulation of ILC3: A key driver of intestinal inflammation. Gut Microbes 2025, 17, 2461210. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef]
- Cao, S.; Fachi, J.L.; Ma, K.; Ulezko Antonova, A.; Wang, Q.; Cai, Z.; Kaufman, R.J.; Ciorba, M.A.; Deepak, P.; Colonna, M. The IRE1α/XBP1 pathway sustains cytokine responses of group 3 innate lymphoid cells in inflammatory bowel disease. J. Clin. Investig. 2024, 134, e174198. [Google Scholar] [CrossRef]
- Shao, F.; Liu, Z.; Wei, Q.; Yu, D.; Zhao, M.; Zhang, X.; Gao, X.; Fan, Z.; Wang, S. FOXO1 orchestrates the intestinal homeostasis via neuronal signaling in group 3 innate lymphoid cells. J. Exp. Med. 2023, 220, e20230133. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, T.; Wang, Y.; Liu, R.; Chang, M.; Wang, X. Effects of oral vitamin D supplementation on inflammatory bowel disease: A systematic review and meta-analysis. Food Funct. 2021, 12, 7588–7606. [Google Scholar] [CrossRef]
- Dan, L.; Wang, S.; Chen, X.; Sun, Y.; Fu, T.; Deng, M.; Chen, J.; Du, Z.; Wang, X. Circulating 25-hydroxyvitamin D concentration can predict bowel resection risk among individuals with inflammatory bowel disease in a longitudinal cohort with 13 years of follow-up. Int. J. Surg. 2024, 110, 4275–4285. [Google Scholar] [CrossRef]
- Konya, V.; Czarnewski, P.; Forkel, M.; Rao, A.; Kokkinou, E.; Villablanca, E.J.; Almer, S.; Lindforss, U.; Friberg, D.; Höög, C.; et al. Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J. Allergy Clin. Immunol. 2018, 141, 279–292. [Google Scholar] [CrossRef]
- Spencer, S.P.; Wilhelm, C.; Yang, Q.; Hall, J.A.; Bouladoux, N.; Boyd, A.; Nutman, T.B.; Urban, J.F., Jr.; Wang, J.; Ramalingam, T.R.; et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 2014, 343, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Doan, H.T.; Cheng, L.-C.; Chiu, Y.-L.; Cheng, Y.-K.; Hsu, C.-C.; Chen, Y.-C.; Lo, H.-J.; Chiang, H.-S. Candida tropicalis-derived vitamin B3 exerts protective effects against intestinal inflammation by promoting IL-17A/IL-22-dependent epithelial barrier function. Gut Microbes 2024, 16, 2416922. [Google Scholar] [CrossRef]
- Liu, H.; Huang, R.; Shen, B.; Huang, C.; Zhou, Q.; Xu, J.; Chen, S.; Lin, X.; Wang, J.; Zhao, X.; et al. Live Akkermansia muciniphila boosts dendritic cell retinoic acid synthesis to modulate IL-22 activity and mitigate colitis in mice. Microbiome 2024, 12, 275. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Yan, X.; Liu, Y.; Huang, L.; Zhu, Y.; He, J.; Gao, R.; Kalady, M.F.; Goel, A.; Qin, H.; et al. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct Target Ther. 2021, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Diwakarla, S.; Chatzis, R.; Artaiz, O.; Macowan, M.; Zhang, S.; Garnham, A.; Morgan, P.K.; Mellett, N.A.; Meikle, P.J.; et al. Acute exposure to high-fat diet impairs ILC3 functions and gut homeostasis. Immunity 2025, 58, 1185–1200. [Google Scholar] [CrossRef]
- Xiong, L.; Helm, E.Y.; Dean, J.W.; Sun, N.; Jimenez-Rondan, F.R.; Zhou, L. Nutrition impact on ILC3 maintenance and function centers on a cell-intrinsic CD71-iron axis. Nat. Immunol. 2023, 24, 1671–1684. [Google Scholar] [CrossRef]
- Sivori, S.; Pende, D.; Quatrini, L. NK cells and ILCs in tumor immunotherapy. Mol. Asp. Med. 2020, 80, 100870. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, H.; Jounaidi, Y. Comprehensive snapshots of natural killer cells functions, signaling, molecular mechanisms and clinical utilization. Signal Transduct Target Ther. 2024, 9, 302. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Serafini, N.; Vosshenrich, C.A.; Santo, J.P. Transcriptional regulation of innate lymphoid cell fate. Nat. Rev. Immunol. 2015, 15, 415–428. [Google Scholar] [CrossRef]
- Kennedy, M.K.; Glaccum, M.; Brown, S.N.; Butz, E.A.; Viney, J.L.; Embers, M.; Matsuki, N.; Charrier, K.; Sedger, L.; Willis, C.R.; et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 2000, 191, 771–780. [Google Scholar] [CrossRef]
- Takayama, T.; Kamada, N.; Chinen, H.; Okamoto, S.; Kitazume, M.T.; Chang, J.; Matuzaki, Y.; Suzuki, S.; Sugita, A.; Koganei, K.; et al. Imbalance of NKp44+NKp46− and NKp44−NKp46+ natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology 2010, 139, 882–892.e3. [Google Scholar] [CrossRef] [PubMed]
- Baumdick, M.E.; Niehrs, A.; Degenhardt, F.; Schwerk, M.; Hinrichs, O.; Jordan-Paiz, A.; Padoan, B.; Wegner, L.H.M.; Schloer, S.; Zecher, B.F.; et al. HLA-DP on Epithelial Cells Enables Tissue Damage by NKp44(+) Natural Killer Cells in Ulcerative Colitis. Gastroenterology 2023, 165, 946–962.e913. [Google Scholar] [CrossRef] [PubMed]
- Samarani, S.; Sagala, P.; Jantchou, P. Phenotypic and functional changes in peripheral blood natural killer cells in Crohn disease patients. Med. Inflamm. 2020, 2020, 6401969. [Google Scholar] [CrossRef]
- Zaiatz Bittencourt, V.; Jones, F.; Tosetto, M.; Doherty, G.A.; Ryan, E.J. Dysregulation of Metabolic Pathways in Circulating Natural Killer Cells Isolated from Inflammatory Bowel Disease Patients. J. Crohn’s Colitis 2021, 15, 1316–1325. [Google Scholar] [CrossRef]
- Bank, U.; Deiser, K.; Plaza-Sirvent, C.; Osbelt, L.; Witte, A.; Knop, L.; Labrenz, R.; Jänsch, R.; Richter, F.; Biswas, A.; et al. c-FLIP is crucial for IL-7/IL-15-dependent NKp46(+) ILC development and protection from intestinal inflammation in mice. Nat. Commun. 2020, 11, 1056. [Google Scholar] [CrossRef]
- Wagner, J.A.; Rosario, M.; Romee, R.; Berrien-Elliott, M.M.; Schneider, S.E.; Leong, J.W.; Sullivan, R.P.; Jewell, B.A.; Becker-Hapak, M.; Schappe, T.; et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J. Clin. Investig. 2017, 127, 4042–4058. [Google Scholar] [CrossRef]
- Liu, Z.; Geboes, K.; Colpaert, S.; D’Haens, G.R.; Rutgeerts, P.; Ceuppens, J.L. IL-15 is highly expressed in inflammatory bowel disease and regulates local T cell-dependent cytokine production. J. Immunol. 2000, 164, 3608–3615. [Google Scholar] [CrossRef]
- Monteleone, G.; Monteleone, I.; Fina, D.; Vavassori, P.; Del Vecchio Blanco, G.; Caruso, R.; Tersigni, R.; Alessandroni, L.; Biancone, L.; Naccari, G.C.; et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology 2005, 128, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, L.; Cui, Y.; Wang, X.; Guo, C.; Huang, Z.; Kan, Q.; Liu, Z.; Liu, Y. Il-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1133–1144. [Google Scholar] [CrossRef]
- Gaifem, J.; Rodrigues, C.S.; Petralia, F.; Alves, I.; Leite-Gomes, E.; Cavadas, B.; Dias, A.M.; Moreira-Barbosa, C.; Revés, J.; Laird, R.M.; et al. A unique serum IgG glycosylation signature predicts development of Crohn’s disease and is associated with pathogenic antibodies to mannose glycan. Nat. Immunol. 2024, 25, 1692–1703. [Google Scholar] [CrossRef]
- Cobb, L.M.; Verneris, M.R. Therapeutic manipulation of innate lymphoid cells. JCI Insight 2021, 6, e146006. [Google Scholar] [CrossRef] [PubMed]
- Lo Pizzo, M.; La Barbera, L.; Rizzo, C.; Mohammadnezhad, L.; Camarda, F.; Ciccia, F.; Guggino, G. JAK/STAT inhibition modifies the ILC1 immune response in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2024, 42, 593–600. [Google Scholar] [CrossRef]
- Eken, A.; Yetkin, M.F.; Vural, A.; Okus, F.Z.; Erdem, S.; Azizoglu, Z.B.; Haliloglu, Y.; Cakir, M.; Turkoglu, E.M.; Kilic, O.; et al. Fingolimod Alters Tissue Distribution and Cytokine Production of Human and Murine Innate Lymphoid Cells. Front. Immunol. 2019, 10, 217. [Google Scholar] [CrossRef]
- Xie, J.; Tian, S.; Liu, J.; Huang, S.; Yang, M.; Yang, X.; Xu, R.; Lin, J.; Han, L.; Zhang, D. Combination therapy with indigo and indirubin for ulcerative colitis via reinforcing intestinal barrier function. Oxidative Med. Cell. Longev. 2023, 2023, 2894695. [Google Scholar] [CrossRef]
- Mazzurana, L.; Forkel, M.; Rao, A.; Van Acker, A.; Kokkinou, E.; Ichiya, T.; Almer, S.; Höög, C.; Friberg, D.; Mjösberg, J. Suppression of Aiolos and Ikaros expression by lenalidomide reduces human ILC3-ILC1/NK cell transdifferentiation. Eur. J. Immunol. 2019, 49, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Becker, C.; Ivanov, I.I.; Hirth, S.; Wirtz, S.; Neufert, C.; Pouly, S.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 2009, 136, 257–267. [Google Scholar] [CrossRef]
- Vuyyuru, S.K.; Shackelton, L.M.; Hanzel, J.; Ma, C.; Jairath, V.; Feagan, B.G. Targeting IL-23 for IBD: Rationale and Progress to Date. Drugs 2023, 83, 873–891. [Google Scholar] [CrossRef]
- Stabile, H.; Scarno, G.; Fionda, C.; Gismondi, A.; Santoni, A.; Gadina, M.; Sciumè, G. JAK/STAT signaling in regulation of innate lymphoid cells: The gods before the guardians. Immunol. Rev. 2018, 286, 148–159. [Google Scholar] [CrossRef]
- Virtanen, A.; Spinelli, F.R.; Telliez, J.B.; O’Shea, J.J.; Silvennoinen, O.; Gadina, M. JAK inhibitor selectivity: New opportunities, better drugs? Nat. Rev. Rheumatol. 2024, 20, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Abo, H.; Flannigan, K.L.; Geem, D.; Ngo, V.L.; Harusato, A.; Denning, T.L. Combined IL-2 Immunocomplex and Anti-IL-5 mAb Treatment Expands Foxp3(+) Treg Cells in the Absence of Eosinophilia and Ameliorates Experimental Colitis. Front. Immunol. 2019, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Yang, Z.; Zhou, Y.; Yue, H.; Hua, L.; Liu, Z.; Lin, G.; Cai, H.; Chen, Y.; Hu, W.; et al. Antagonizing interleukin-5 receptor ameliorates dextran sulfate sodium-induced experimental colitis in mice through reducing NLRP3 inflammasome activation. Eur. J. Pharmacol. 2024, 965, 176331. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, W.; de Villiers, W.; Bene, L.; Simon, L.; Rácz, I.; Katz, S.; Altorjay, I.; Feagan, B.; Riff, D.; Bernstein, C.N. Fontolizumab in moderate to severe Crohn’s disease: A phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm. Bowel Dis. 2010, 16, 233–242. [Google Scholar] [CrossRef]
- Reinisch, W.; Panés, J.; Khurana, S.; Toth, G.; Hua, F.; Comer, G.M.; Hinz, M.; Page, K.; O’Toole, M.; Moorehead, T.M.; et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut 2015, 64, 894–900. [Google Scholar] [CrossRef]
- Danese, S.; Rudziński, J.; Brandt, W.; Dupas, J.L.; Peyrin-Biroulet, L.; Bouhnik, Y.; Kleczkowski, D.; Uebel, P.; Lukas, M.; Knutsson, M.; et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut 2015, 64, 243–249. [Google Scholar] [CrossRef]
- Roth, L.; MacDonald, J.K.; McDonald, J.W.; Chande, N. Sargramostim (GM-CSF) for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Krueger, J.G.; Wharton, K.A.; Schlitt, T.; Suprun, M.; Torene, R.I.; Jiang, X.; Wang, C.Q.; Fuentes-Duculan, J.; Hartmann, N.; Peters, T.; et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J. Allergy Clin. Immunol. 2019, 144, 750–763. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef]
- Wagner, F.; Mansfield, J.C.; Lekkerkerker, A.N.; Wang, Y.; Keir, M.; Dash, A.; Butcher, B.; Harder, B.; Orozco, L.D.; Mar, J.S.; et al. Dose escalation randomised study of efmarodocokin alfa in healthy volunteers and patients with ulcerative colitis. Gut 2023, 72, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Furfaro, F.; Vetrano, S. Targeting S1P in inflammatory bowel disease: New avenues for modulating intestinal leukocyte migration. J. Crohn’s Colitis 2018, 12, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Karuppuchamy, T.; Behrens, E.h.; González-Cabrera, P.; Sarkisyan, G.; Gima, L.; Boyer, J.D.; Bamias, G.; Jedlicka, P.; Veny, M.; Clark, D.; et al. Sphingosine-1-phosphate receptor-1 (S1P1) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017, 10, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Dutton, E.E.; Gajdasik, D.W.; Willis, C.; Fiancette, R.; Bishop, E.L.; Camelo, A.; Sleeman, M.A.; Coccia, M.; Didierlaurent, A.M.; Tomura, M.; et al. Peripheral lymph nodes contain migratory and resident innate lymphoid cell populations. Sci. Immunol. 2019, 4, eaau8082. [Google Scholar] [CrossRef]
- Huang, Y.; Mao, K.; Chen, X.; Sun, M.A.; Kawabe, T.; Li, W.; Usher, N.; Zhu, J.; Urban, J.F., Jr.; Paul, W.E.; et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Vermeire, S.; Peyrin-Biroulet, L.; Dubinsky, M.C.; Panes, J.; Yarur, A.; Ritter, T.; Baert, F.; Schreiber, S.; Sloan, S.; et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): Two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet 2023, 401, 1159–1171. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; D’Haens, G.; Wolf, D.C.; Jovanovic, I.; Hanauer, S.B.; Ghosh, S.; Petersen, A.; Hua, S.Y.; Lee, J.H.; et al. Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2021, 385, 1280–1291. [Google Scholar] [CrossRef]
- Sands, B.E.; Feagan, B.G.; Peyrin-Biroulet, L.; Danese, S.; Rubin, D.T.; Laurent, O.; Luo, A.; Nguyen, D.D.; Lu, J.; Yen, M.; et al. Phase 2 Trial of Anti-TL1A Monoclonal Antibody Tulisokibart for Ulcerative Colitis. N. Engl. J. Med. 2024, 391, 1119–1129. [Google Scholar] [CrossRef]
- Bamias, G.; Martin, C.; Marini, M.; Hoang, S.; Mishina, M.; Ross, W.G.; Sachedina, M.A.; Friel, C.M.; Mize, J.; Bickston, S.J.; et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J. Immunol. 2003, 171, 4868–4874. [Google Scholar] [CrossRef]
- Jin, S.; Chin, J.; Seeber, S.; Niewoehner, J.; Weiser, B.; Beaucamp, N.; Woods, J.; Murphy, C.; Fanning, A.; Shanahan, F.; et al. TL1A/TNFSF15 directly induces proinflammatory cytokines, including TNFα, from CD3+CD161+ T cells to exacerbate gut inflammation. Mucosal Immunol. 2013, 6, 886–899. [Google Scholar] [CrossRef]
- Bamias, G.; Menghini, P.; Pizarro, T.T.; Cominelli, F. Targeting TL1A and DR3: The new frontier of anti-cytokine therapy in IBD. Gut 2025, 74, 652–668. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Pappu, R.; Ramirez-Carrozzi, V.; Ota, N.; Caplazi, P.; Zhang, J.; Yan, D.; Xu, M.; Lee, W.P.; Grogan, J.L. TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers. Mucosal Immunol. 2014, 7, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.O.; Weeres, M.A.; Neulen, M.L.; Choi, J.; Kang, S.H.; Heo, D.S.; Bergerson, R.; Blazar, B.R.; Miller, J.S.; Verneris, M.R. Human group3 innate lymphoid cells express DR3 and respond to TL1A with enhanced IL-22 production and IL-2-dependent proliferation. Eur. J. Immunol. 2015, 45, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lin, C.; Tan, H.-Y.; Bian, Z.-x. The double-edged sword effect of indigo naturalis. Food Chem. Toxicol. 2024, 185, 114476. [Google Scholar] [CrossRef]
- Naganuma, M.; Sugimoto, S.; Mitsuyama, K.; Kobayashi, T.; Yoshimura, N.; Ohi, H.; Tanaka, S.; Andoh, A.; Ohmiya, N.; Saigusa, K.; et al. Efficacy of Indigo Naturalis in a Multicenter Randomized Controlled Trial of Patients With Ulcerative Colitis. Gastroenterology 2018, 154, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Boguniewicz, M.; Quintana, F.J.; Clark, R.A.; Gross, L.; Hirano, I.; Tallman, A.M.; Brown, P.M.; Fredericks, D.; Rubenstein, D.S.; et al. Tapinarof validates the aryl hydrocarbon receptor as a therapeutic target: A clinical review. J. Allergy Clin. Immunol. 2024, 154, 1–10. [Google Scholar] [CrossRef]
- Paller, A.S.; Stein Gold, L.; Soung, J.; Tallman, A.M.; Rubenstein, D.S.; Gooderham, M. Efficacy and patient-reported outcomes from a phase 2b, randomized clinical trial of tapinarof cream for the treatment of adolescents and adults with atopic dermatitis. J. Am. Acad. Dermatol. 2021, 84, 632–638. [Google Scholar] [CrossRef]
- Mears, K.S.; Denny, J.E.; Maslanka, J.R.; Mdluli, N.V.; Hulit, E.N.; Matsuda, R.; Furth, E.E.; Buffie, C.G.; Abt, M.C. Therapeutic activation of IL-22-producing innate lymphoid cells enhances host defenses to Clostridioides difficile infection. Cell Rep. 2025, 44, 115438. [Google Scholar] [CrossRef]
- Troch, K.F.; Jakob, M.O.; Forster, P.M.; Jarick, K.J.; Schreiber, J.; Preusser, A.; Guerra, G.M.; Durek, P.; Tizian, C.; Sterczyk, N.; et al. Group 2 innate lymphoid cells are a non-redundant source of interleukin-5 required for development and function of murine B1 cells. Nat. Commun. 2024, 15, 10566. [Google Scholar] [CrossRef]
- Withers, D.R.; Hepworth, M.R.; Wang, X.; Mackley, E.C.; Halford, E.E.; Dutton, E.E.; Marriott, C.L.; Brucklacher-Waldert, V.; Veldhoen, M.; Kelsen, J.; et al. Transient inhibition of ROR-γt therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat. Med. 2016, 22, 319–323. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Treatments | Mechanism | Effects on Humans or Mice | Circulating or Tissue-Resident | Impacts on ILCs | ||
|---|---|---|---|---|---|---|
| Increased | Decreased | |||||
| Approved | Infliximab or Adalimumab [61] | anti-TNFα | Humans | Intestinal | NCR+ ILC3s | ILC1s |
| Circulating | NCR- ILC3s | ILC1s | ||||
| Vedolizumab [61] | anti-α4β7 integrin | Humans | Intestinal | NCR+ILC3s | ILC1s | |
| Circulating | NCR− ILC3 | - | ||||
| Ustekinumab [61] | anti-IL-12/IL-23 | Humans | Intestinal | NCR+ ILC3s | NCR- ILC3 | |
| Circulating | ILC1s | - | ||||
| Tofacitinib [196] (rheumatoid arthritis) | JAK1/JAK3 inhibitor | Humans | Circulating | - | IFN-γ+ ILC1s | |
| Fingolimod [197] (multiple sclerosis) | S1PR modulator | Mice | Intestinal | - | ILC2s and ILC3s | |
| Circulating | - | Total ILCs | ||||
| Humans | Tonsillar | - | IFN-γ+ ILC1s GM-CSF+ ILC3s | |||
| Clinical trials | DR3-Fc [165] | Anti-TL1A | Mice | Intestinal | Restored ILC3s | GM-CSF+ ILC3s |
| Indigo naturalis [198] | AHR agonist | Mice | Intestinal | NK cells | ILC2s and ILC3s | |
| Lenalidomide [199] | Degrades Ikaros and Aiolos | Mice | Tonsillar | Restored ILC3s | ILC1s and NK cells | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, X.; Ma, K.; Zhu, Y.; Cao, S. Innate Lymphoid Cells in Inflammatory Bowel Disease. Cells 2025, 14, 825. https://doi.org/10.3390/cells14110825
Yao X, Ma K, Zhu Y, Cao S. Innate Lymphoid Cells in Inflammatory Bowel Disease. Cells. 2025; 14(11):825. https://doi.org/10.3390/cells14110825
Chicago/Turabian StyleYao, Xin, Kaiming Ma, Yangzhuangzhuang Zhu, and Siyan Cao. 2025. "Innate Lymphoid Cells in Inflammatory Bowel Disease" Cells 14, no. 11: 825. https://doi.org/10.3390/cells14110825
APA StyleYao, X., Ma, K., Zhu, Y., & Cao, S. (2025). Innate Lymphoid Cells in Inflammatory Bowel Disease. Cells, 14(11), 825. https://doi.org/10.3390/cells14110825