
Integrated Analysis of DNA Methylome and Transcriptome Reveals Regulatory Mechanism in the Longissimus Dorsi of Duroc Pigs

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. RNA Extraction, Strand-Specific Library Construction, and Sequencing
2.3. DNA Isolation, BS-Seq Library Construction, and Sequencing
2.4. Correlation of DNA Methylation and Gene Expression in Samples
2.5. Correlation of DNA Methylation and Gene Expression Between H and L Groups
2.6. Functional Enrichment Analysis of Differentially Methylated and Differentially Expressed Genes
2.7. RNA Interference and Overexpression of LPAR1
2.8. Cell Culture and 5-Aza Treatment
2.9. Quantitative Real-Time PCR
2.10. Western Blotting
2.11. Immunofluorescence Assay
2.12. Luciferase Reporter Assay
2.13. Electrophoretic Mobility Shift Assay
2.14. Statistical Analysis
3. Results
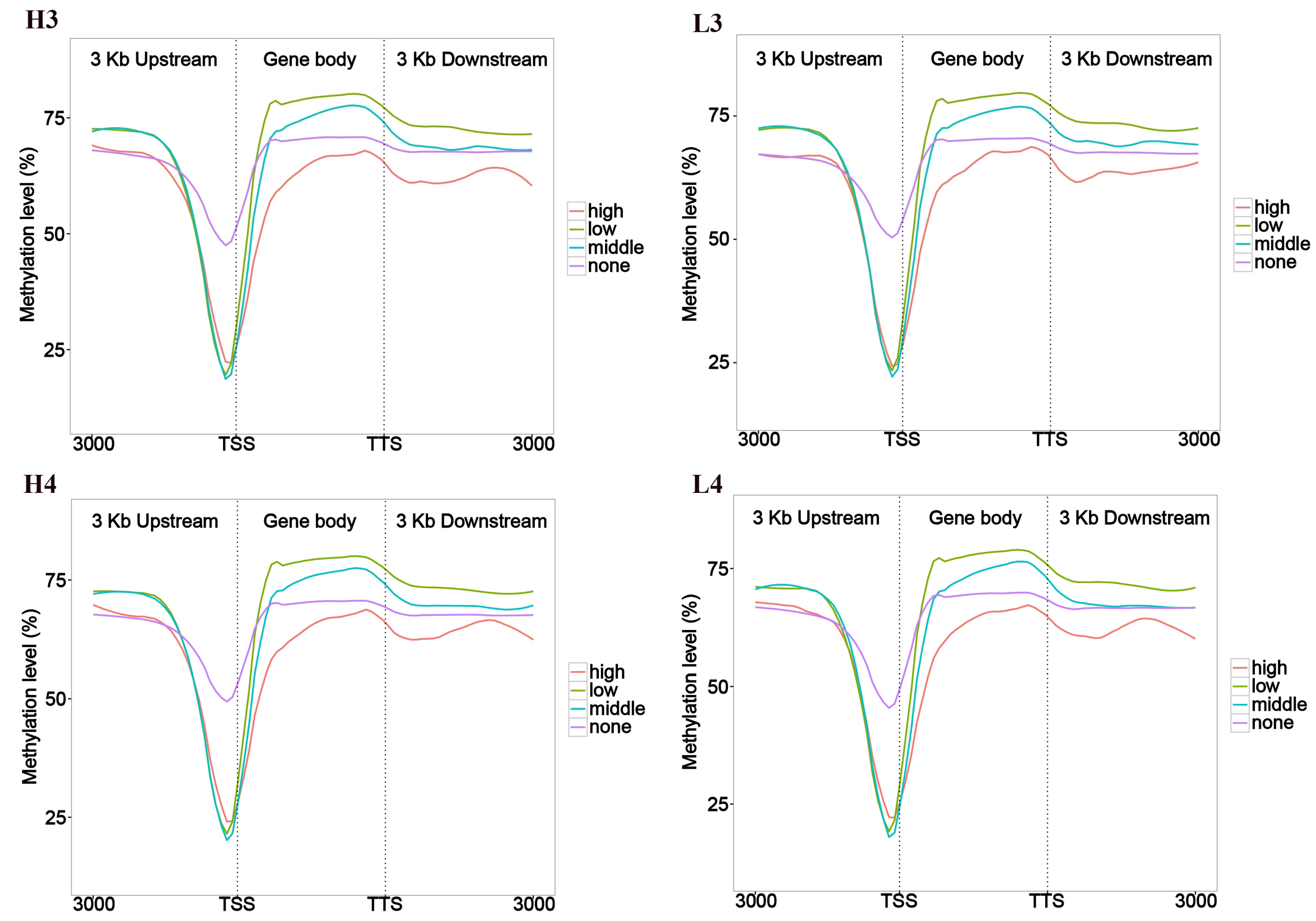
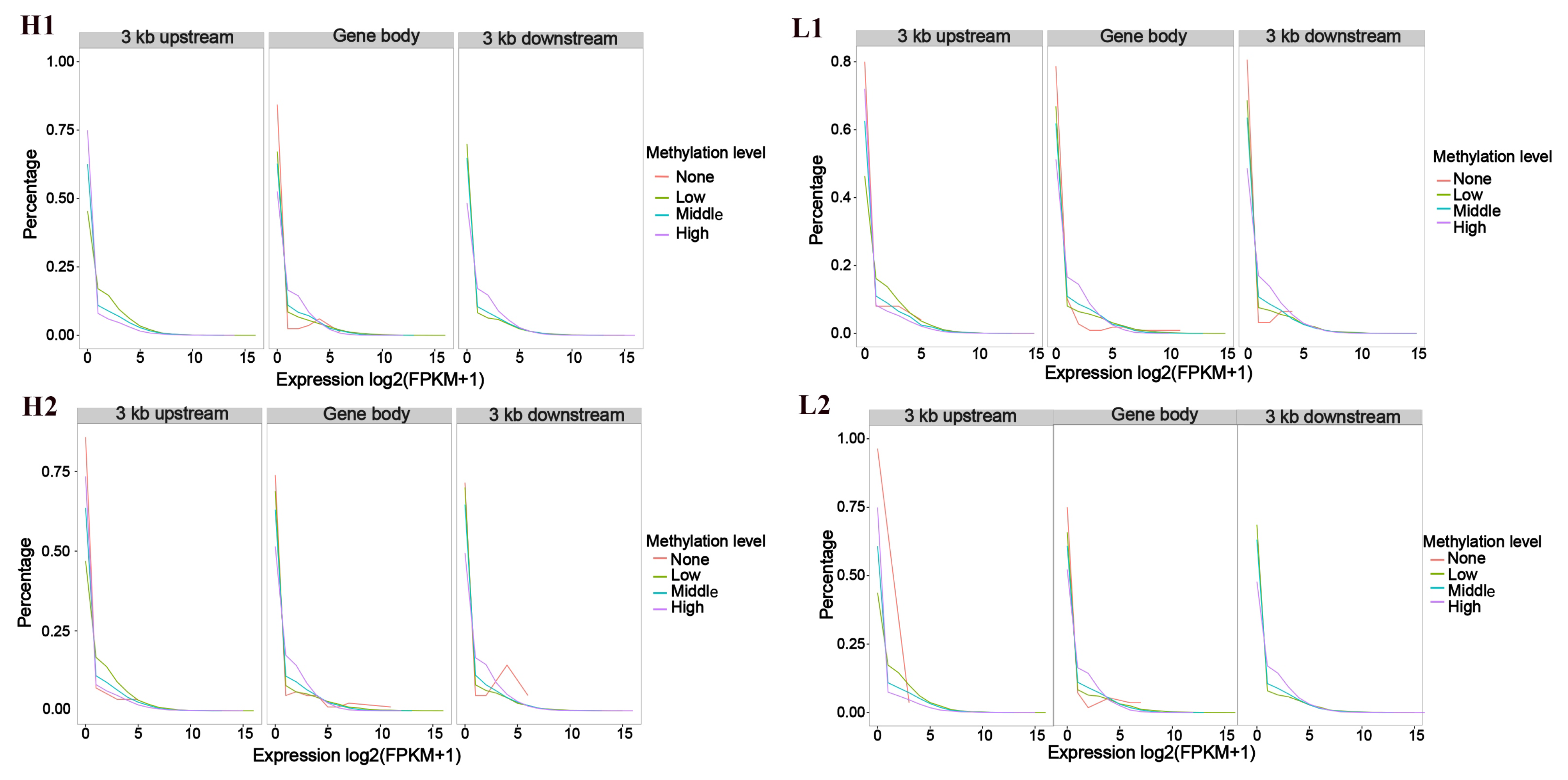
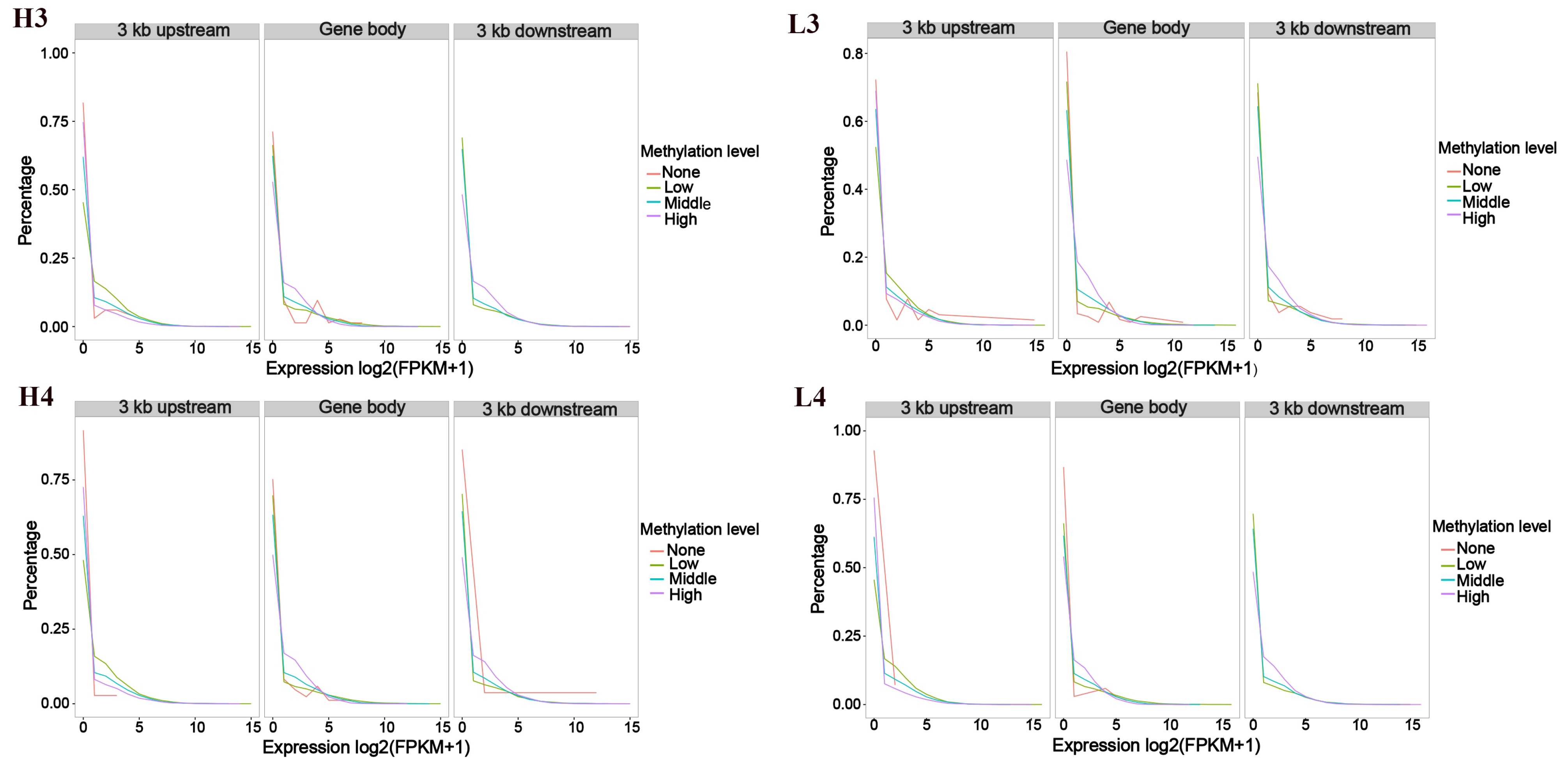
3.1. Association Analysis Between Gene Expression and Methylation Rates Within the Sample
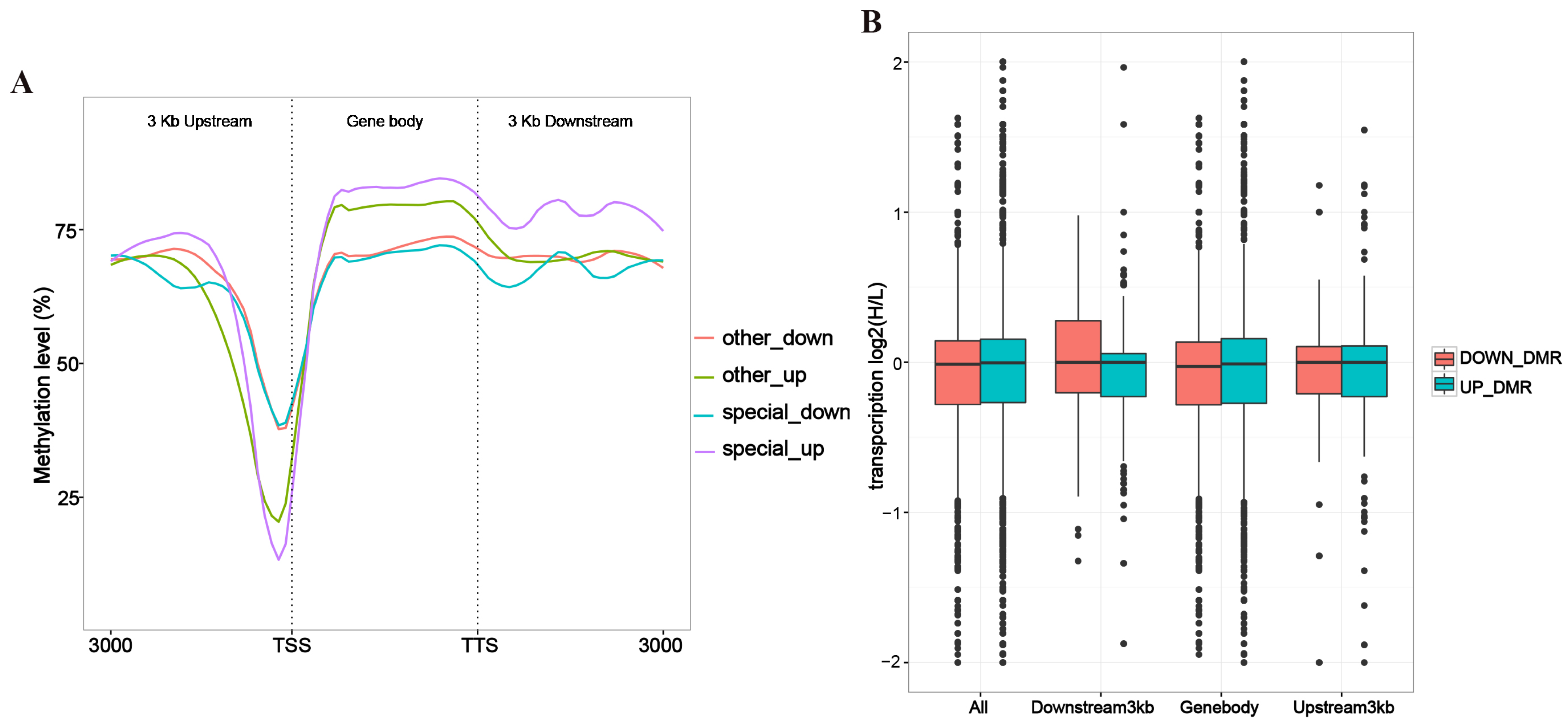
3.2. Correlation Analysis of Gene Expression and Methylation Rates Between L vs. H Groups
3.3. Analysis of Changes in Gene Expression Levels
3.4. GO Enrichment Analysis of DMEGs
3.5. KEGG Enrichment Analysis of DMEGs
3.6. DNA Methylation Influences the Development of Skeletal Muscle Through Its Impact on Gene Expression
3.7. DNA Methylation Regulates LPAR1 by Modulating SP1 Binding in Skeletal Muscle
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Yu, B.; Mao, X.; Zheng, P.; He, J.; Yu, J.; He, Y.; Reecy, J.; Chen, D. Lean and obese pig breeds exhibit differences in prenatal gene expression profiles of muscle development. Animal 2015, 9, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Du, Z.Q. Advances in the discovery of genetic elements underlying longissimus dorsi muscle growth and development in the pig. Anim. Genet. 2023, 54, 709–720. [Google Scholar] [CrossRef]
- Vicente-García, C.; Hernández-Camacho, J.D.; Carvajal, J.J. Regulation of myogenic gene expression. Exp. Cell Res. 2022, 419, 113299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, G.; Niu, G.; Zhang, Y.; Zhou, R.; Wang, Y.; Mu, Y.; Tang, Z.; Li, K. Comparative analysis of DNA methylome and transcriptome of skeletal muscle in lean-, obese-, and mini-type pigs. Sci. Rep. 2017, 7, 39883. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, X.; Li, C.; Qu, C.; Shi, Y.; Li, C.J.; Kang, X. Integrative analysis of whole genome bisulfite and transcriptome sequencing reveals the effect of sodium butyrate on DNA methylation in the differentiation of bovine skeletal muscle satellite cells. Genomics 2024, 116, 110959. [Google Scholar] [CrossRef]
- Li, J.W.; Shen, Z.K.; Lin, Y.S.; Wang, Z.Y.; Li, M.L.; Sun, H.X.; Wang, Q.; Zhao, C.; Xu, J.S.; Lu, X.; et al. DNA methylation of skeletal muscle function-related secretary factors identifies FGF2 as a potential biomarker for sarcopenia. J. Cachexia Sarcopenia Muscle 2024, 15, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Carrio, E.; Díez-Villanueva, A.; Lois, S.; Mallona, I.; Cases, I.; Forn, M.; Peinado, M.A.; Suelves, M. Deconstruction of DNA methylation patterns during myogenesis reveals specific epigenetic events in the establishment of the skeletal muscle lineage. Stem Cells 2015, 33, 2025–2036. [Google Scholar] [CrossRef]
- Carrió, E.; Magli, A.; Muñoz, M.; Peinado, M.A.; Perlingeiro, R.; Suelves, M. Muscle cell identity requires Pax7-mediated lineage-specific DNA demethylation. BMC Biol. 2016, 14, 30. [Google Scholar] [CrossRef]
- Miyata, K.; Miyata, T.; Nakabayashi, K.; Okamura, K.; Naito, M.; Kawai, T.; Takada, S.; Kato, K.; Miyamoto, S.; Hata, K.; et al. DNA methylation analysis of human myoblasts during in vitro myogenic differentiation: De novo methylation of promoters of muscle-related genes and its involvement in transcriptional down-regulation. Hum. Mol. Genet. 2015, 24, 410–423. [Google Scholar] [CrossRef]
- Kim, W.; Park, H.; Seo, K.S.; Seo, S. Characterization and functional inferences of a genome-wide DNA methylation profile in the loin (longissimus dorsi) muscle of swine. Asian-Australas. J. Anim. Sci. 2017, 31, 3. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, R.; Mu, Y.; Hou, X.; Tang, Z.; Li, K. Genome-wide analysis of DNA methylation in obese, lean and miniature pig breeds. Sci. Rep. 2016, 6, 30160. [Google Scholar] [CrossRef] [PubMed]
- Héberlé, É.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, P.; Magotra, A.; Sindhu, V.; Chaudhary, P. Unravelling the impact of epigenetic mechanisms on offspring growth, production, reproduction and disease susceptibility. Zygote 2024, 32, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Wang, Y.Z.; Chen, W.; Ma, L.X.; Zhang, J.M.; Zhang, Y.L.; Zeng, Y.Q. Integrated analysis of the DNA methylome and RNA transcriptome during the development of skeletal muscle in Duroc pigs. BMC Genom. 2024, 25, 504. [Google Scholar] [CrossRef]
- Ma, L.; Qin, M.; Zhang, Y.; Xue, H.; Li, S.; Chen, W.; Zeng, Y. Identification and functional prediction of long non-coding RNAs related to skeletal muscle development in Duroc pigs. Anim. Biosci. 2022, 35, 1512. [Google Scholar] [CrossRef]
- Fan, Y.; Liang, Y.; Deng, K.; Zhang, Z.; Zhang, G.; Zhang, Y.; Wang, F. Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genom. 2020, 21, 327. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, X.; Zheng, X.; Wang, F.; Sun, R.; Wei, L.; Zhang, Y.; Liu, H.; Lin, Y.; Hong, L.; et al. Diverse WGBS profiles of longissimus dorsi muscle in Hainan black goats and hybrid goats. BMC Genom. Data 2023, 24, 77. [Google Scholar] [CrossRef]
- Wu, W.; Ren, Z.; Liu, H.; Wang, L.; Huang, R.; Chen, J.; Zhang, L.; Li, P.; Xiong, Y. Core promoter analysis of porcine Six1 gene and its regulation of the promoter activity by CpG methylation. Gene 2013, 529, 238–244. [Google Scholar] [CrossRef]
- Tan, B.; Zhou, C.; Zang, X.; Zhao, X.; Xiao, L.; Zeng, J.; Hong, L.; Wu, Z.; Gu, T. Integrated analysis of DNA methylation and gene expression in porcine placental development. Int. J. Mol. Sci. 2023, 24, 5169. [Google Scholar] [CrossRef]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene body methylation in cancer: Molecular mechanisms and clinical applications. Clin. Epigenetics 2022, 14, 154. [Google Scholar] [CrossRef]
- Sarda, S.; Zeng, J.; Hunt, B.G.; Yi, S.V. The evolution of invertebrate gene body methylation. Mol. Biol. Evol. 2012, 29, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Jjingo, D.; Conley, A.B.; Soojin, V.Y.; Lunyak, V.V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462. [Google Scholar] [CrossRef]
- Deaton, A.M.; Webb, S.; Kerr, A.R.; Illingworth, R.S.; Guy, J.; Andrews, R.; Bird, A. Cell type–specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011, 21, 1074–1086. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef]
- Schroeder, D.I.; Jayashankar, K.; Douglas, K.C.; Thirkill, T.L.; York, D.; Dickinson, P.J.; Williams, L.E.; Samollow, P.B.; Ross, P.J.; Bannasch, D.L.; et al. Early developmental and evolutionary origins of gene body DNA methylation patterns in mammalian placentas. PLoS Genet. 2015, 11, e1005442. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Sung, K.W.K.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, C.; Liu, Q.; Peng, M.; Li, Q.; Wang, H.; Chen, X.; Zhang, B.; Feng, P.; Chen, T.; et al. Integrated analysis of DNA methylome and transcriptome reveals epigenetic regulation of cold tolerance in Litopenaeus vannamei. Int. J. Mol. Sci. 2023, 24, 11573. [Google Scholar] [CrossRef]
- Mohammadabadi, M.; Bordbar, F.; Jensen, J.; Du, M.; Guo, W. Key genes regulating skeletal muscle development and growth in farm animals. Animals 2021, 11, 835. [Google Scholar] [CrossRef] [PubMed]
- Kai, W.; Ping-Xian, W.; Shu-Jie, W.; Xiang, J.; Dong, C.; An-An, J.; Wei-Hang, X.; Yan-Zhi, J.; Li, Z.; Yang-Shuang, Z.; et al. Epigenome-wide DNA methylation analysis reveals differentially methylation patterns in skeletal muscle between Chinese Chenghua and Qingyu pigs. J. Integr. Agric. 2022, 21, 1731–1739. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, S.; Xu, Y.; Ma, Y.; Zhang, D.; Li, X.; Zhao, S. The DNA methylation status of Wnt and Tgfβ signals is a key factor on functional regulation of skeletal muscle satellite cell development. Front. Genet. 2019, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Li, A.; Raza, S.H.A. Genetic Regulation of Meat Quality Traits in Livestock Species. Front. Genet. 2023, 13, 1092562. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wen, H.; Zhang, M.; Hu, N.; Si, Y.; Li, S.; He, F. The DNA methylation status of MyoD and IGF-I genes are correlated with muscle growth during different developmental stages of Japanese flounder (Paralichthys olivaceus). Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2018, 219, 33–43. [Google Scholar] [CrossRef]
- Ma, X.; Jia, C.; Chu, M.; Fu, D.; Lei, Q.; Ding, X.; Wu, X.; Guo, X.; Pei, J.; Bao, P.; et al. Transcriptome and DNA methylation analyses of the molecular mechanisms underlying with longissimus dorsi muscles at different stages of development in the polled yak. Genes 2019, 10, 970. [Google Scholar] [CrossRef]
- Zhao, Y.; Xia, X.; Wang, Q.; Hu, D.; Zhang, L.; Li, X.; Ding, X.; Guo, H.; Guo, Y. Myostatin mutation enhances bovine myogenic differentiation through PI3K/AKT/mTOR signalling via removing DNA methylation of RACK1. Cells 2022, 12, 59. [Google Scholar] [CrossRef]
- Zhou, X.; Ding, Y.; Yang, C.; Li, C.; Su, Z.; Xu, J.; Qu, C.; Shi, Y.; Kang, X. FHL3 gene regulates bovine skeletal muscle cell growth through the PI3K/Akt/mTOR signaling pathway. Comp. Biochem. Physiol. Part D Genom. Proteom. 2024, 52, 101356. [Google Scholar] [CrossRef]
- Zhao, T.L.; Qi, Y.; Wang, Y.F.; Wang, Y.; Liang, H.; Pu, Y.B. 5-methoxytryptophan induced apoptosis and PI3K/Akt/FoxO3a phosphorylation in colorectal cancer. World J. Gastroenterol. 2023, 29, 6148. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, Y.; He, P.; Li, J.; Liu, W.; Yuan, J.; Zhao, H.; Liu, Z.; Shen, C.; Shi, B. Mechanisms by which Mettl3 regulates pericyte-myofibroblast transdifferentiation through PI3K/AKT signaling pathway. Zhonghua Xin Xue Guan Bing Za Zhi 2024, 52, 814–826. [Google Scholar] [CrossRef]
- Sun, R.; Wu, Y.; Wang, Y.; Zang, K.; Wei, H.; Wang, F.; Yu, M. DNA methylation regulates bromodomain-containing protein 2 expression during adipocyte differentiation. Mol. Cell. Biochem. 2015, 402, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Davegårdh, C.; Hall Wedin, E.; Broholm, C.; Henriksen, T.I.; Pedersen, M.; Pedersen, B.K.; Scheele, C.; Ling, C. Sex influences DNA methylation and gene expression in human skeletal muscle myoblasts and myotubes. Stem Cell Res. Ther. 2019, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Kreibich, E.; Krebs, A.R. Relevance of DNA methylation at enhancers for the acquisition of cell identities. FEBS Lett. 2023, 597, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; Nimwegen, E.V.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Ray, R.; Sinha, S.; Aidinis, V.; Rai, V. Atx regulates skeletal muscle regeneration via LPAR1 and promotes hypertrophy. Cell Rep. 2021, 34, 108809. [Google Scholar] [CrossRef] [PubMed]
- Cordova-Casanova, A.; Cruz-Soca, M.; Chun, J.; Casar, J.C.; Brandan, E. Activation of the ATX/LPA/LPARs axis induces a fibrotic response in skeletal muscle. Matrix Biol. 2022, 109, 121–139. [Google Scholar] [CrossRef]
- Messmer, T.; Klevernic, I.; Furquim, C.; Ovchinnikova, E.; Dogan, A.; Cruz, H.; Post, M.J.; Flack, J.E. A serum-free media formulation for cultured meat production supports bovine satellite cell differentiation in the absence of serum starvation. Nat. Food 2022, 3, 74–85. [Google Scholar] [CrossRef]
- Xiong, X.; Liu, X.; Zhou, L.; Yang, J.; Yang, B.; Ma, H.; Xie, X.; Huang, Y.; Fang, S.; Xiao, S.; et al. Genome-wide association analysis reveals genetic loci and candidate genes for meat quality traits in Chinese Laiwu pigs. Mamm. Genome 2015, 26, 181–190. [Google Scholar] [CrossRef]
- He, Y.; Hariharan, M.; Gorkin, D.U.; Dickel, D.E.; Luo, C.; Castanon, R.G.; Nery, J.R.; Lee, A.Y.; Zhao, Y.; Huang, H.; et al. Spatiotemporal DNA methylome dynamics of the developing mouse fetus. Nature 2020, 583, 752–759. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Ma, L.; Bao, Q.; Liang, C.; Chu, M.; Guo, X.; Bao, P.; Yan, P. DNA methylation dynamics during yak adipocyte differentiation. Int. J. Biol. Macromol. 2024, 261, 129715. [Google Scholar] [CrossRef]
- Angeloni, A.; Bogdanovic, O. Enhancer DNA methylation: Implications for gene regulation. Essays Biochem. 2019, 63, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Fabyanic, E.B.; Hu, P.; Qiu, Q.; Berríos, K.N.; Connolly, D.R.; Wang, T.; Flournoy, J.; Zhou, Z.; Kohli, R.M.; Wu, H. Joint single-cell profiling resolves 5mC and 5hmC and reveals their distinct gene regulatory effects. Nat. Biotechnol. 2024, 42, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Pacalin, N.M.; Steinhart, Z.; Shi, Q.; Belk, J.A.; Dorovskyi, D.; Kraft, K.; Parker, K.R.; Shy, B.R.; Marson, A.; Chang, H.Y. Bidirectional epigenetic editing reveals hierarchies in gene regulation. Nat. Biotechnol. 2024, 43, 355–368. [Google Scholar] [CrossRef]
- Cappelluti, M.A.; Mollica Poeta, V.; Valsoni, S.; Quarato, P.; Merlin, S.; Merelli, I.; Lombardo, A. Durable and efficient gene silencing in vivo by hit-and-run epigenome editing. Nature 2024, 627, 416–423. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Gao, Y.; Ma, L.; Chen, W.; Ma, Z.; Ren, Z.; Wang, Y.; Zeng, Y. Integrated Analysis of DNA Methylome and Transcriptome Reveals Regulatory Mechanism in the Longissimus Dorsi of Duroc Pigs. Cells 2025, 14, 786. https://doi.org/10.3390/cells14110786
Li S, Gao Y, Ma L, Chen W, Ma Z, Ren Z, Wang Y, Zeng Y. Integrated Analysis of DNA Methylome and Transcriptome Reveals Regulatory Mechanism in the Longissimus Dorsi of Duroc Pigs. Cells. 2025; 14(11):786. https://doi.org/10.3390/cells14110786
Chicago/Turabian StyleLi, Shiyin, Yarui Gao, Lixia Ma, Wei Chen, Zhao Ma, Zhanchi Ren, Yunzhou Wang, and Yongqing Zeng. 2025. "Integrated Analysis of DNA Methylome and Transcriptome Reveals Regulatory Mechanism in the Longissimus Dorsi of Duroc Pigs" Cells 14, no. 11: 786. https://doi.org/10.3390/cells14110786
APA StyleLi, S., Gao, Y., Ma, L., Chen, W., Ma, Z., Ren, Z., Wang, Y., & Zeng, Y. (2025). Integrated Analysis of DNA Methylome and Transcriptome Reveals Regulatory Mechanism in the Longissimus Dorsi of Duroc Pigs. Cells, 14(11), 786. https://doi.org/10.3390/cells14110786