
The Generation of Two Induced Pluripotent Cell Lines from Patients with an Atypical Familial Form of Lung Fibrosis
 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reprogramming
2.2. Pluripotency Detection
2.3. Mitochondrial Integrity Measurements and Intracellular Calcium
2.4. Measurement of Inflammatory Mediators
2.5. Direct Differentiation into Lung Epithelial Cells
2.6. Statistical Analysis
3. Results
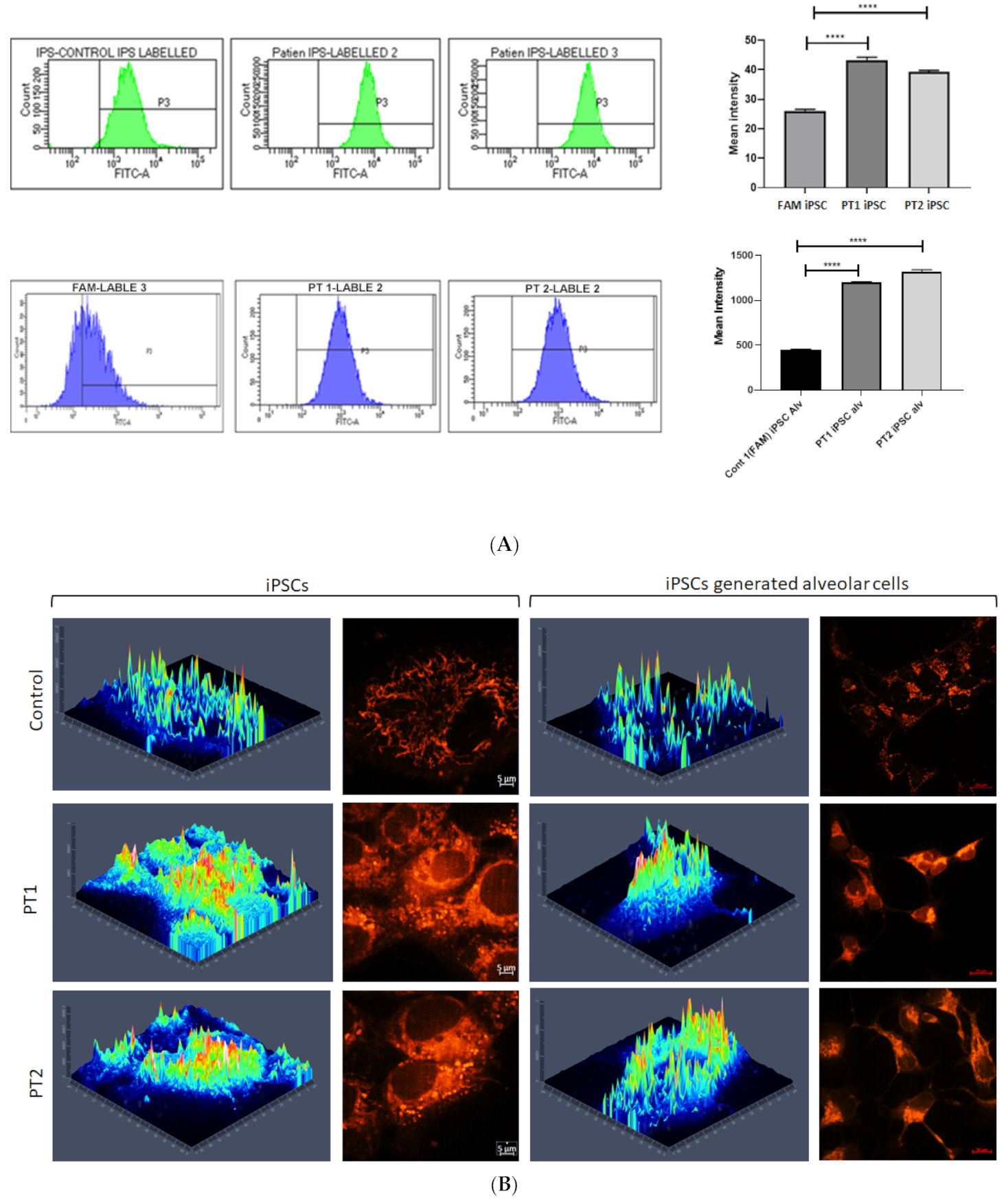
3.1. Generated iPSC Cell Lines
3.2. Effects of S100A3 and S100A13 Mutations on Mitochondria and Intracellular Calcium Release
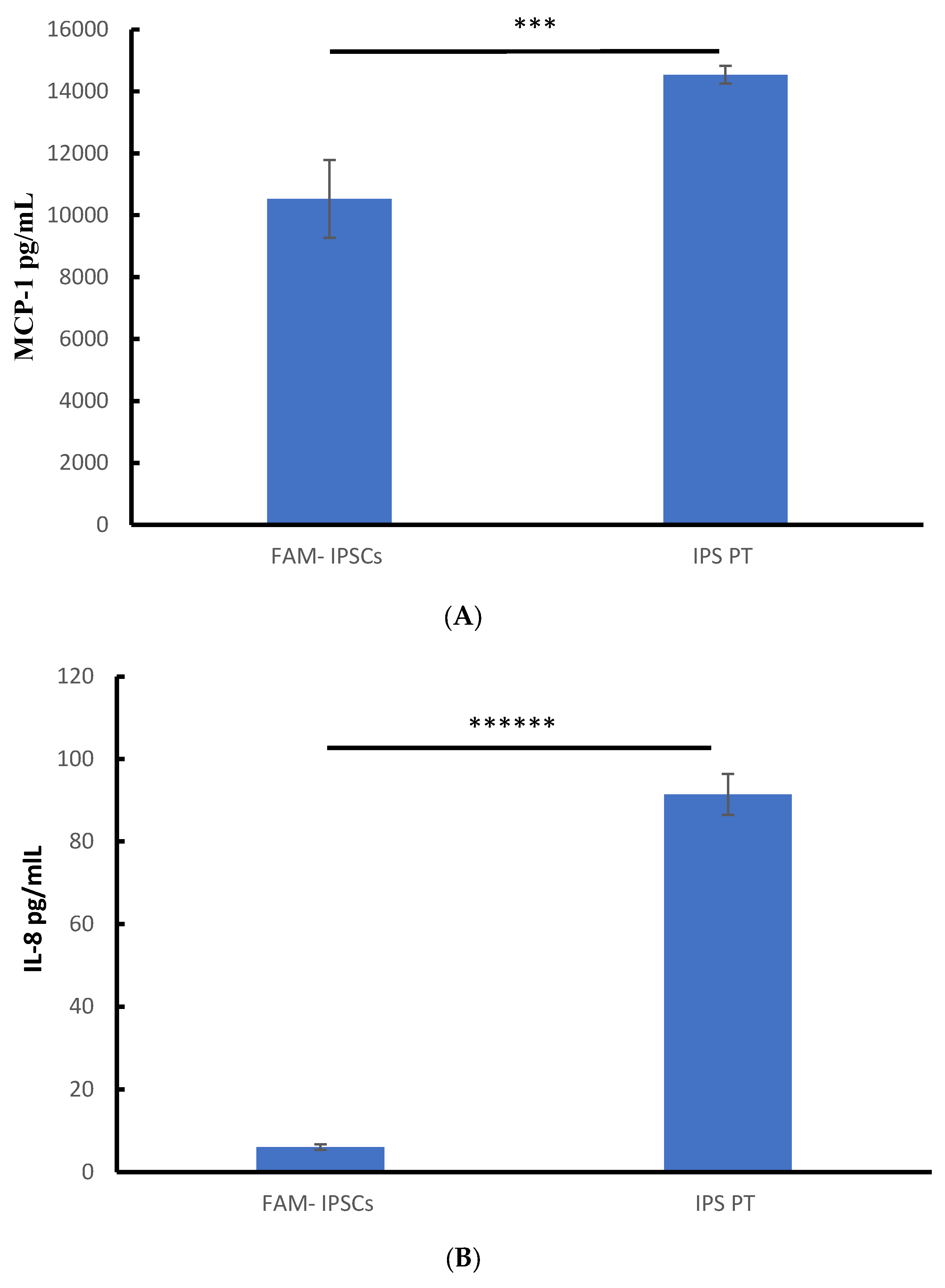
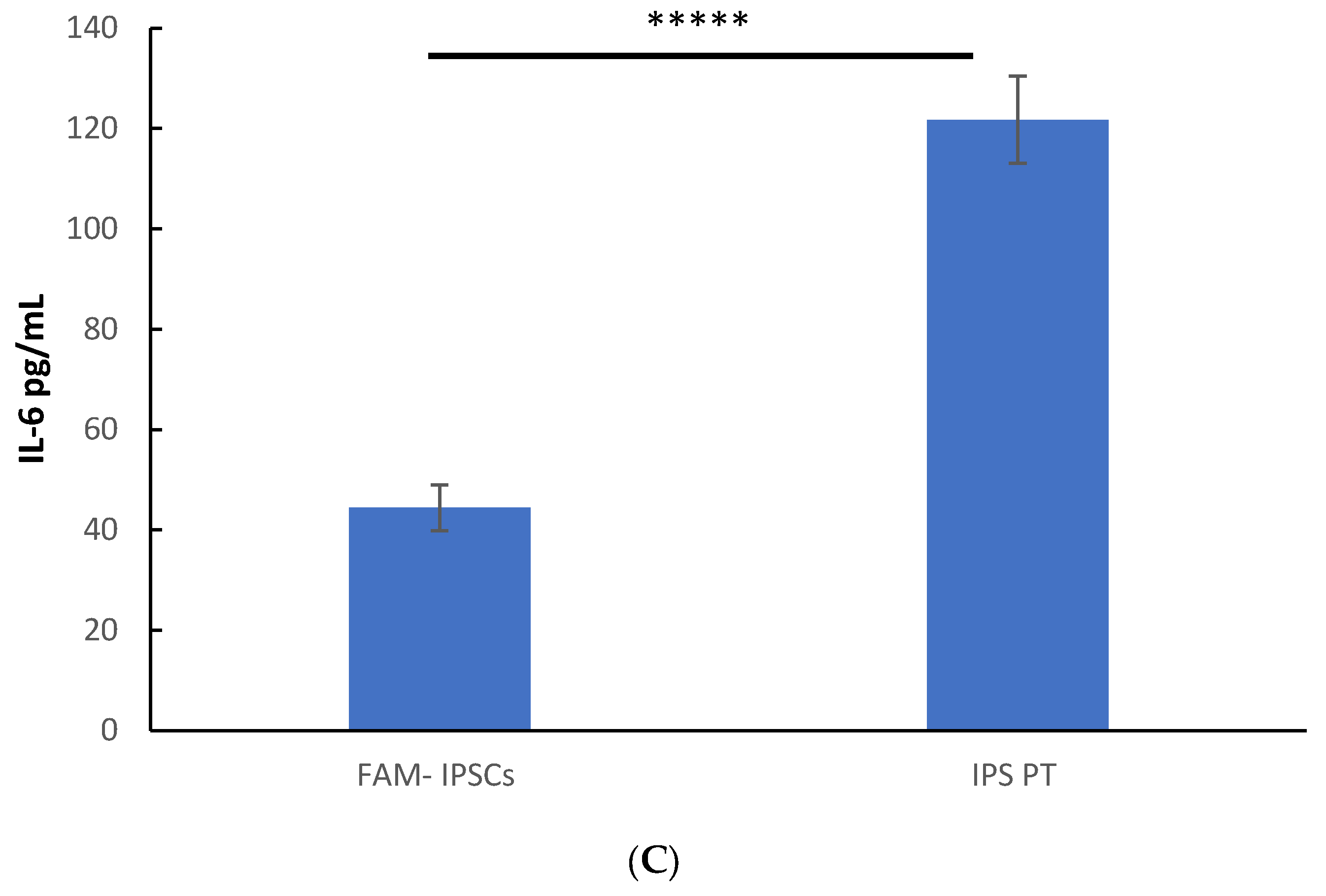
3.3. Constitutive Secretion of IL-8, IL-6 and MCP-1 in Patient-Derived Fibroblasts
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lo, B.; Parham, L. Ethical issues in stem cell research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Kimbrel, E.A.; Lanza, R. Current status of pluripotent stem cells: Moving the first therapies to the clinic. Nat. Rev. Drug Discov. 2015, 14, 681–692. [Google Scholar] [CrossRef]
- Scudellari, M. How iPS cells changed the world. Nature 2016, 534, 310–312. [Google Scholar] [CrossRef]
- Soldner, F.; Jaenisch, R. Medicine. iPSC disease modeling. Science 2012, 338, 1155–1156. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Nagy, K. The mysteries of induced pluripotency: Where will they lead? Nat. Methods 2010, 7, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Gulisano, W.; Palmeri, A.; Arancio, O. Rodent models for Alzheimer’s disease drug discovery. Expert. Opin. Drug Discov. 2015, 10, 703–711. [Google Scholar] [CrossRef]
- Ben-David, U.; Kopper, O.; Benvenisty, N. Expanding the boundaries of embryonic stem cells. Cell Stem Cell 2012, 10, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Hardie, W.D.; Hagood, J.S.; Dave, V.; Perl, A.K.; Whitsett, J.A.; Korfhagen, T.R.; Glasser, S. Signaling pathways in the epithelial origins of pulmonary fibrosis. Cell Cycle 2010, 9, 2769–2776. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ambalavanan, N.; Carlo, W.A. Bronchopulmonary dysplasia: New insights. Clin. Perinatol. 2004, 31, 613–628. [Google Scholar] [CrossRef]
- Postma, D.S.; Timens, W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 434–439. [Google Scholar] [CrossRef]
- Rennard, S.I. Chronic obstructive pulmonary disease: Linking outcomes and pathobiology of disease modification. Proc. Am. Thorac. Soc. 2006, 3, 276–280. [Google Scholar] [CrossRef]
- Weitzenblum, E.; Chaouat, A.; Canuet, M.; Kessler, R. Pulmonary hypertension in chronic obstructive pulmonary disease and interstitial lung diseases. Semin. Respir. Crit. Care Med. 2009, 30, 458–470. [Google Scholar] [CrossRef]
- Kim, R.; Meyer, K.C. Therapies for interstitial lung disease: Past, present and future. Ther. Adv. Respir. Dis. 2008, 2, 319–338. [Google Scholar] [CrossRef]
- Olson, A.L.; Swigris, J.J.; Lezotte, D.C.; Norris, J.M.; Wilson, C.G.; Brown, K.K. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am. J. Respir. Crit. Care Med. 2007, 176, 277–284. [Google Scholar] [CrossRef]
- Yusen, R.D.; Christie, J.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Dipchand, A.I.; Dobbels, F.; Kirk, R.; Lund, L.H.; Rahmel, A.O.; et al. The Registry of the International Society for Heart and Lung Transplantation: Thirtieth Adult Lung and Heart-Lung Transplant Report—2013; focus theme: Age. J. Heart Lung Transplant. 2013, 32, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Veldhuizen, R.; Possmayer, F. Phospholipid metabolism in lung surfactant. Subcell. Biochem. 2004, 37, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, J.J. Surfactant phospholipids: Synthesis and storage. Am. J. Physiol. 1992, 262 Pt 1, L367–L385. [Google Scholar] [CrossRef]
- Olmeda, B.; Martínez-Calle, M.; Pérez-Gil, J. Pulmonary surfactant metabolism in the alveolar airspace: Biogenesis, extracellular conversions, recycling. Ann. Anat. 2017, 209, 78–92. [Google Scholar] [CrossRef]
- Doyle, I.R.; Barr, H.A.; Nicholas, T.E. Distribution of surfactant protein A in rat lung. Am. J. Respir. Cell Mol. Biol. 1994, 11, 405–415. [Google Scholar] [CrossRef]
- Hobi, N.; Siber, G.; Bouzas, V.; Ravasio, A.; Pérez-Gil, J.; Haller, T. Physiological variables affecting surface film formation by native lamellar body-like pulmonary surfactant particles. Biochim. Biophys. Acta 2014, 1838, 1842–1850. [Google Scholar] [CrossRef]
- Galván, E.M.; Chen, H.; Schifferli, D.M. The Psa fimbriae of Yersinia pestis interact with phosphatidylcholine on alveolar epithelial cells and pulmonary surfactant. Infect. Immun. 2007, 75, 1272–1279. [Google Scholar] [CrossRef]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- da Cunha, M.J.; da Cunha, A.A.; Scherer, E.B.; Machado, F.R.; Loureiro, S.O.; Jaenisch, R.B.; Guma, F.; Lago, P.D.; Wyse, A.T. Experimental lung injury promotes alterations in energy metabolism and respiratory mechanics in the lungs of rats: Prevention by exercise. Mol. Cell. Biochem. 2014, 389, 229–238. [Google Scholar] [CrossRef]
- Kovacs, T.; Csongei, V.; Feller, D.; Ernszt, D.; Smuk, G.; Sarosi, V.; Jakab, L.; Kvell, K.; Bartis, D.; Pongracz, J.E. Alteration in the Wnt microenvironment directly regulates molecular events leading to pulmonary senescence. Aging Cell 2014, 13, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Wert, S.E.; Whitsett, J.A.; Nogee, L.M. Genetic disorders of surfactant dysfunction. Pediatr. Dev. Pathol. 2009, 12, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hyatt, B.A.; Mucenski, M.L.; Mason, R.J.; Shannon, J.M. Identification and characterization of a lysophosphatidylcholine acyltransferase in alveolar type II cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11724–11729. [Google Scholar] [CrossRef]
- Kasper, M.; Barth, K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci. Rep. 2017, 37, BSR20171301. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giacomelli, C.; Piccarducci, R.; Marchetti, L.; Romei, C.; Martini, C. Pulmonary fibrosis from molecular mechanisms to therapeutic interventions: Lessons from post-COVID-19 patients. Biochem. Pharmacol. 2021, 193, 114812. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lindner, T.H.; Hoffmann, K. easyLINKAGE: A PERL script for easy and automated two-/multi- point linkage analyses. Bioinformatics 2005, 21, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W.; Ackermann, G.E.; Galichet, A. Pathologies involving the S100 proteins and RAGE. Subcell. Biochem. 2007, 45, 93–138. [Google Scholar] [CrossRef] [PubMed]
- Al-Mutairy, E.A.; Imtiaz, F.A.; Khalid, M.; Al Qattan, S.; Saleh, S.; Mahmoud, L.M.; Al-Saif, M.M.; Al-Haj, L.; Al-Enazi, A.; AlJebreen, A.M.; et al. An atypical pulmonary fibrosis is associated with co-inheritance of mutations in the calcium binding protein genes S100A3 and S100A13. Eur. Respir. J. 2019, 54, 1802041. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Al-Mutairy, E.A.; Al Qattan, S.; Khalid, M.; Al-Enazi, A.A.; Al-Saif, M.M.; Imtiaz, F.; Ramzan, K.; Raveendran, V.; Alaiya, A.; Meyer, B.F.; et al. Wild-type S100A3 and S100A13 restore calcium homeostasis and mitigate mitochondrial dysregulation in pulmonary fibrosis patient-derived cells. Front. Cell Dev. Biol. 2023, 11, 1282868. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Emberley, E.D.; Murphy, L.C.; Watson, P.H. S100 proteins and their influence on pro-survival pathways in cancer. Biochem. Cell Biol. 2004, 82, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W.; Fritz, G.; Schäfer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci. 2002, 7, d1356–d1368. [Google Scholar] [CrossRef]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, G.Q.; Qu, J.; Che, C.Y.; Lin, J.; Jiang, N.; Zhao, H.; Wang, X.J. Expression of S100B during the innate immune of corneal epithelium against fungi invasion. Int. J. Ophthalmol. 2016, 9, 191–197. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, H.; Xu, G.; Wen, H.; Gu, B.; Liu, J.; Mao, S.; Na, R.; Jing, Y.; Ding, Q.; et al. Proteins S100A8 and S100A9 are potential biomarkers for renal cell carcinoma in the early stages: Results from a proteomic study integrated with bioinformatics analysis. Mol. Med. Rep. 2015, 11, 4093–4100. [Google Scholar] [CrossRef]
- Kleiman, R.J.; Engle, S.J. Human inducible pluripotent stem cells: Realization of initial promise in drug discovery. Cell Stem Cell 2021, 28, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Sama, D.M.; Norris, C.M. Calcium dysregulation and neuroinflammation: Discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Luzina, I.G.; Todd, N.W.; Sundararajan, S.; Atamas, S.P. The cytokines of pulmonary fibrosis: Much learned, much more to learn. Cytokine 2015, 74, 88–100. [Google Scholar] [CrossRef] [PubMed]
- She, Y.X.; Yu, Q.Y.; Tang, X.X. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.I.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Campo, I.; Zorzetto, M.; Mariani, F.; Kadija, Z.; Morbini, P.; Dore, R.; Kaltenborn, E.; Frixel, S.; Zarbock, R.; Liebisch, G.; et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir. Res. 2014, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Daba, M.H.; El-Tahir, K.E.; Al-Arifi, M.N.; Gubara, O.A. Drug-induced pulmonary fibrosis. Saudi Med. J. 2004, 25, 700–706. [Google Scholar]
- Takebe, T.; Wells, J.M. Organoids by design. Science 2019, 364, 956–959. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Corsini, N.S.; Knoblich, J.A. Human organoids: New strategies and methods for analyzing human development and disease. Cell 2022, 185, 2756–2769. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Toshimitsu, K.; Takano, A.; Mikami, Y.; Nishikori, S.; Sugimoto, S.; Sato, T. Human intestinal organoids maintain self-renewal capacity and cellular diversity in niche-inspired culture condition. Cell Stem Cell 2018, 23, 787–793. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.J.; Krotenberg Garcia, A.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879.e11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kretzschmar, K.; Clevers, H. Organoids: Modeling development and the stem cell niche in a dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
- Hartl, D.; Griese, M.; Nicolai, T.; Zissel, G.; Prell, C.; Reinhardt, D.; Schendel, D.J.; Krauss-Etschmann, S. A role for MCP-1/CCR2 in interstitial lung disease in children. Respir. Res. 2005, 6, 93. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Papiris, S.A.; Tomos, I.P.; Karakatsani, A.; Spathis, A.; Korbila, I.; Analitis, A.; Kolilekas, L.; Kagouridis, K.; Loukides, S.; Karakitsos, P.; et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2018, 102, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhai, H.; Zhang, T.; Li, S.; Li, N.; Chen, J.; Gu, M.; Qin, Z.; Liu, X. New therapeutic strategies for IPF: Based on the “phagocytosis-secretion-immunization” network regulation mechanism of pulmonary macrophages. Biomed. Pharmacother. 2019, 118, 109230. [Google Scholar] [CrossRef]
- Papiris, S.A.; Manali, E.D.; Kolilekas, L.; Kagouridis, K.; Triantafillidou, C.; Tsangaris, I.; Roussos, C. Clinical review: Idiopathic pulmonary fibrosis acute exacerbations--unravelling Ariadne’s thread. Crit. Care 2010, 14, 246. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef]
- Collard, H.R.; Calfee, C.S.; Wolters, P.J.; Song, J.W.; Hong, S.B.; Brady, S.; Ishizaka, A.; Jones, K.D.; King, T.E., Jr.; Matthay, M.A.; et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L3–L7. [Google Scholar] [CrossRef]
- Gui, X.; Qiu, X.; Tian, Y.; Xie, M.; Li, H.; Gao, Y.; Zhuang, Y.; Cao, M.; Ding, H.; Ding, J.; et al. Prognostic value of IFN-γ, sCD163, CCL2 and CXCL10 involved in acute exacerbation of idiopathic pulmonary fibrosis. Int. Immunopharmacol. 2019, 70, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.R.; Qiu, H.; Wu, Q.; Du, Y.K.; Yin, Z.F.; Chen, S.S.; Jin, Y.P.; Zhao, M.M.; Wang, C.; Weng, D.; et al. Establishment of the mouse model of acute exacerbation of idiopathic pulmonary fibrosis. Exp. Lung Res. 2016, 42, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Ashok, A.; Naing, M.W. Commercialization of Organoids. Trends Mol. Med. 2020, 26, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, G.; Comi, G.P.; Corti, S. Advancing Drug Discovery for Neurological Disorders Using iPSC-Derived Neural Organoids. Int. J. Mol. Sci. 2021, 22, 2659. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Mutairy, E.; Al Qattan, S.M.; Imtiaz, F.; AlAnazi, A.; Inglis, A.; Al-Rabiah, R.; Al-Hejailan, R.S. The Generation of Two Induced Pluripotent Cell Lines from Patients with an Atypical Familial Form of Lung Fibrosis. Cells 2025, 14, 781. https://doi.org/10.3390/cells14110781
Al-Mutairy E, Al Qattan SM, Imtiaz F, AlAnazi A, Inglis A, Al-Rabiah R, Al-Hejailan RS. The Generation of Two Induced Pluripotent Cell Lines from Patients with an Atypical Familial Form of Lung Fibrosis. Cells. 2025; 14(11):781. https://doi.org/10.3390/cells14110781
Chicago/Turabian StyleAl-Mutairy, Eid, Somaya M. Al Qattan, Faiqa Imtiaz, Azizah AlAnazi, Angela Inglis, Rana Al-Rabiah, and Reem S. Al-Hejailan. 2025. "The Generation of Two Induced Pluripotent Cell Lines from Patients with an Atypical Familial Form of Lung Fibrosis" Cells 14, no. 11: 781. https://doi.org/10.3390/cells14110781
APA StyleAl-Mutairy, E., Al Qattan, S. M., Imtiaz, F., AlAnazi, A., Inglis, A., Al-Rabiah, R., & Al-Hejailan, R. S. (2025). The Generation of Two Induced Pluripotent Cell Lines from Patients with an Atypical Familial Form of Lung Fibrosis. Cells, 14(11), 781. https://doi.org/10.3390/cells14110781