
Targeting the Cargo Receptor TMED9 as a Therapeutic Strategy Against Brain Tumors

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. GSC and Neural Cell Cultures
2.2. DIPG Cultures
2.3. Real-Time PCR
2.4. TMED9 Silencing
2.5. Western Blot and SDS-PAGE Gel Electrophoresis
2.6. Transwell Migration Assay
2.7. Neurosphere Formation (Self-Renewal) Assay
2.8. Cell Viability and Cell Death
2.9. Image Analysis
2.10. Propidium Iodide Uptake
2.11. LDH Assay
2.12. TCGA Data Analysis
2.13. Statistical Analysis
3. Results
3.1. High TMED9 Expression Is Associated with Aggressive Tumors and Poor Prognosis
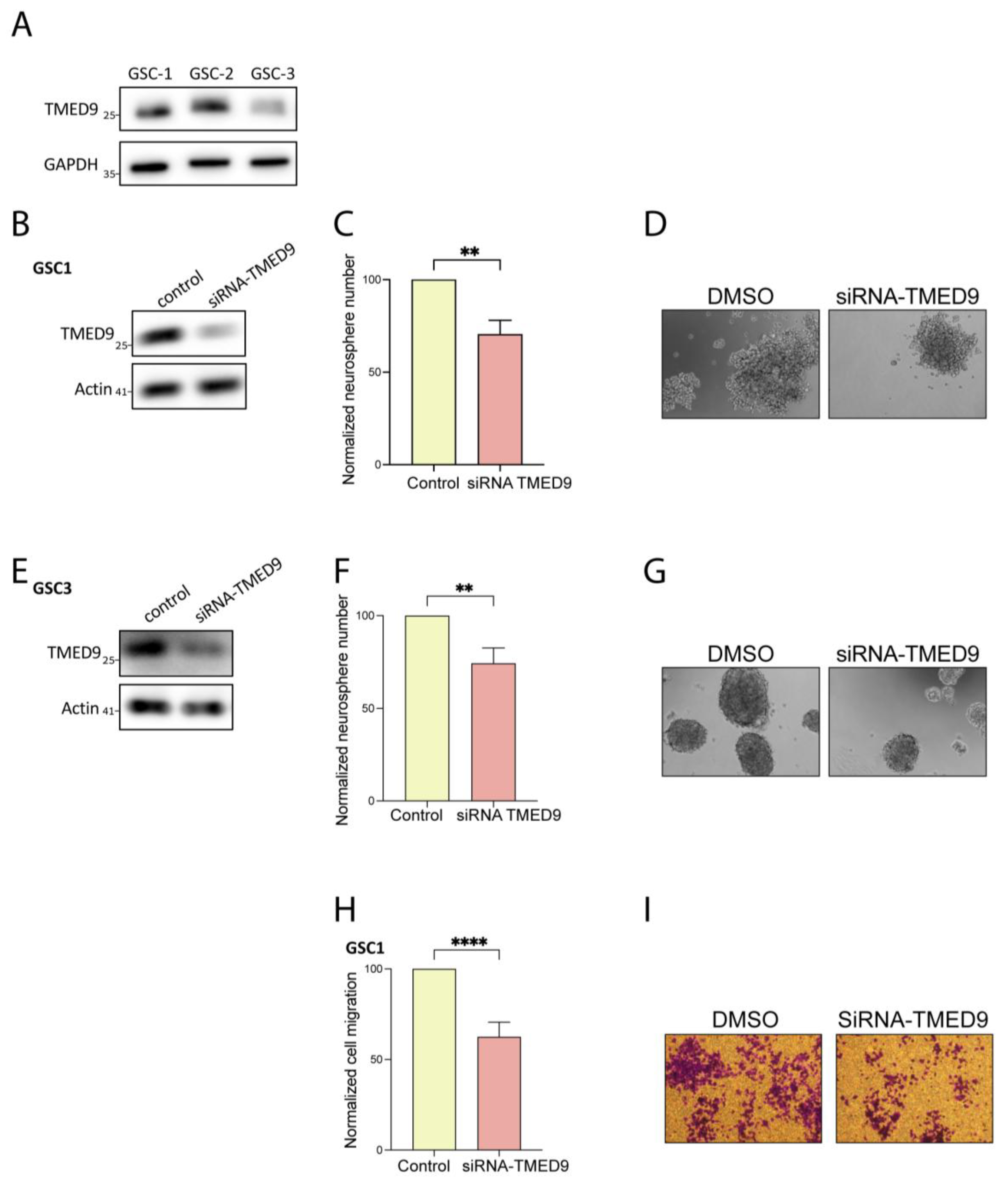
3.2. Genetic Removal of TMED9 Inhibits Self-Renewal and Migration of Patient-Derived GSCs
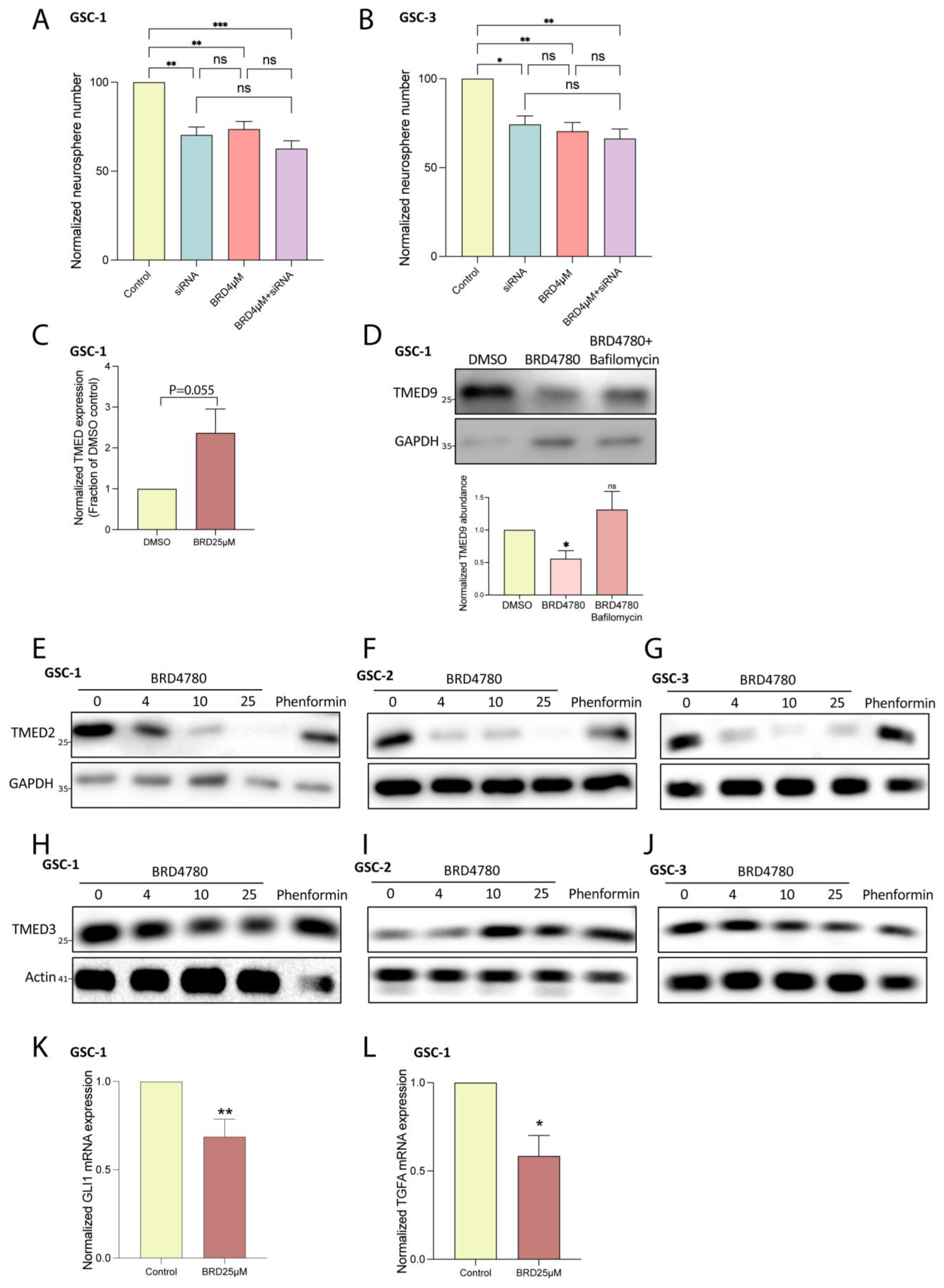
3.3. BRD4780 Targets and Removes TMED9 from Patient-Derived GSCs and Induces Anti-Tumor Effects
3.4. BRD4780 Enhances the Effect of TMZ
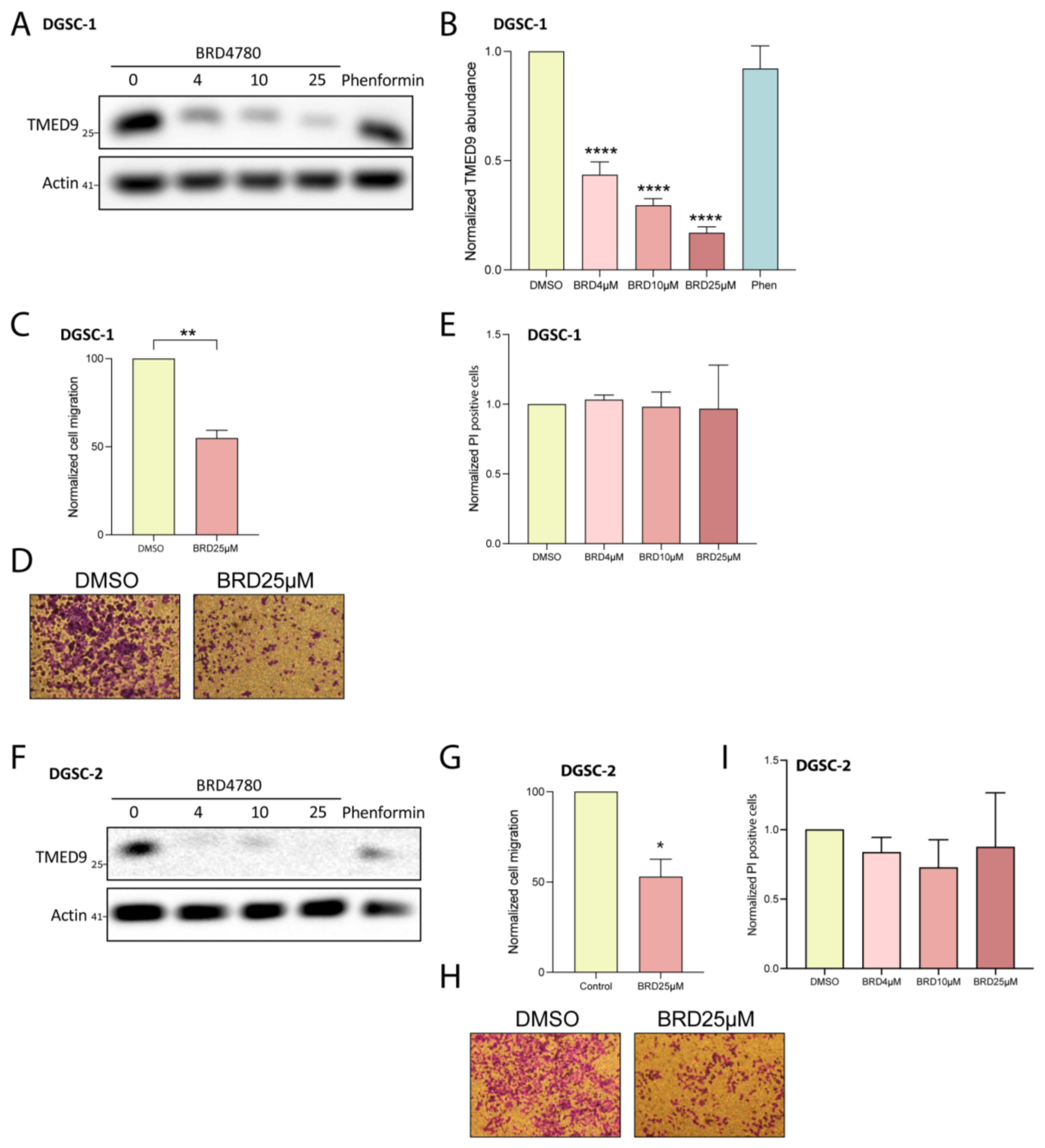
3.5. BRD4780 Targets and Removes TMED9 from Differentiated Tumor Cell Progeny (DGSCs) and Promotes Anti-Tumor Effects
3.6. BRD4780 Drives Its Anti-Tumor Effects Through the Lysosomal Removal of TMED9
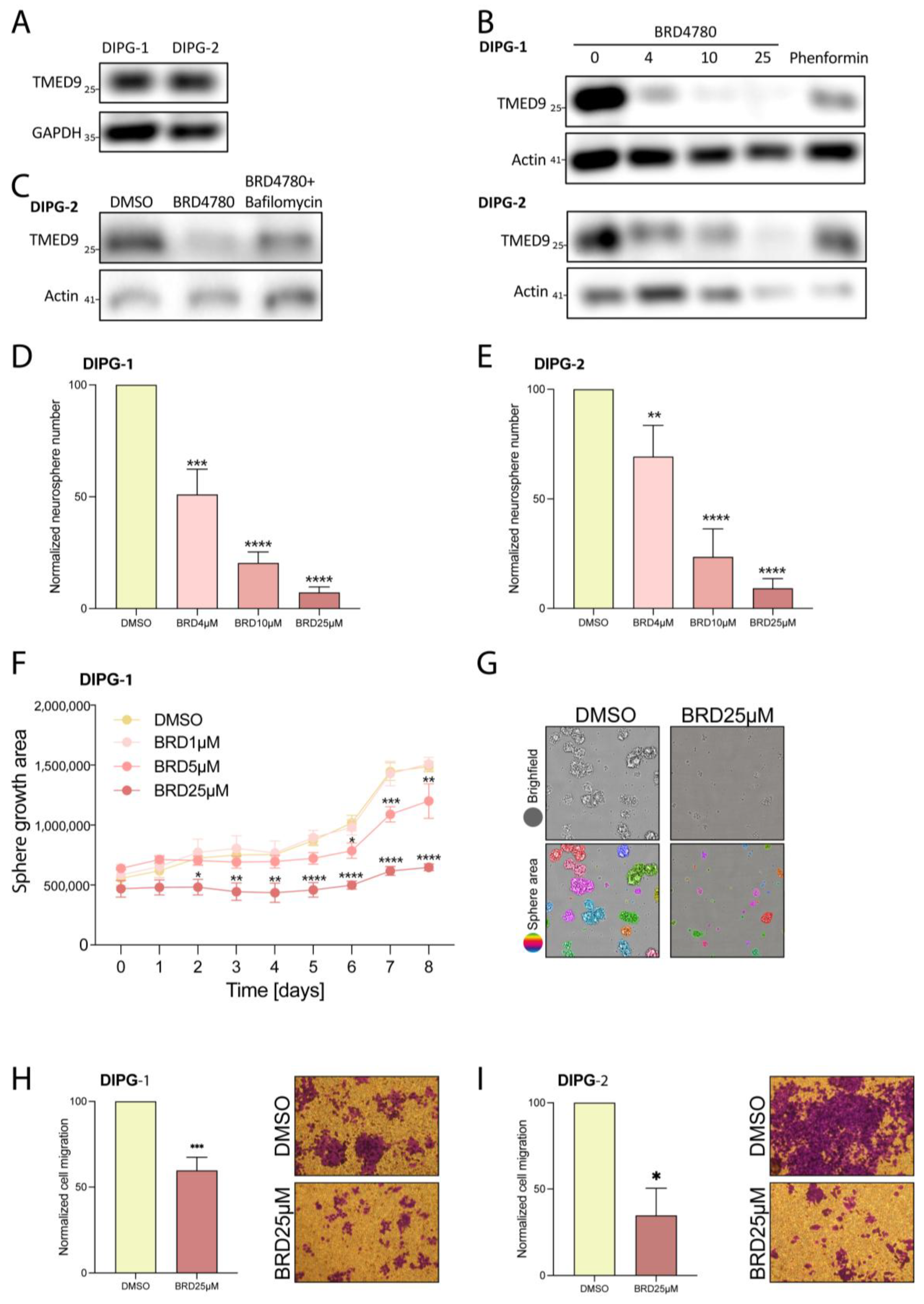
3.7. BRD4780’s Anti-Tumor Effect Is Also Observed in Pediatric Brain Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- McKinnon, C.; Nandhabalan, M.; Murray, S.A.; Plaha, P. Glioblastoma: Clinical Presentation, Diagnosis, and Management. BMJ 2021, 374, n1560. [Google Scholar] [CrossRef] [PubMed]
- Comba, A.; Faisal, S.M.; Varela, M.L.; Hollon, T.; Al-Holou, W.N.; Umemura, Y.; Nunez, F.J.; Motsch, S.; Castro, M.G.; Lowenstein, P.R. Uncovering Spatiotemporal Heterogeneity of High-Grade Gliomas: From Disease Biology to Therapeutic Implications. Front. Oncol. 2021, 11, 703764. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, B.T.; Huse, J.T. Classification of Adult-Type Diffuse Gliomas: Impact of the World Health Organization 2021 Update. Brain Pathol. 2022, 32, e13062. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Steponaitis, G.; Tamasauskas, A. Mesenchymal and Proneural Subtypes of Glioblastoma Disclose Branching Based on GSC Associated Signature. Int. J. Mol. Sci. 2021, 22, 4964. [Google Scholar] [CrossRef]
- Sales, A.H.A.; Beck, J.; Schnell, O.; Fung, C.; Meyer, B.; Gempt, J. Surgical Treatment of Glioblastoma: State-of-the-Art and Future Trends. J. Clin. Med. 2022, 11, 5354. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma Stem Cells: Lessons from the Tumor Hierarchy in a Lethal Cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.P.; Laks, D.R.; Sun, D.; Ganbold, M.; Wang, Z.; Pedraza, A.M.; Bale, T.; Tabar, V.; Brennan, C.; Zhou, X.; et al. Quiescent Human Glioblastoma Cancer Stem Cells Drive Tumor Initiation, Expansion, and Recurrence Following Chemotherapy. Dev. Cell 2022, 57, 32–46.e8. [Google Scholar] [CrossRef] [PubMed]
- Bayik, D.; Lathia, J.D. Cancer Stem Cell-Immune Cell Crosstalk in Tumour Progression. Nat. Rev. Cancer 2021, 21, 526–536. [Google Scholar] [CrossRef]
- Strating, J.R.P.M.; Martens, G.J.M. The P24 Family and Selective Transport Processes at the ER-Golgi Interface. Biol. Cell 2009, 101, 495–509. [Google Scholar] [CrossRef]
- Pastor-Cantizano, N.; Montesinos, J.C.; Bernat-Silvestre, C.; Marcote, M.J.; Aniento, F. P24 Family Proteins: Key Players in the Regulation of Trafficking along the Secretory Pathway. Protoplasma 2016, 253, 967–985. [Google Scholar] [CrossRef] [PubMed]
- Belden, W.J.; Barlowe, C. Deletion of Yeast P24 Genes Activates the Unfolded Protein Response. Mol. Biol. Cell 2001, 12, 957–969. [Google Scholar] [CrossRef]
- Elrod-Erickson, M.J.; Kaiser, C.A. Genes That Control the Fidelity of Endoplasmic Reticulum to Golgi Transport Identified as Suppressors of Vesicle Budding Mutations. Mol. Biol. Cell 1996, 7, 1043–1058. [Google Scholar] [CrossRef]
- D’Arcangelo, J.G.; Crissman, J.; Pagant, S.; Copic, A.; Latham, C.F.; Snapp, E.L.; Miller, E.A. Traffic of P24 Proteins and COPII Coat Composition Mutually Influence Membrane Scaffolding. Curr. Biol. 2015, 25, 1296–1305. [Google Scholar] [CrossRef]
- Aber, R.; Chan, W.; Mugisha, S.; Jerome-Majewska, L.A. Transmembrane Emp24 Domain Proteins in Development and Disease. Genet. Res. 2019, 101, e14. [Google Scholar] [CrossRef]
- Zhou, L.; Li, H.; Yao, H.; Dai, X.; Gao, P.; Cheng, H. TMED Family Genes and Their Roles in Human Diseases. Int. J. Med. Sci. 2023, 20, 1732–1743. [Google Scholar] [CrossRef]
- Roberts, B.S.; Satpute-Krishnan, P. The Many Hats of Transmembrane Emp24 Domain Protein TMED9 in Secretory Pathway Homeostasis. Front. Cell Dev. Biol. 2023, 10, 1096899. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Lin, Y.; Zhang, W.; He, F.; Xu, Y.; Chen, Z. Bioinformatics Analysis of LMAN1 Expression, Clinical Characteristics, and Its Effects on Cell Proliferation and Invasion in Glioma. Brain Res. 2022, 1789, 147952. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, B.; Liu, Y.; Weng, X.; Wang, S.; Li, Y.; Deng, S.-Z.; Cheng, B. Study on the Role and Mechanism of TMED2 in Oral Squamous Cell Carcinoma. Int. J. Biol. Macromol. 2025, 289, 138805. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Bernal, C.; Silvano, M.; Anand, S.; Ruiz i Altaba, A. The Protein Secretion Modulator TMED9 Drives CNIH4/TGFα/GLI Signaling Opposing TMED3-WNT-TCF to Promote Colon Cancer Metastases. Oncogene 2019, 38, 5817. [Google Scholar] [CrossRef]
- Yang, X.-Y.; Ren, C.-P.; Wang, L.; Li, H.; Jiang, C.-J.; Zhang, H.-B.; Zhao, M.; Yao, K.-T. Identification of Differentially Expressed Genes in Metastatic and Non-Metastatic Nasopharyngeal Carcinoma Cells by Suppression Subtractive Hybridization. Cell. Oncol. 2005, 27, 215–223. [Google Scholar] [CrossRef]
- Tashima, Y.; Hirata, T.; Maeda, Y.; Murakami, Y.; Kinoshita, T. Differential Use of P24 Family Members as Cargo Receptors for the Transport of Glycosylphosphatidylinositol-Anchored Proteins and Wnt1. J. Biochem. 2022, 171, 75–83. [Google Scholar] [CrossRef]
- Yang, J.; Huang, H.; Xiao, D.; Duan, Y.; Zheng, Y.; Chen, Z. Knockdown of TMED3 Inhibits Cell Viability and Migration and Increases Apoptosis in Human Chordoma Cells. Int. J. Oncol. 2021, 58, 15. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, Y. Depleting TMED3 Alleviates the Development of Endometrial Carcinoma. Cancer Cell Int. 2022, 22, 231. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, Y.; Li, Q. TMED3 Promotes Proliferation and Migration in Breast Cancer Cells by Activating Wnt/β-Catenin Signaling. Onco Targets Ther. 2020, 13, 5819–5830. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, Y.; Wang, Z.; Chen, J.; Zhang, J.; Gu, Y. TMED2 Promotes Glioma Tumorigenesis by Being Involved in EGFR Recycling Transport. Int. J. Biol. Macromol. 2024, 262 Pt 2, 130055. [Google Scholar] [CrossRef]
- Oprita, A.; Baloi, S.-C.; Staicu, G.-A.; Alexandru, O.; Tache, D.E.; Danoiu, S.; Micu, E.S.; Sevastre, A.-S. Updated Insights on EGFR Signaling Pathways in Glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.A.; Tabassum, T.; Farzana, M.; Moin, A.T.; Zohora, U.S.; Rahman, M.S. Expression Analysis, Molecular Characterization and Prognostic Evaluation on TMED4 and TMED9 Gene Expression in Glioma. Biomed. Signal Process. Control. 2022, 78, 103922. [Google Scholar] [CrossRef]
- Dvela-Levitt, M.; Kost-Alimova, M.; Emani, M.; Kohnert, E.; Thompson, R.; Sidhom, E.H.; Rivadeneira, A.; Sahakian, N.; Roignot, J.; Papagregoriou, G.; et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 2019, 178, 521–535.e23. [Google Scholar] [CrossRef]
- Dvela-Levitt, M.; Shaw, J.L.; Greka, A. A Rare Kidney Disease to Cure Them All? Towards Mechanism-Based Therapies for Proteinopathies. Trends Mol. Med. 2021, 27, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Hasselbach, L.A.; Irtenkauf, S.M.; Lemke, N.W.; Nelson, K.K.; Berezovsky, A.D.; Carlton, E.T.; Transou, A.D.; Mikkelsen, T.; deCarvalho, A.C. Optimization of High Grade Glioma Cell Culture from Surgical Specimens for Use in Clinically Relevant Animal Models and 3D Immunochemistry. J. Vis. Exp. 2014, 83, e51088. [Google Scholar] [CrossRef]
- Giladi, N.D.; Ziv-Av, A.; Lee, H.K.; Finniss, S.; Cazacu, S.; Xiang, C.; Waldman Ben-Asher, H.; deCarvalho, A.; Mikkelsen, T.; Poisson, L.; et al. RTVP-1 Promotes Mesenchymal Transformation of Glioma via a STAT-3/IL-6-Dependent Positive Feedback Loop. Oncotarget 2015, 6, 22680–22697. [Google Scholar] [CrossRef]
- Bier, A.; Giladi, N.; Kronfeld, N.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Poisson, L.; deCarvalho, A.C.; Slavin, S.; et al. MicroRNA-137 Is Downregulated in Glioblastoma and Inhibits the Stemness of Glioma Stem Cells by Targeting RTVP-1. Oncotarget 2013, 4, 665–676. [Google Scholar] [CrossRef]
- Penning, D.H.; Cazacu, S.; Brodie, A.; Jevtovic-Todorovic, V.; Kalkanis, S.N.; Lewis, M.; Brodie, C. Neuron-Glia Crosstalk Plays a Major Role in the Neurotoxic Effects of Ketamine via Extracellular Vesicles. Front. Cell Dev. Biol. 2021, 9, 691648. [Google Scholar] [CrossRef]
- Zats, L.P.; Ahmad, L.; Casden, N.; Lee, M.J.; Belzer, V.; Adato, O.; Bar Cohen, S.; Ko, S.-H.B.; Filbin, M.G.; Unger, R.; et al. An Affinity for Brainstem Microglia in Pediatric High-Grade Gliomas of Brainstem Origin. Neuro Oncol. Adv. 2022, 4, vdac117. [Google Scholar] [CrossRef]
- Bazua-Valenti, S.; Brown, M.R.; Zavras, J.; Riedl Khursigara, M.; Grinkevich, E.; Sidhom, E.-H.; Keller, K.H.; Racette, M.; Dvela-Levitt, M.; Quintanova, C.; et al. Disrupted Uromodulin Trafficking Is Rescued by Targeting TMED Cargo Receptors. J. Clin. Investig. 2024, 134, e180347. [Google Scholar] [CrossRef]
- Kinstlinger, S.; Dvela-Levitt, M. Opening New Routes for Kidney Therapy. J. Am. Soc. Nephrol. 2024, 36, 519–521. [Google Scholar] [CrossRef]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing Phenformin for the Targeting of Glioma Stem Cells and the Treatment of Glioblastoma. Oncotarget 2016, 7, 56456–56470. [Google Scholar] [CrossRef]
- Mirzaei, S.; Paskeh, M.D.A.; Entezari, M.; Mirmazloomi, S.R.; Hassanpoor, A.; Aboutalebi, M.; Rezaei, S.; Hejazi, E.S.; Kakavand, A.; Heidari, H.; et al. SOX2 Function in Cancers: Association with Growth, Invasion, Stemness and Therapy Response. Biomed. Pharmacother. 2022, 156, 113860. [Google Scholar] [CrossRef] [PubMed]
- Mamun, M.A.; Mannoor, K.; Cao, J.; Qadri, F.; Song, X. SOX2 in Cancer Stemness: Tumor Malignancy and Therapeutic Potentials. J. Mol. Cell Biol. 2020, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.; Lengerke, C. SOX2 Protein Biochemistry in Stemness, Reprogramming, and Cancer: The PI3K/AKT/SOX2 Axis and Beyond. Oncogene 2019, 39, 278–292. [Google Scholar] [CrossRef]
- Di Bonaventura, R.; Martini, M.; Cenci, T.; Caccavella, V.M.; Barresi, V.; Gessi, M.; Albanese, A.; Lauretti, L.; Pallini, R.; D′ Alessandris, Q.G.; et al. Dissecting Stemness in Aggressive Intracranial Meningiomas: Prognostic Role of SOX2 Expression. Int. J. Mol. Sci. 2022, 23, 11690. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Nejad, R.; Karabork, M.; Ekinci, C.; Solaroglu, I.; Aldape, K.D.; Zadeh, G. Sox2: Regulation of Expression and Contribution to Brain Tumors. CNS Oncol. 2016, 5, 159. [Google Scholar] [CrossRef]
- Batista, K.M.P.; de Eulate-Beramendi, S.A.; de Pińa, K.Y.Á.R.; Figueira, P.R.; Canal, A.F.; Chasin, J.M.A.; Meilan, Á.; Ugalde, R.; Vega, I.F. Mesenchymal/Proangiogenic Factor YKL-40 Related to Glioblastomas and Its Relationship with the Subventricular Zone. Folia Neuropathol. 2017, 55, 14–22. [Google Scholar] [CrossRef]
- Jefri, M.; Huang, Y.-N.; Huang, W.-C.; Tai, C.-S.; Chen, W.-L. YKL-40 Regulated Epithelial-Mesenchymal Transition and Migration/Invasion Enhancement in Non-Small Cell Lung Cancer. BMC Cancer 2015, 15, 590. [Google Scholar] [CrossRef]
- Chen, W.-J.; Zhang, X.; Han, H.; Lv, J.-N.; Kang, E.-M.; Zhang, Y.-L.; Liu, W.-P.; He, X.-S.; Wang, J.; Wang, G.-H.; et al. The Different Role of YKL-40 in Glioblastoma Is a Function of MGMT Promoter Methylation Status. Cell Death Dis. 2020, 11, 668. [Google Scholar] [CrossRef]
- Emery, G.; Gruenberg, J.; Rojo, M. The P24 Family of Transmembrane Proteins at the Interface between Endoplasmic Reticulum and Golgi Apparatus. Protoplasma 1999, 207, 24–30. [Google Scholar] [CrossRef]
- Jenne, N.; Frey, K.; Brugger, B.; Wieland, F.T. Oligomeric State and Stoichiometry of P24 Proteins in the Early Secretory Pathway. J. Biol. Chem. 2002, 277, 46504–46511. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Pi, X.; Goss, A.C.; El-Baba, T.; Ehrmann, J.F.; Grinkevich, E.; Bazua-Valenti, S.; Padovano, V.; Alper, S.L.; Carey, D.; et al. Molecular Basis of TMED9 Oligomerization and Entrapment of Misfolded Protein Cargo in the Early Secretory Pathway. Sci. Adv. 2024, 10, eadp2221. [Google Scholar] [CrossRef]
- Weisbrod, L.J.; Thiraviyam, A.; Vengoji, R.; Shonka, N.; Jain, M.; Ho, W.; Batra, S.K.; Salehi, A. Diffuse Intrinsic Pontine Glioma (DIPG): A Review of Current and Emerging Treatment Strategies. Cancer Lett. 2024, 590, 216876. [Google Scholar] [CrossRef]
- Mandorino, M.; Maitra, A.; Armenise, D.; Baldelli, O.M.; Miciaccia, M.; Ferorelli, S.; Perrone, M.G.; Scilimati, A. Pediatric Diffuse Midline Glioma H3K27-Altered: From Developmental Origins to Therapeutic Challenges. Cancers 2024, 16, 1814. [Google Scholar] [CrossRef]
- Vredevoogd, D.W.; Apriamashvili, G.; Levy, P.L.; Sinha, S.; Huinen, Z.R.; Visser, N.L.; De Bruijn, B.; Boshuizen, J.; Van Hal-van Veen, S.E.; Ligtenberg, M.A.; et al. TMED Inhibition Suppresses Cell Surface PD-1 Expression and Overcomes T Cell Dysfunction. J. Immunother. Cancer 2024, 12, e010145. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Chien, M.-H.; Lai, T.-C.; Tung, M.-C.; Jan, Y.-H.; Chang, W.-M.; Jung, S.-M.; Chen, M.-H.; Yeh, C.-N.; Hsiao, M. Proteomics-Based Identification of TMED9 Is Linked to Vascular Invasion and Poor Prognoses in Patients with Hepatocellular Carcinoma. J. Biomed. Sci. 2021, 28, 29. [Google Scholar] [CrossRef]
- Goga, A.; Yagabasan, B.; Herrmanns, K.; Godbersen, S.; Silva, P.N.; Denzler, R.; Zünd, M.; Furter, M.; Schwank, G.; Sunagawa, S.; et al. miR-802 Regulates Paneth Cell Function and Enterocyte Differentiation in the Mouse Small Intestine. Nat. Commun. 2021, 12, 3339. [Google Scholar] [CrossRef] [PubMed]
- Buechling, T.; Chaudhary, V.; Spirohn, K.; Weiss, M.; Boutros, M. P24 Proteins Are Required for Secretion of Wnt Ligands. EMBO Rep. 2011, 12, 1265–1272. [Google Scholar] [CrossRef]
- Nakano, N.; Tsuchiya, Y.; Kako, K.; Umezaki, K.; Sano, K.; Ikeno, S.; Otsuka, E.; Shigeta, M.; Nakagawa, A.; Sakata, N.; et al. TMED10 Protein Interferes with Transforming Growth Factor (TGF)-β Signaling by Disrupting TGF-β Receptor Complex Formation. J. Biol. Chem. 2017, 292, 4099–4112. [Google Scholar] [CrossRef]
- Di Minin, G.; Holzner, M.; Grison, A.; Dumeau, C.E.; Chan, W.; Monfort, A.; Jerome-Majewska, L.A.; Roelink, H.; Wutz, A. TMED2 Binding Restricts SMO to the ER and Golgi Compartments. PLoS Biol. 2022, 20, e3001596. [Google Scholar] [CrossRef] [PubMed]
- Izzy, S.; Yahya, T.; Albastaki, O.; Abou-El-Hassan, H.; Aronchik, M.; Cao, T.; De Oliveira, M.G.; Lu, K.-J.; Moreira, T.G.; da Silva, P.; et al. Nasal Anti-CD3 Monoclonal Antibody Ameliorates Traumatic Brain Injury, Enhances Microglial Phagocytosis and Reduces Neuroinflammation via IL-10-Dependent Treg–Microglia Crosstalk. Nat. Neurosci. 2025, 28, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xue, Y.; Markovic, T.; Li, H.; Wang, S.; Zhong, Y.; Du, S.; Zhang, Y.; Hou, X.; Yu, Y.; et al. Blood–brain-barrier-crossing lipid nanoparticles for mRNA delivery to the central nervous system. Nat. Mater. 2025, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Valenteen, F.; Mikhael, S.; Wang, H.; Sims, J.; Taguiam, M.; Teh, J.; Sances, S.; Wong, M.; Miao, T.; Srinivas, D.; et al. Systemic HER3 Ligand-Mimicking Nanobioparticles Enter the Brain and Reduce Intracranial Tumour Growth. Nat. Nanotechnol. 2025, 20, 683–696. [Google Scholar] [CrossRef]
- Chu, L.; Sun, Y.; Zhao, Y.; Wang, A.; Sun, Y.; Duan, X.; Li, N.; Xia, H.; Liu, W.; Sun, K. Exosome-Mediated Delivery Platform of Biomacromolecules into the Brain: Cetuximab in Combination with Doxorubicin for Glioblastoma Therapy. Int. J. Pharm. 2024, 660, 124262. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daoud Sarsour, A.; Kinstlinger, S.; Nizar, R.; Amos, N.; Arbeli, N.; Kazimirsky, G.; Bronshtein-Berger, I.; Fried, I.; Unger, R.; Brodie, C.; et al. Targeting the Cargo Receptor TMED9 as a Therapeutic Strategy Against Brain Tumors. Cells 2025, 14, 772. https://doi.org/10.3390/cells14110772
Daoud Sarsour A, Kinstlinger S, Nizar R, Amos N, Arbeli N, Kazimirsky G, Bronshtein-Berger I, Fried I, Unger R, Brodie C, et al. Targeting the Cargo Receptor TMED9 as a Therapeutic Strategy Against Brain Tumors. Cells. 2025; 14(11):772. https://doi.org/10.3390/cells14110772
Chicago/Turabian StyleDaoud Sarsour, Alaa, Sara Kinstlinger, Rephael Nizar, Naama Amos, Narkis Arbeli, Gila Kazimirsky, Irena Bronshtein-Berger, Iris Fried, Ron Unger, Chaya Brodie, and et al. 2025. "Targeting the Cargo Receptor TMED9 as a Therapeutic Strategy Against Brain Tumors" Cells 14, no. 11: 772. https://doi.org/10.3390/cells14110772
APA StyleDaoud Sarsour, A., Kinstlinger, S., Nizar, R., Amos, N., Arbeli, N., Kazimirsky, G., Bronshtein-Berger, I., Fried, I., Unger, R., Brodie, C., & Dvela-Levitt, M. (2025). Targeting the Cargo Receptor TMED9 as a Therapeutic Strategy Against Brain Tumors. Cells, 14(11), 772. https://doi.org/10.3390/cells14110772