
RNF213 Acts as a Molecular Switch for Cav-1 Ubiquitination and Phosphorylation in Human Cells

 , ,
, ,  , ,
, ,  , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Human Clones and Plasmids
2.3. siRNA-Directed Gene Silencing
2.4. Immunoprecipitation
2.5. Immunoblotting
2.6. Cell Imaging
2.7. Depletion and Measurement of Intracellular ATP Levels
2.8. Mass Spectrometry
2.9. NF-κB Dual Luciferase Assays
2.10. HPAEC H2O2 Treatments and Nitric Oxide (NO) Measurements in Live Cells
2.11. Statistical Analysis
3. Results
3.1. RNF213 Co-Localizes and Interacts with Cav-1 in Endothelial Cells
3.2. Cav-1 Specifically Binds Within the Functional AAA+ Domains of RNF213
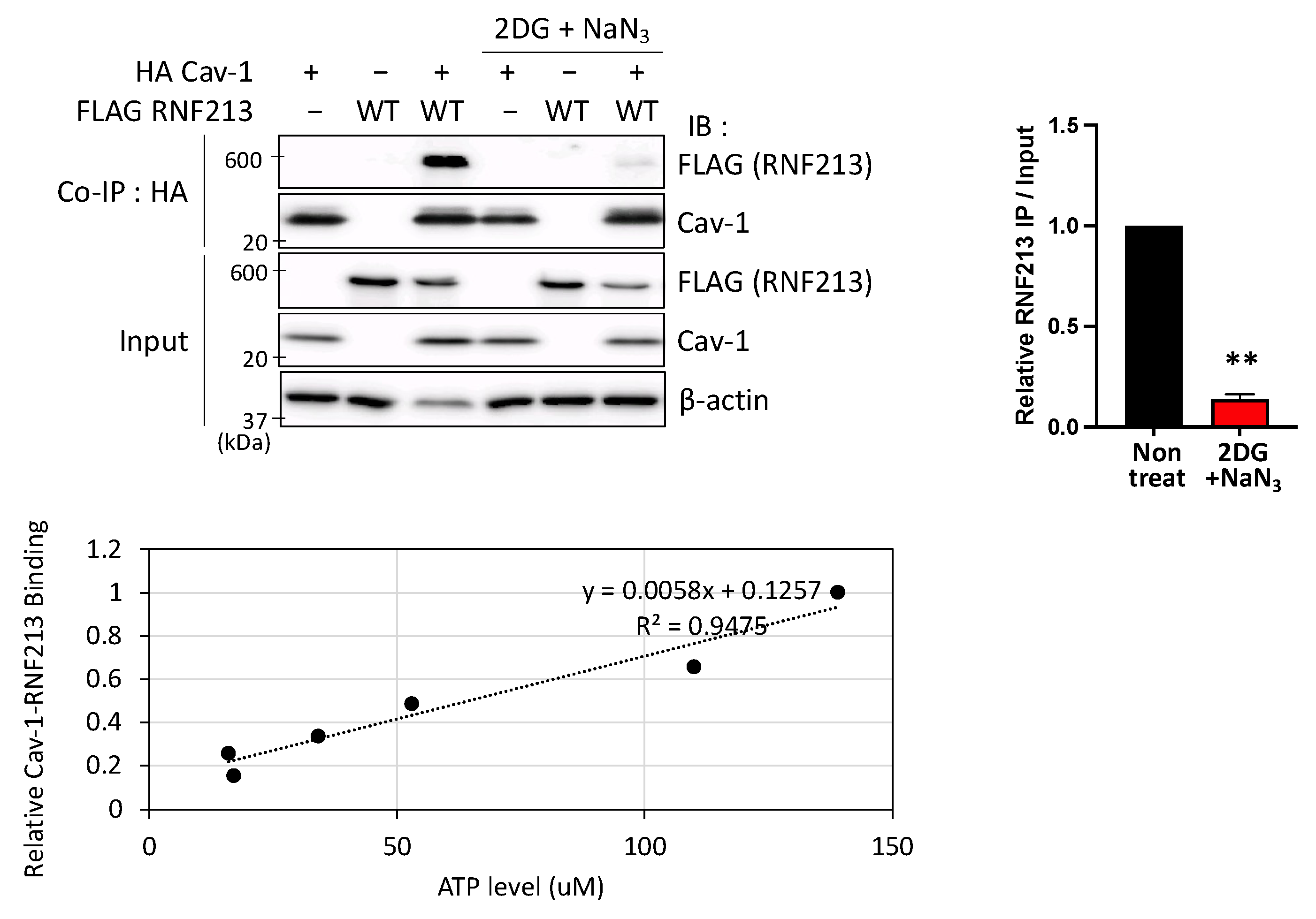
3.3. Interactions Between RNF213 and Cav-1 Are ATP-Dependent
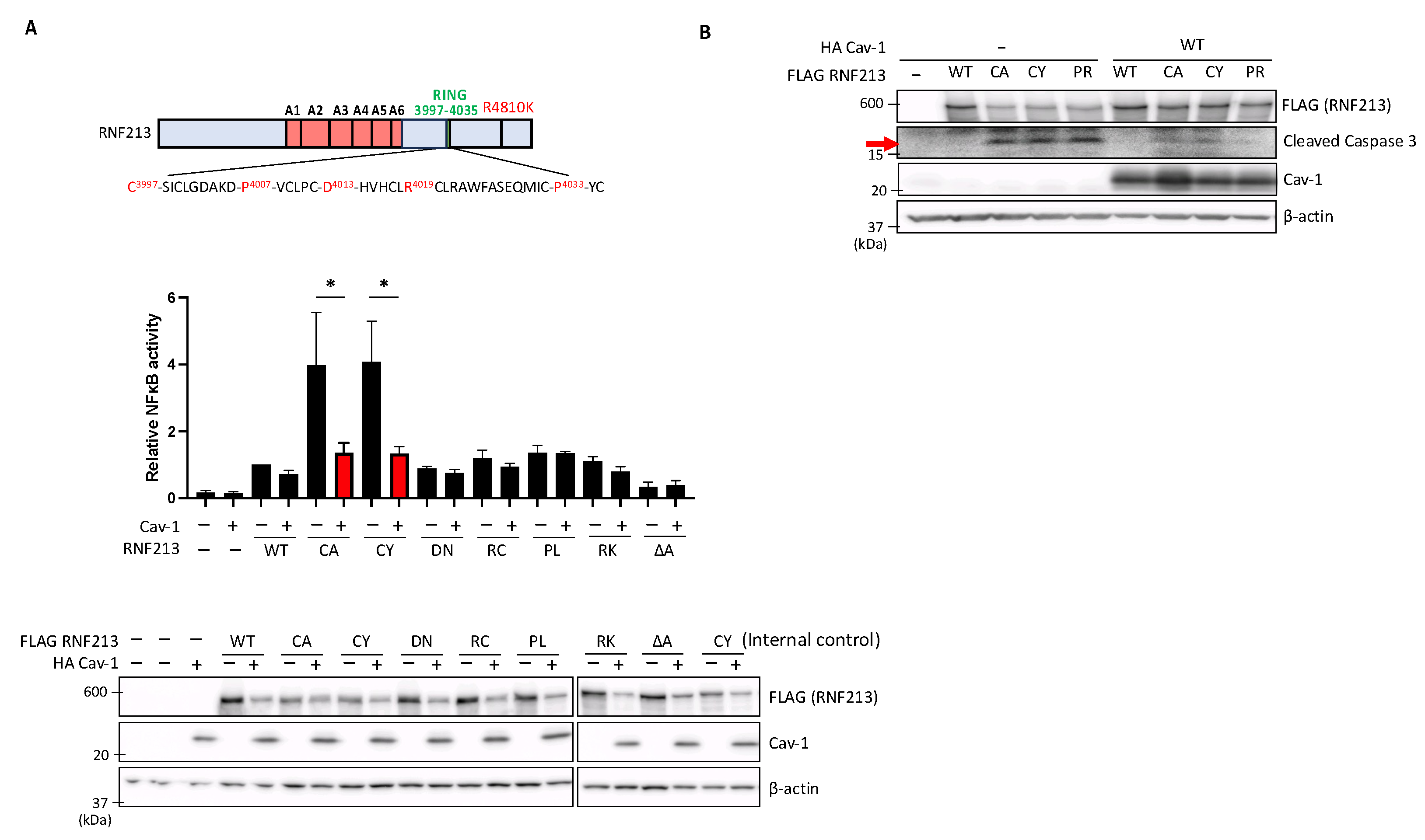
3.4. Cav-1 Suppresses Enhanced NF-κB Activity and Apoptosis Induced by RNF213 RING Mutations
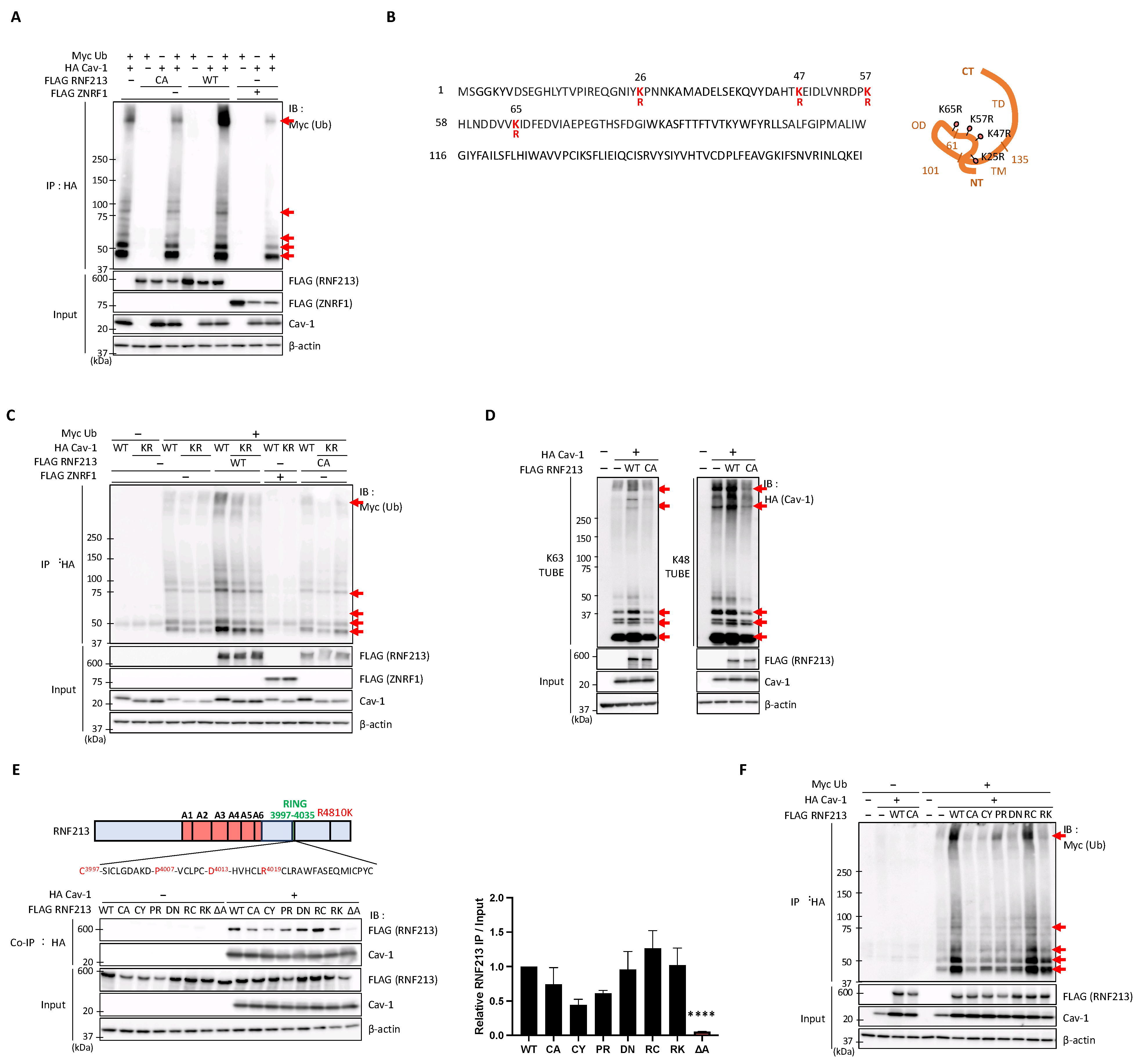
3.5. RNF213 Mediates the Ubiquitination of Cav-1 at Multiple Lysine Residues but Is Constrained by Several MMD-Related RNF213 Mutations
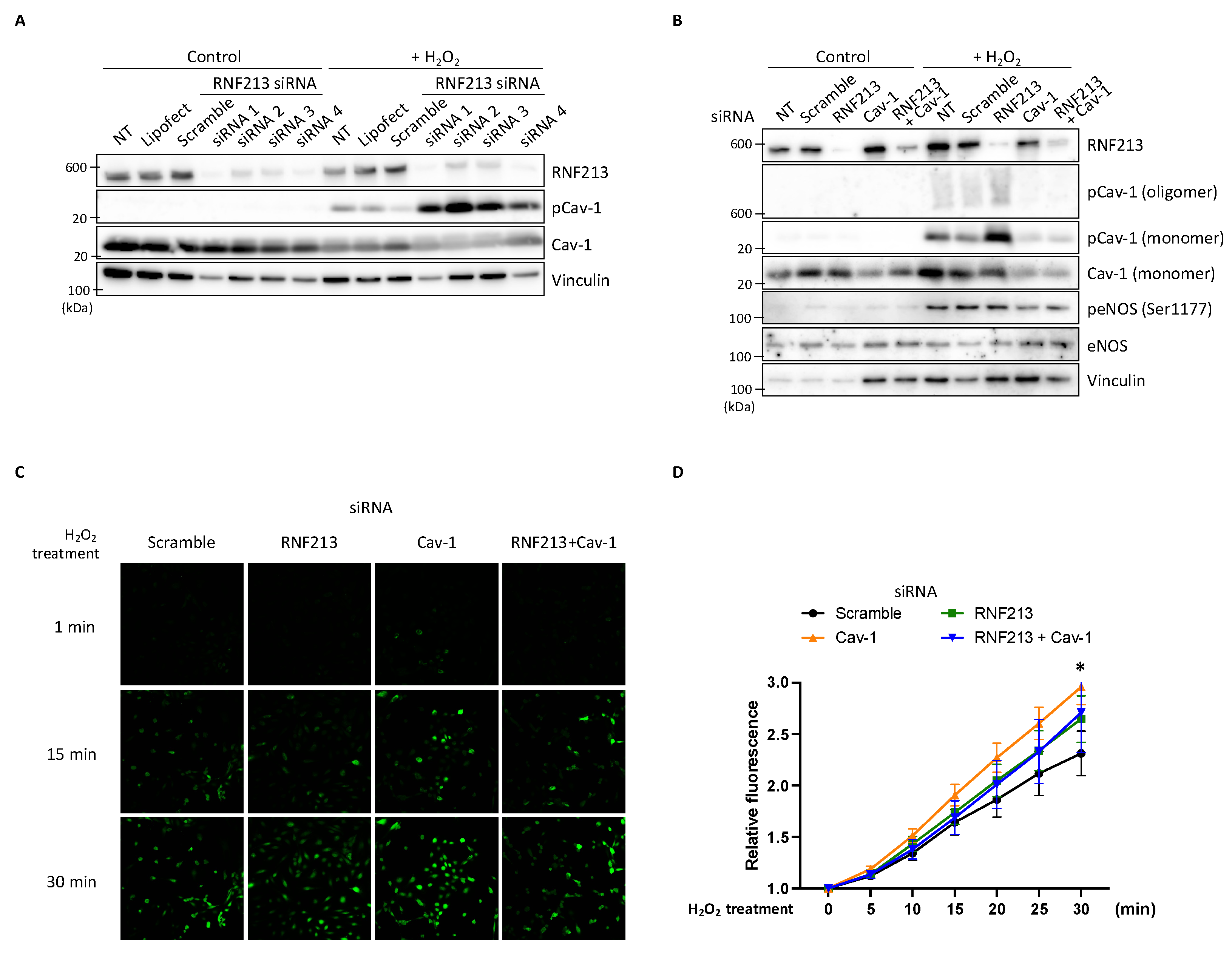
3.6. RNF213 Knockdown Enhances Cav-1 Y14 Phosphorylation Under H2O2 and Affects Nitric Oxide Stimulation in HPAECs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, A.; Kobayashi, H.; Hitomi, T.; Harada, K.H.; Habu, T.; Youssefian, S. A new horizon of moyamoya disease and associated health risks explored through RNF213. Environ. Health Prev. Med. 2016, 21, 55–70. [Google Scholar] [CrossRef]
- Ihara, M.; Yamamoto, Y.; Hattori, Y.; Liu, W.; Kobayashi, H.; Ishiyama, H.; Yoshimoto, T.; Miyawaki, S.; Clausen, T.; Bang, O.Y.; et al. Moyamoya disease: Diagnosis and interventions. Lancet Neurol. 2022, 21, 747–758. [Google Scholar] [CrossRef]
- Fukushima, H.; Takenouchi, T.; Kosaki, K. Homozygosity for moyamoya disease risk allele leads to moyamoya disease with extracranial systemic and pulmonary vasculopathy. Am. J. Med. Genet. A 2016, 170, 2453–2456. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kabata, R.; Kinoshita, H.; Morimoto, T.; Ono, K.; Takeda, M.; Choi, J.; Okuda, H.; Liu, W.; Harada, K.H.; et al. Rare variants in RNF213, a susceptibility gene for moyamoya disease, are found in patients with pulmonary hypertension and aggravate hypoxia-induced pulmonary hypertension in mice. Pulm. Circ. 2018, 8, 2045894018778155. [Google Scholar] [CrossRef]
- Morimoto, T.; Mineharu, Y.; Ono, K.; Nakatochi, M.; Ichihara, S.; Kabata, R.; Takagi, Y.; Cao, Y.; Zhao, L.; Kobayashi, H.; et al. Significant association of RNF213 p.R4810K, a moyamoya susceptibility variant, with coronary artery disease. PLoS ONE 2017, 12, e0175649. [Google Scholar] [CrossRef]
- Yamada, I.; Himeno, Y.; Matsushima, Y.; Shibuya, H. Renal artery lesions in patients with moyamoya disease: Angiographic findings. Stroke 2000, 31, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Zhao, H.; Liu, C.; Tan, X.; Luo, C.; Yang, S. Research progress of moyamoya disease combined with renovascular hypertension. Front. Surg. 2022, 9, 969090. [Google Scholar] [CrossRef]
- Banh, R.S.; Iorio, C.; Marcotte, R.; Xu, Y.; Cojocari, D.; Rahman, A.A.; Pawling, J.; Zhang, W.; Sinha, A.; Rose, C.M.; et al. PTP1B controls non-mitochondrial oxygen consumption by regulating RNF213 to promote tumour survival during hypoxia. Nat. Cell Biol. 2016, 18, 803–813. [Google Scholar] [CrossRef]
- Ahel, J.; Lehner, A.; Vogel, A.; Schleiffer, A.; Meinhart, A.; Haselbach, D.; Clausen, T. Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. Elife 2020, 9, e56185. [Google Scholar] [CrossRef]
- Bhabha, G.; Cheng, H.-C.; Zhang, N.; Moeller, A.; Liao, M.; Speir, J.A.; Cheng, Y.; Vale, R.D. Allosteric Communication in the Dynein Motor Domain. Cell 2014, 159, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Morito, D.; Nishikawa, K.; Hoseki, J.; Kitamura, A.; Kotani, Y.; Kiso, K.; Kinjo, M.; Fujiyoshi, Y.; Nagata, K. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci. Rep. 2014, 4, 4442. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, J.J.; Franklin, M.C.; Kamtekar, S.; Im, Y.J.; Rho, S.H.; Seong, I.S.; Lee, C.S.; Chung, C.H.; Eom, S.H. Crystal Structures of the HslVU Peptidase–ATPase Complex Reveal an ATP-Dependent Proteolysis Mechanism. Structure 2001, 9, 177–184. [Google Scholar] [CrossRef]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Tezuka, T.; Kim, M.; Choi, J.; Oichi, Y.; Kobayashi, H.; Harada, K.H.; Mizushima, T.; Taketani, S.; Koizumi, A.; et al. Moyamoya disease patient mutations in the RING domain of RNF213 reduce its ubiquitin ligase activity and enhance NFkappaB activation and apoptosis in an AAA+ domain-dependent manner. Biochem. Biophys. Res. Commun. 2020, 525, 668–674. [Google Scholar] [CrossRef]
- Habu, T.; Harada, K.H. UBC13 is an RNF213-associated E2 ubiquitin-conjugating enzyme, and Lysine 63-linked ubiquitination by the RNF213-UBC13 axis is responsible for angiogenic activity. FASEB Bioadv. 2021, 3, 243–258. [Google Scholar] [CrossRef]
- Tian, H.; Yu, K.; He, L.; Xu, H.; Han, C.; Zhang, X.; Wang, X.; Zhang, X.; Zhang, L.; Gao, G.; et al. RNF213 modulates γ-herpesvirus infection and reactivation via targeting the viral Replication and Transcription Activator. Proc. Natl. Acad. Sci. USA 2023, 120, e2218825120. [Google Scholar] [CrossRef]
- Otten, E.G.; Werner, E.; Crespillo-Casado, A.; Boyle, K.B.; Dharamdasani, V.; Pathe, C.; Santhanam, B.; Randow, F. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 2021, 594, 111–116. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, X.; Sheng, J.; Fu, Y.; Nie, D.; You, X.; Chen, Y.; Yang, X.; Ling, Q.; Zhang, H.; et al. RNF213 promotes Treg cell differentiation by facilitating K63-linked ubiquitination and nuclear translocation of FOXO1. Nat. Commun. 2024, 15, 5961. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Panepinto, M.C.; Ueberheide, B.; Neel, B.G. A mechanism for hypoxia-induced inflammatory cell death in cancer. Nature 2025, 637, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Couet, J.; Li, S.; Okamoto, T.; Ikezu, T.; Lisanti, M.P. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 1997, 272, 6525–6533. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.G.; Kamen, B.A.; Rothberg, K.G.; Lacey, S.W. Potocytosis: Sequestration and transport of small molecules by caveolae. Science 1992, 255, 410–411. [Google Scholar] [CrossRef] [PubMed]
- Quest, A.F.; Gutierrez-Pajares, J.L.; Torres, V.A. Caveolin-1: An ambiguous partner in cell signalling and cancer. J. Cell Mol. Med. 2008, 12, 1130–1150. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Chevrot, G.; Khandelia, H. The role of caveolin-1 in lipid droplets and their biogenesis. Chem. Phys. Lipids 2018, 211, 93–99. [Google Scholar] [CrossRef]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef]
- Frank, P.G.; Woodman, S.E.; Park, D.S.; Lisanti, M.P. Caveolin, caveolae, and endothelial cell function. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1161–1168. [Google Scholar] [CrossRef]
- Glenney, J.R., Jr.; Soppet, D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10517–10521. [Google Scholar] [CrossRef]
- Bernatchez, P.; Sharma, A.; Bauer, P.M.; Marin, E.; Sessa, W.C. A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J. Clin. Investig. 2011, 121, 3747–3755. [Google Scholar] [CrossRef]
- Chen, Z.; D S Oliveira, S.; Zimnicka, A.M.; Jiang, Y.; Sharma, T.; Chen, S.; Lazarov, O.; Bonini, M.G.; Haus, J.M.; Minshall, R.D. Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Mol. Biol. Cell 2018, 29, 1190–1202. [Google Scholar] [CrossRef]
- Mathew, R.; Huang, J.; Gewitz, M.H. Pulmonary artery hypertension: Caveolin-1 and eNOS interrelationship: A new perspective. Cardiol. Rev. 2007, 15, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Troiano, J.A.; Potje, S.R.; Graton, M.E.; Gonçalves, E.T.; Tostes, R.C.; Antoniali, C. Caveolin-1/Endothelial Nitric Oxide Synthase Interaction Is Reduced in Arteries From Pregnant Spontaneously Hypertensive Rats. Front. Physiol. 2021, 12, 760237. [Google Scholar] [CrossRef]
- Forrester, S.J.; Elliott, K.J.; Kawai, T.; Obama, T.; Boyer, M.J.; Preston, K.J.; Yan, Z.; Eguchi, S.; Rizzo, V. Caveolin-1 Deletion Prevents Hypertensive Vascular Remodeling Induced by Angiotensin II. Hypertension 2017, 69, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Gairhe, S.; Awad, K.S.; Dougherty, E.J.; Ferreyra, G.A.; Wang, S.; Yu, Z.X.; Takeda, K.; Demirkale, C.Y.; Torabi-Parizi, P.; Austin, E.D.; et al. Type I interferon activation and endothelial dysfunction in caveolin-1 insufficiency-associated pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2021, 118, e2010206118. [Google Scholar] [CrossRef]
- Bang, O.Y.; Chung, J.W.; Kim, S.J.; Oh, M.J.; Kim, S.Y.; Cho, Y.H.; Cha, J.; Yeon, J.Y.; Kim, K.H.; Kim, G.M.; et al. Caveolin-1, Ring finger protein 213, and endothelial function in Moyamoya disease. Int. J. Stroke 2016, 11, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Kiko, T.; Asano, R.; Ishibashi, T.; Endo, H.; Nishi, N.; Hayashi, H.; Ueda, J.; Aoki, T.; Tsuji, A.; Nakaoka, Y.; et al. Prevalence and Clinical Characteristics of Heterozygous RNF213 p.Arg4810Lys Variant Carriers Diagnosed With Chronic Thromboembolic Pulmonary Hypertension. J. Am. Heart Assoc. 2024, 13, e035009. [Google Scholar] [CrossRef]
- Mineharu, Y.; Miyamoto, S. RNF213 and GUCY1A3 in Moyamoya Disease: Key Regulators of Metabolism, Inflammation, and Vascular Stability. Front. Neurol. 2021, 12, 687088. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, Y.; Jiang, L.; Liu, Y.; Zhang, L. The emerging role of E3 ubiquitin ligase RNF213 as an antimicrobial host determinant. Front. Cell Infect. Microbiol. 2023, 13, 1205355. [Google Scholar] [CrossRef]
- Kobayashi, H.; Matsuda, Y.; Hitomi, T.; Okuda, H.; Shioi, H.; Matsuda, T.; Imai, H.; Sone, M.; Taura, D.; Harada, K.H.; et al. Biochemical and Functional Characterization of RNF213 (Mysterin) R4810K, a Susceptibility Mutation of Moyamoya Disease, in Angiogenesis In Vitro and In Vivo. J. Am. Heart Assoc. 2015, 4, e002146. [Google Scholar] [CrossRef]
- Kobayashi, H.; Brozman, M.; Kyselová, K.; Viszlayová, D.; Morimoto, T.; Roubec, M.; Školoudík, D.; Petrovičová, A.; Juskanič, D.; Strauss, J.; et al. RNF213 Rare Variants in Slovakian and Czech Moyamoya Disease Patients. PLoS ONE 2016, 11, e0164759. [Google Scholar] [CrossRef]
- Sugihara, M.; Morito, D.; Ainuki, S.; Hirano, Y.; Ogino, K.; Kitamura, A.; Hirata, H.; Nagata, K. The AAA+ ATPase/ubiquitin ligase mysterin stabilizes cytoplasmic lipid droplets. J. Cell Biol. 2019, 218, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Copeland, C.A.; Kawano, Y.; Rosenzweig, E.B.; Austin, E.D.; Shahmirzadi, L.; Tang, S.; Raghunathan, K.; Chung, W.K.; Kenworthy, A.K. Characterization of a caveolin-1 mutation associated with both pulmonary arterial hypertension and congenital generalized lipodystrophy. Traffic 2016, 17, 1297–1312. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, C.; Xiao, X.; Li, A.; Liu, Y.; Zhao, J.; Fan, L.; Liang, Z.; Pang, W.; Yao, W.; et al. Endothelial discoidin domain receptor 1 senses flow to modulate YAP activation. Nat. Commun. 2023, 14, 6457. [Google Scholar] [CrossRef]
- Słońska, A.; Miedzińska, A.; Chodkowski, M.; Bąska, P.; Mielnikow, A.; Bartak, M.; Bańbura, M.W.; Cymerys, J. Human Adenovirus Entry and Early Events during Infection of Primary Murine Neurons: Immunofluorescence Studies In Vitro. Pathogens 2024, 13, 158. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Banh, R.S.; Zhang, W.; Sidhu, S.S.; Neel, B.G. MMD-associated RNF213 SNPs encode dominant-negative alleles that globally impair ubiquitylation. Life Sci. Alliance 2022, 5, e202000807. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Ai, H.W. Genetically encoded green fluorescent biosensors for monitoring UDP-GlcNAc in live cells. ACS Cent. Sci. 2021, 7, 1763–1770. [Google Scholar] [CrossRef]
- Poothong, J.; Pottekat, A.; Siirin, M.; Campos, A.R.; Paton, A.W.; Paton, J.C.; Lagunas-Acosta, J.; Chen, Z.; Swift, M.; Volkmann, N.; et al. Factor VIII exhibits chaperone-dependent and glucose-regulated reversible amyloid formation in the endoplasmic reticulum. bioRxiv 2020. 2020.2001.2013.905190. [Google Scholar] [CrossRef]
- Burana, D.; Yoshihara, H.; Tanno, H.; Yamamoto, A.; Saeki, Y.; Tanaka, K.; Komada, M. The Ankrd13 Family of Ubiquitin-interacting Motif-bearing Proteins Regulates Valosin-containing Protein/p97 Protein-mediated Lysosomal Trafficking of Caveolin 1. J. Biol. Chem. 2016, 291, 6218–6231. [Google Scholar] [CrossRef] [PubMed]
- Bang, O.Y.; Chung, J.W.; Kim, D.H.; Won, H.H.; Yeon, J.Y.; Ki, C.S.; Shin, H.J.; Kim, J.S.; Hong, S.C.; Kim, D.K.; et al. Moyamoya Disease and Spectrums of RNF213 Vasculopathy. Transl. Stroke Res. 2020, 11, 580–589. [Google Scholar] [CrossRef]
- Kuroda, S.; Houkin, K. Moyamoya disease: Current concepts and future perspectives. Lancet Neurol. 2008, 7, 1056–1066. [Google Scholar] [CrossRef]
- Scott, R.M.; Smith, E.R. Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Takaku, A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 1969, 20, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Copeland, C.A.; Han, B.; Tiwari, A.; Austin, E.D.; Loyd, J.E.; West, J.D.; Kenworthy, A.K. A disease-associated frameshift mutation in caveolin-1 disrupts caveolae formation and function through introduction of a de novo ER retention signal. Mol. Biol. Cell 2017, 28, 3095–3111. [Google Scholar] [CrossRef]
- Couet, J.; Sargiacomo, M.; Lisanti, M.P. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Biol. Chem. 1997, 272, 30429–30438. [Google Scholar] [CrossRef]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Porta, J.C.; Han, B.; Gulsevin, A.; Chung, J.M.; Peskova, Y.; Connolly, S.; McHaourab, H.S.; Meiler, J.; Karakas, E.; Kenworthy, A.K.; et al. Molecular architecture of the human caveolin-1 complex. Sci. Adv. 2022, 8, eabn7232. [Google Scholar] [CrossRef]
- Shu, Y.; Jin, S. Caveolin-1 in endothelial cells: A potential therapeutic target for atherosclerosis. Heliyon 2023, 9, e18653. [Google Scholar] [CrossRef]
- Lee, C.Y.; Lai, T.Y.; Tsai, M.K.; Chang, Y.C.; Ho, Y.H.; Yu, I.S.; Yeh, T.W.; Chou, C.C.; Lin, Y.S.; Lawrence, T.; et al. The ubiquitin ligase ZNRF1 promotes caveolin-1 ubiquitination and degradation to modulate inflammation. Nat. Commun. 2017, 8, 15502. [Google Scholar] [CrossRef]
- Hodge, C.D.; Spyracopoulos, L.; Glover, J.N. Ubc13: The Lys63 ubiquitin chain building machine. Oncotarget 2016, 7, 64471–64504. [Google Scholar] [CrossRef]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258. [Google Scholar] [CrossRef]
- Guey, S.; Kraemer, M.; Herve, D.; Ludwig, T.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.C.; Choi, S.; Broseus, L.; Callebaut, I.; et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, G.; Zhang, X.; Minshall, R.D. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ. Res. 2009, 105, 676–685. [Google Scholar] [CrossRef]
- Thomas, S.R.; Chen, K.; Keaney, J.F., Jr. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J. Biol. Chem. 2002, 277, 6017–6024. [Google Scholar] [CrossRef]
- Chen, Z.; Bakhshi, F.R.; Shajahan, A.N.; Sharma, T.; Mao, M.; Trane, A.; Bernatchez, P.; van Nieuw Amerongen, G.P.; Bonini, M.G.; Skidgel, R.A.; et al. Nitric oxide-dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol. Biol. Cell 2012, 23, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kotamraju, S.; Zielonka, J.; Harder, D.R.; Kalyanaraman, B. Hydrogen peroxide induces nitric oxide and proteosome activity in endothelial cells: A bell-shaped signaling response. Free Radic. Biol. Med. 2007, 42, 1049–1061. [Google Scholar] [CrossRef]
- Yue, L.; Bian, J.T.; Grizelj, I.; Cavka, A.; Phillips, S.A.; Makino, A.; Mazzone, T. Apolipoprotein E enhances endothelial-NO production by modulating caveolin 1 interaction with endothelial NO synthase. Hypertension 2012, 60, 1040–1046. [Google Scholar] [CrossRef]
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macaluso, F.; Russell, R.G.; Li, M.; Pestell, R.G.; et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 2001, 276, 38121–38138. [Google Scholar] [CrossRef] [PubMed]
- Ahel, J.; Fletcher, A.; Grabarczyk, D.B.; Roitinger, E.; Deszcz, L.; Lehner, A.; Virdee, S.; Clausen, T. E3 ubiquitin ligase RNF213 employs a non-canonical zinc finger active site and is allosterically regulated by ATP. bioRxiv 2021. 2021.2005.2010.443411. [Google Scholar] [CrossRef]
- Kirchner, P.; Bug, M.; Meyer, H. Ubiquitination of the N-terminal region of caveolin-1 regulates endosomal sorting by the VCP/p97 AAA-ATPase. J. Biol. Chem. 2013, 288, 7363–7372. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Dósa, A.; Csizmadia, T. The role of K63-linked polyubiquitin in several types of autophagy. Biol. Futur. 2022, 73, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, Z.J. Expanding role of ubiquitination in NF-κB signaling. Cell Res. 2011, 21, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell. Mol. Biol. Lett. 2021, 26, 1. [Google Scholar] [CrossRef]
- Hernandez, D.; Walsh, S.; Saavedra Sanchez, L.; Dickinson, M.S.; Coers, J. Interferon-Inducible E3 Ligase RNF213 Facilitates Host-Protective Linear and K63-Linked Ubiquitylation of Toxoplasma gondii Parasitophorous Vacuoles. mBio 2022, 13, e0188822. [Google Scholar] [CrossRef]
- Kim, Y.-C.; Snoberger, A.; Schupp, J.; Smith, D.M. ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat. Commun. 2015, 6, 8520. [Google Scholar] [CrossRef]
- Flood, D.; Lee, E.S.; Taylor, C.T. Intracellular energy production and distribution in hypoxia. J. Biol. Chem. 2023, 299, 105103. [Google Scholar] [CrossRef]
- Nagahama, M.; Yamazoe, T.; Hara, Y.; Tani, K.; Tsuji, A.; Tagaya, M. The AAA-ATPase NVL2 is a component of pre-ribosomal particles that interacts with the DExD/H-box RNA helicase DOB1. Biochem. Biophys. Res. Commun. 2006, 346, 1075–1082. [Google Scholar] [CrossRef]
- Lo, Y.H.; Romes, E.M.; Pillon, M.C.; Sobhany, M.; Stanley, R.E. Structural Analysis Reveals Features of Ribosome Assembly Factor Nsa1/WDR74 Important for Localization and Interaction with Rix7/NVL2. Structure 2017, 25, 762–772.e764. [Google Scholar] [CrossRef] [PubMed]
- Blythe, E.E.; Olson, K.C.; Chau, V.; Deshaies, R.J. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. USA 2017, 114, E4380–E4388. [Google Scholar] [CrossRef]
- Thery, F.; Martina, L.; Asselman, C.; Zhang, Y.; Vessely, M.; Repo, H.; Sedeyn, K.; Moschonas, G.D.; Bredow, C.; Teo, Q.W.; et al. Ring finger protein 213 assembles into a sensor for ISGylated proteins with antimicrobial activity. Nat. Commun. 2021, 12, 5772. [Google Scholar] [CrossRef]
- Lim, J.E.; Bernatchez, P.; Nabi, I.R. Scaffolds and the scaffolding domain: An alternative paradigm for caveolin-1 signaling. Biochem. Soc. Trans. 2024, 52, 947–959. [Google Scholar] [CrossRef] [PubMed]
- García-Cardeña, G.; Martasek, P.; Masters, B.S.; Skidd, P.M.; Couet, J.; Li, S.; Lisanti, M.P.; Sessa, W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 1997, 272, 25437–25440. [Google Scholar] [CrossRef] [PubMed]
- Gratton, J.P.; Fontana, J.; O’Connor, D.S.; Garcia-Cardena, G.; McCabe, T.J.; Sessa, W.C. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J. Biol. Chem. 2000, 275, 22268–22272. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Fonseca, F.V.; Kumar, S.; Pardo, D.; Darragh, C.; Elms, S.; Fulton, D.; Black, S.M. eNOS activation and NO function: Structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J. Endocrinol. 2011, 210, 271–284. [Google Scholar] [CrossRef]
- Zimnicka, A.M.; Husain, Y.S.; Shajahan, A.N.; Sverdlov, M.; Chaga, O.; Chen, Z.; Toth, P.T.; Klomp, J.; Karginov, A.V.; Tiruppathi, C.; et al. Src-dependent phosphorylation of caveolin-1 Tyr-14 promotes swelling and release of caveolae. Mol. Biol. Cell 2016, 27, 2090–2106. [Google Scholar] [CrossRef]
- Chen, F.; Kumar, S.; Yu, Y.; Aggarwal, S.; Gross, C.; Wang, Y.; Chakraborty, T.; Verin, A.D.; Catravas, J.D.; Lucas, R.; et al. PKC-Dependent Phosphorylation of eNOS at T495 Regulates eNOS Coupling and Endothelial Barrier Function in Response to G+ -Toxins. PLoS ONE 2014, 9, e99823. [Google Scholar] [CrossRef]
- Sanguinetti, A.R.; Cao, H.; Corley Mastick, C. Fyn is required for oxidative- and hyperosmotic-stress-induced tyrosine phosphorylation of caveolin-1. Biochem. J. 2003, 376, 159–168. [Google Scholar] [CrossRef]
- Hao, L.; Wei, X.; Guo, P.; Zhang, G.; Qi, S. Neuroprotective Effects of Inhibiting Fyn S-Nitrosylation on Cerebral Ischemia/Reperfusion-Induced Damage to CA1 Hippocampal Neurons. Int. J. Mol. Sci. 2016, 17, 1100. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, H.B.; Kwon, H.S. RNF213 Polymorphism in Intracranial Artery Dissection. J. Stroke 2018, 20, 404–406. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.; Inoue, R.; Masuo, Y.; Shimizu, Y.; Sonomura, K.; Kim, M.; Kobayashi, H.; Harada, K.H.; Mineharu, Y.; Koizumi, A.; et al. RNF213 Acts as a Molecular Switch for Cav-1 Ubiquitination and Phosphorylation in Human Cells. Cells 2025, 14, 775. https://doi.org/10.3390/cells14110775
Choi J, Inoue R, Masuo Y, Shimizu Y, Sonomura K, Kim M, Kobayashi H, Harada KH, Mineharu Y, Koizumi A, et al. RNF213 Acts as a Molecular Switch for Cav-1 Ubiquitination and Phosphorylation in Human Cells. Cells. 2025; 14(11):775. https://doi.org/10.3390/cells14110775
Chicago/Turabian StyleChoi, Jungmi, Ryoichi Inoue, Yuki Masuo, Yukiko Shimizu, Kazuhiro Sonomura, Minsoo Kim, Hatasu Kobayashi, Kouji H. Harada, Yohei Mineharu, Akio Koizumi, and et al. 2025. "RNF213 Acts as a Molecular Switch for Cav-1 Ubiquitination and Phosphorylation in Human Cells" Cells 14, no. 11: 775. https://doi.org/10.3390/cells14110775
APA StyleChoi, J., Inoue, R., Masuo, Y., Shimizu, Y., Sonomura, K., Kim, M., Kobayashi, H., Harada, K. H., Mineharu, Y., Koizumi, A., Tezuka, T., & Youssefian, S. (2025). RNF213 Acts as a Molecular Switch for Cav-1 Ubiquitination and Phosphorylation in Human Cells. Cells, 14(11), 775. https://doi.org/10.3390/cells14110775