

IL-6 as a Mediator of Platelet Hyper-Responsiveness

Abstract

1. Introduction
2. Primary Mechanisms of Platelet Activation
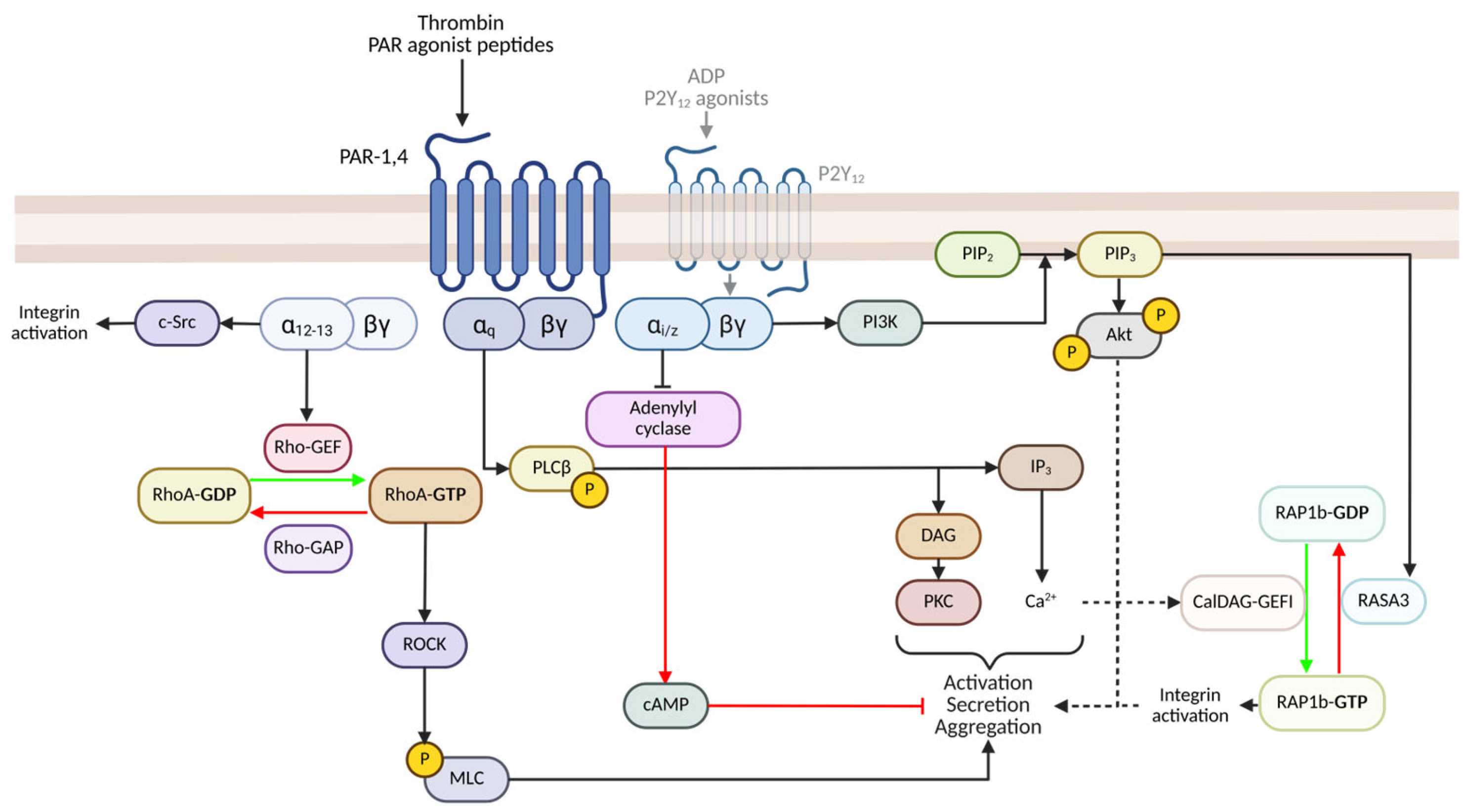
2.1. PAR-Mediated Signalling and Activation
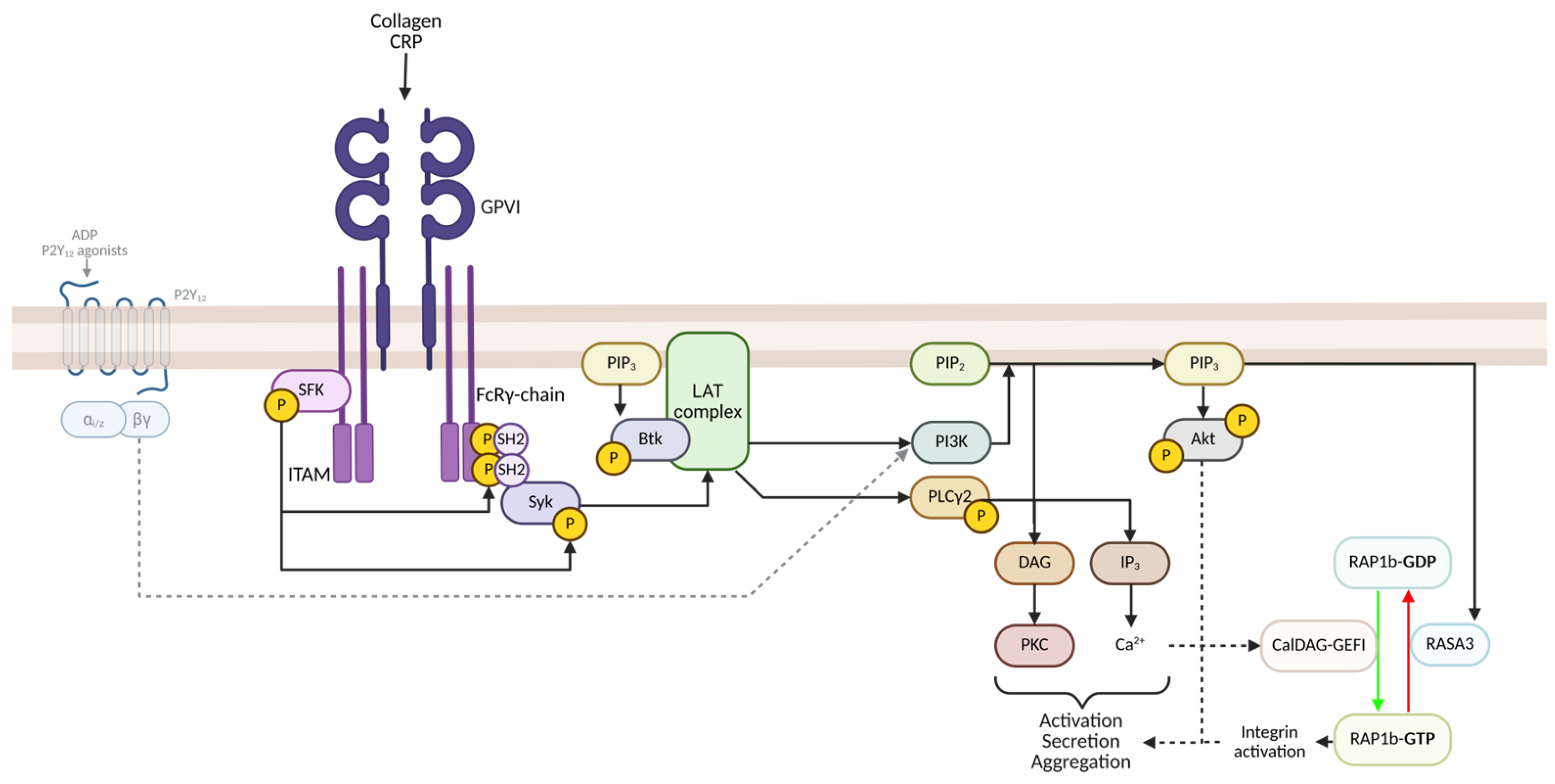
2.2. GPVI-Mediated Signalling and Activation
3. Mechanisms of Platelet Priming
3.1. Synergism Between Circulating Primers
3.2. Primers and ‘Anti-Platelet Resistance’
4. Interleukin-6 Structure and Signalling
4.1. IL-6 and IL-6-Associated Receptors Structure and Expression
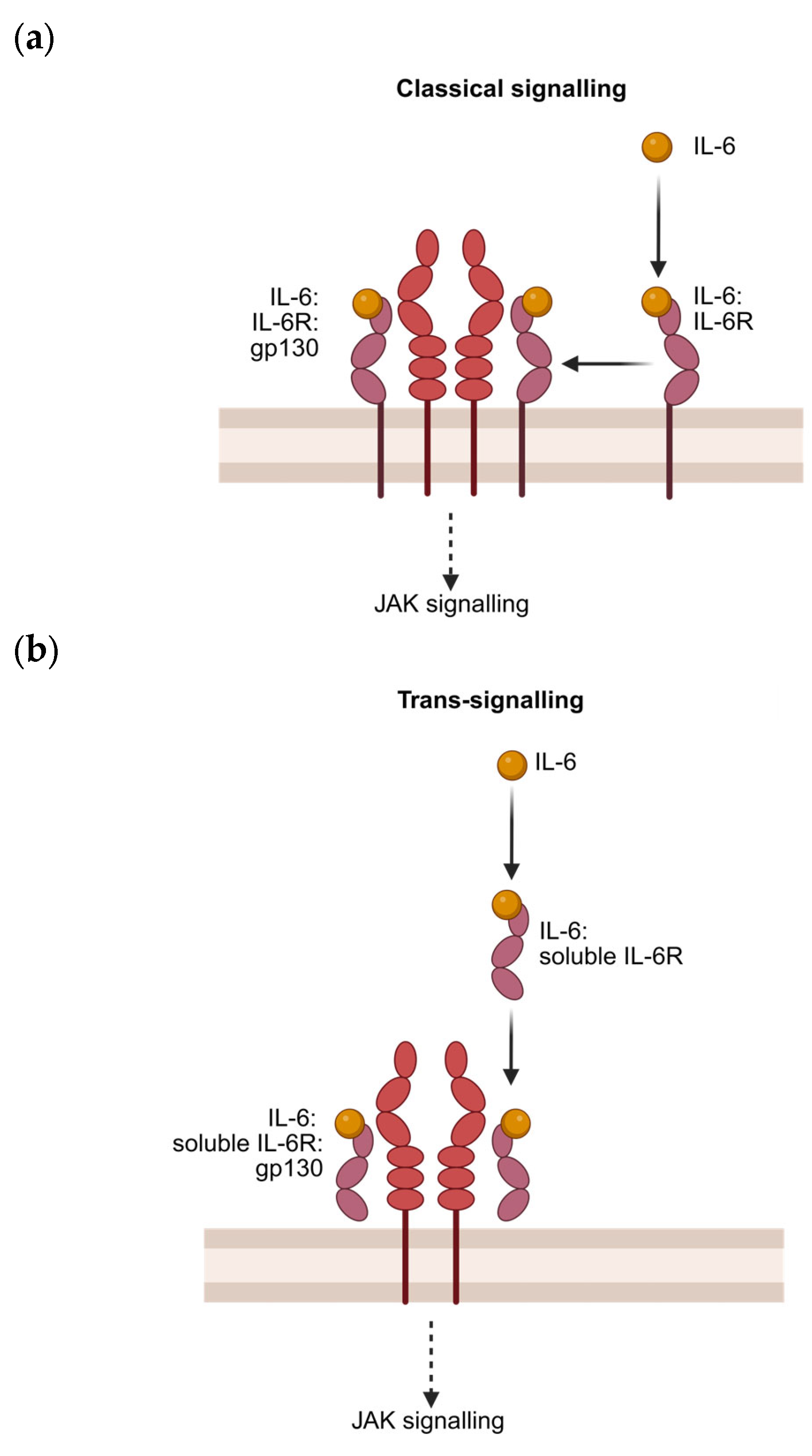
4.2. Canonical Signalling
4.3. Trans-Signalling and Presentation
4.4. Comparing the Inflammatory Effects Based on Signalling Mechanism
4.5. Modulation of IL-6 and IL-6 Signalling
5. Effects of IL-6 on Megakaryocytes and Platelets
5.1. Megakaryopoesis and Thrombopoesis
5.2. Soluble IL-6R Is Required for Platelet Priming
5.3. IL-6 Priming May Be Limited to GPVI-Mediated Signalling
5.4. IL-6 May Potentiate Multiple Intracellular Cascades Following Platelet Activation
6. Evidence of IL-6-Mediated Platelet Hyper-Responsiveness in Disease
6.1. Cardiovascular and Thrombotic Diseases
6.2. Infectious Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-HT | 5-Hydroxytryptamine (Serotonin) |
| ADP | Adenosine Diphosphate |
| Akt | Protein Kinase B |
| ALI | Acute Lung Injury |
| APACHE II | Acute Physiology and Chronic Health Evaluation II |
| APS | Antiphospholipid Syndrome |
| C-Mpl | Thrombopoietin Receptor |
| CD40L | CD40 Ligand |
| CRP | C-Reactive Protein |
| CSS | Cytokine Storm Syndrome |
| CXCL | C-X-C Motif Chemokine Ligand |
| cPLA2 | Cytosolic Phospholipase A2 |
| DAMPs | Damage-Associated Molecular Patterns |
| DIC | Disseminated Intravascular Coagulation |
| EP3-4 | Prostaglandin E Receptors 3 and 4 |
| Fcɣ | Fc-Receptor Gamma Chain |
| Gab1 | GRB2-Associated Binding Protein 1 |
| Gas6 | Growth Arrest-Specific 6 |
| GPVI | Glycoprotein VI |
| Grb2 | Growth Factor Receptor-Bound Protein 2 |
| GSK-3 | Glycogen Synthase Kinase 3 |
| HSC | Hematopoietic Stem Cell |
| IGF-1 | Insulin-Like Growth Factor-1 |
| IL-1β | Interleukin-1 Beta |
| IL-6 | Interleukin-6 |
| IP3 | Inositol Triphosphate |
| ITP | Immune Thrombocytopenia |
| ITAM | Immunoreceptor Tyrosine-Based Activation Motif |
| JAK | Janus Kinase |
| LDBM | Low-Density Bone Marrow |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-Activated Protein Kinase |
| MEK | Mitogen-Activated Protein Kinase |
| MODS | Multiple Organ Dysfunction Syndrome |
| MPO | Myeloperoxidase |
| MMP2 | Matrix Metalloproteinase 2 |
| NF-κB | Nuclear Factor Kappa B |
| NO | Nitric Oxide |
| Ox-LDL | Oxidized Low-Density Lipoprotein |
| PAR | Protease-Activated Receptor |
| PECAM | Platelet Endothelial Cell Adhesion Molecule |
| PDK1 | 3-Phosphoinositide-Dependent Protein Kinase 1 |
| PF4 | Platelet Factor 4 |
| PI3K | Phosphoinositide 3-Kinase |
| PIP2 | Phosphatidylinositol 4,5-Bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)-Triphosphate |
| PKC | Protein Kinase C |
| PLC | Phospholipase C |
| PLA | Platelet–Leukocyte Aggregate |
| PGE2 | Prostaglandin E2 |
| PRP | Platelet-Rich Plasma |
| RANTES | Regulated on Activation, Normal T Cell Expressed and Secreted |
| RAP1b | Ras-Related Protein 1b |
| RASA3 | RAS P21 Protein Activator 3 |
| ROCK | Rho-Associated Protein Kinase |
| ROS | Reactive Oxygen Species |
| S1P | Sphingosine-1-Phosphate |
| S1PR1, 4-5 | Sphingosine-1-Phosphate Receptors 1, 4, and 5 |
| SCF | Stem Cell Factor |
| gp130 | Soluble Glycoprotein 130 |
| SHP-2 | Src Homology 2 Domain-Containing Phosphatase-2 |
| SLE | Systemic Lupus Erythematosus |
| SOFA | Sequential Organ Failure Assessment |
| SOS1 | Son of Sevenless Homolog 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| STEMI | ST-Elevation Myocardial Infarction |
| SUCNR1 | Succinate Receptor 1 |
| Syk | Spleen Tyrosine Kinase |
| TCZ | Tocilizumab |
| TLR | Toll-Like Receptor |
| TNF-α | Tumour Necrosis Factor-Alpha |
| TPO | Thrombopoietin |
| TP | Thromboxane Receptor |
| TxA2 | Thromboxane A2 |
| TxB2 | Thromboxane B2 |
| Tyro3 | Tyrosine-Protein Kinase Receptor 3 |
| UFO | Axl Receptor Tyrosine Kinase |
| VWF | Von Willebrand Factor |
References
- MacKay, W. The Blood-Platelet: Its Clinical Significance. QJM Int. J. Med. 1931, os-24, 285–328. [Google Scholar] [CrossRef]
- Gaarder, A.; Jonsen, J.; Laland, S.; Hellem, A.; Owren, P.A. Adenosine diphosphate in red cells as a factor in the adhesiveness of human blood platelets. Nature 1961, 192, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Haslam, R.J. Role of adenosine diphosphate in the aggregation of human blood-platelets by thrombin and by fatty acids. Nature 1964, 202, 765–768. [Google Scholar] [CrossRef] [PubMed]
- de Gaetano, G. Historical overview of the role of platelets in hemostasis and thrombosis. Haematologica 2001, 86, 349–356. [Google Scholar]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; Arepally, G.M.; French, P.A.; Dauerman, H.L.; Becker, R.C. Platelet functions beyond hemostasis. J. Thromb. Haemost. 2009, 7, 1759–1766. [Google Scholar] [CrossRef]
- Tyagi, T.; Jain, K.; Gu, S.X.; Qiu, M.; Gu, V.W.; Melchinger, H.; Rinder, H.; Martin, K.A.; Gardiner, E.E.; Lee, A.I.; et al. A guide to molecular and functional investigations of platelets to bridge basic and clinical sciences. Nat. Cardiovasc. Res. 2022, 1, 223–237. [Google Scholar] [CrossRef]
- Bo, Y.; Lu, Q.; Li, B.; Sha, R.; Yu, H.; Miao, C. The role of platelets in central hubs of inflammation: A literature review. Medicine 2024, 103, e38115. [Google Scholar] [CrossRef]
- Marcos-Jubilar, M.; Lecumberri, R.; Páramo, J.A. Immunothrombosis: Molecular Aspects and New Therapeutic Perspectives. J. Clin. Med. 2023, 12, 1399. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Sinha, S.; Mohapatra, S.K. Analysis of transcriptomic data sets supports the role of IL-6 in NETosis and immunothrombosis in severe COVID-19. BMC Genom. Data 2021, 22, 49. [Google Scholar] [CrossRef]
- Zhu, J.; Bouzid, R.; Travert, B.; Géri, G.; Cohen, Y.; Picod, A.; Heming, N.; Rottman, M.; Joly-Laffargue, B.; Veyradier, A.; et al. Combined coagulation and inflammation markers as predictors of venous thrombo-embolism and death in COVID-19. Front. Med. 2024, 11, 1399335. [Google Scholar] [CrossRef] [PubMed]
- Ghamri, R.A.; Alghalayini, K.W.; Baig, M. Correlation of cardiovascular risk parameters with serum IL.6 and C-RP in myocardial infarction. Niger. J. Clin. Pract. 2022, 25, 299–303. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Komoto, S.; Russell, J.; Almeida-Paula, L.D.; Yan, L.S.; Zhang, S.; Granger, D.N. Interleukin-6 mediates the platelet abnormalities and thrombogenesis associated with experimental colitis. Am. J. Pathol. 2013, 183, 173–181. [Google Scholar] [CrossRef]
- Mutlu, G.M.; Green, D.; Bellmeyer, A.; Baker, C.M.; Burgess, Z.; Rajamannan, N.; Christman, J.W.; Foiles, N.; Kamp, D.W.; Ghio, A.J.; et al. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J. Clin. Investig. 2007, 117, 2952–2961. [Google Scholar] [CrossRef]
- Peng, J.; Friese, P.; Wolf, R.F.; Harrison, P.; Downs, T.; Lok, S.; Dale, G.L.; Burstein, S.A. Relative reactivity of platelets from thrombopoietin- and interleukin-6-treated dogs. Blood 1996, 87, 4158–4163. [Google Scholar] [CrossRef]
- Oda, A.; Miyakawa, Y.; Druker, B.J.; Ozaki, K.; Yabusaki, K.; Shirasawa, Y.; Handa, M.; Kato, T.; Miyazaki, H.; Shimosaka, A.; et al. Thrombopoietin primes human platelet aggregation induced by shear stress and by multiple agonists. Blood 1996, 87, 4664–4670. [Google Scholar] [CrossRef]
- Oleksowicz, L.; Mrowiec, Z.; Zuckerman, D.; Isaacs, R.; Dutcher, J.; Puszkin, E. Platelet activation induced by interleukin-6: Evidence for a mechanism involving arachidonic acid metabolism. Thromb. Haemost. 1994, 72, 302–308. [Google Scholar]
- Zhou, Z.; Gushiken, F.C.; Bolgiano, D.; Salsbery, B.J.; Aghakasiri, N.; Jing, N.; Wu, X.; Vijayan, K.V.; Rumbaut, R.E.; Adachi, R.; et al. Signal transducer and activator of transcription 3 (STAT3) regulates collagen-induced platelet aggregation independently of its transcription factor activity. Circulation 2013, 127, 476–485. [Google Scholar] [CrossRef]
- Aljuhani, O.; Al Sulaiman, K.; Korayem, G.B.; Altebainawi, A.F.; Alsohimi, S.; Alqahtani, R.; Alfaifi, S.; Alharbi, A.; AlKhayrat, A.; Hattan, A.; et al. The association between tocilizumab therapy and the development of thrombosis in critically ill patients with COVID-19: A multicenter, cohort study. Sci. Rep. 2024, 14, 3037. [Google Scholar] [CrossRef]
- Chu, X.; Ma, Z.; Liu, Y.; Sun, J.; Wang, N.; Li, C.; Feng, X.; Li, J.; Wang, B.; Zhou, C.; et al. IRIS, a randomised, double-blind, placebo-controlled trial of interleukin-6 receptor inhibition undergoing endovascular treatment in acute anterior circulation ischaemic stroke: Study rationale and design. Stroke Vasc. Neurol. 2024, 0, 1–6. [Google Scholar] [CrossRef]
- Estevez, B.; Du, X. New concepts and mechanisms of platelet activation signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling during platelet adhesion and activation. Arter. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.S. Structure and function of the platelet integrin alphaIIbbeta3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef]
- Sharda, A.; Flaumenhaft, R. The life cycle of platelet granules. F1000Research 2018, 7, 236. [Google Scholar] [CrossRef]
- Golebiewska, E.M.; Poole, A.W. Secrets of platelet exocytosis—what do we really know about platelet secretion mechanisms? Br. J. Haematol. 2013, 165, 204–216. [Google Scholar] [CrossRef]
- White, G.C., II; Rompietti, R. Platelet secretion: Indiscriminately spewed forth or highly orchestrated? J. Thromb. Haemost. 2007, 5, 2006–2008. [Google Scholar] [CrossRef]
- De Candia, E. Mechanisms of platelet activation by thrombin: A short history. Thromb. Res. 2012, 129, 250–256. [Google Scholar] [CrossRef]
- Yuan, J.; Slice, L.W.; Gu, J.; Rozengurt, E. Cooperation of Gq, Gi, and G12/13 in Protein Kinase D activation and phosphorylation induced by lysophosphatidic acid. J. Biol. Chem. 2003, 278, 4882–4891. [Google Scholar] [CrossRef]
- Zhang, L.; Brass, L.F.; Manning, D.R. The Gq and G12 families of heterotrimeric G proteins report functional selectivity. Mol. Pharmacol. 2009, 75, 235–241. [Google Scholar] [CrossRef]
- Jin, J.; Mao, Y.; Thomas, D.; Kim, S.; Daniel, J.L.; Kunapuli, S.P. RhoA downstream of G(q) and G(12/13) pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem. Pharmacol. 2009, 77, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Aslan, J.E.; McCarty, O.J. Rho GTPases in platelet function. J. Thromb. Haemost. 2013, 11, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Chikumi, H.; Vázquez-Prado, J.; Servitja, J.M.; Miyazaki, H.; Gutkind, J.S. Potent activation of RhoA by Galpha q and Gq-coupled receptors. J. Biol. Chem. 2002, 277, 27130–27134. [Google Scholar] [CrossRef]
- Offermanns, S. Activation of platelet function through G Protein–Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef]
- Shen, C.; Liu, M.; Xu, R.; Wang, G.; Li, J.; Chen, P.; Ma, W.; Mwangi, J.; Lu, Q.; Duan, Z.; et al. The 14-3-3ζ-c-Src-integrin-β3 complex is vital for platelet activation. Blood 2020, 136, 974–988. [Google Scholar] [CrossRef]
- Gong, H.; Shen, B.; Flevaris, P.; Chow, C.; Lam, S.C.T.; Voyno-Yasenetskaya, T.A.; Kozasa, T.; Du, X. G Protein Subunit Gα13 binds to integrin αIIbβ3 and mediates integrin “outside-in” signaling. Science 2010, 327, 340–343. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Kuliopulos, A.; Tantry, U.S. G-protein-coupled receptors signaling pathways in new antiplatelet drug development. Arter. Thromb. Vasc. Biol. 2015, 35, 500–512. [Google Scholar] [CrossRef]
- Durrant, T.N.; Hutchinson, J.L.; Heesom, K.J.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Marshall, A.J.; Moore, S.F.; Hers, I. In-depth PtdIns(3,4,5)P(3) signalosome analysis identifies DAPP1 as a negative regulator of GPVI-driven platelet function. Blood Adv. 2017, 1, 918–932. [Google Scholar] [CrossRef]
- Battram, A.M.; Durrant, T.N.; Agbani, E.O.; Heesom, K.J.; Paul, D.S.; Piatt, R.; Poole, A.W.; Cullen, P.J.; Bergmeier, W.; Moore, S.F.; et al. The phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) binder Rasa3 regulates phosphoinositide 3-kinase (PI3K)-dependent integrin αIIbβ3 outside-in signaling. J. Biol. Chem. 2017, 292, 1691–1704. [Google Scholar] [CrossRef]
- Stefanini, L.; Paul, D.S.; Robledo, R.F.; Chan, E.R.; Getz, T.M.; Campbell, R.A.; Kechele, D.O.; Casari, C.; Piatt, R.; Caron, K.M.; et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Investig. 2015, 125, 1419–1432. [Google Scholar] [CrossRef]
- Moroi, M.; Jung, S.M. Platelet glycoprotein VI: Its structure and function. Thromb. Res. 2004, 114, 221–233. [Google Scholar] [CrossRef]
- Chen, H.; Locke, D.; Liu, Y.; Liu, C.; Kahn, M.L. The platelet receptor GPVI mediates both adhesion and signaling responses to collagen in a receptor density-dependent fashion. J. Biol. Chem. 2002, 277, 3011–3019. [Google Scholar] [CrossRef] [PubMed]
- Trory, J.S.; Munkacsi, A.; Śledź, K.M.; Vautrinot, J.; Goudswaard, L.J.; Jackson, M.L.; Heesom, K.J.; Moore, S.F.; Poole, A.W.; Nabet, B.; et al. Chemical degradation of BTK/TEC as a novel approach to inhibit platelet function. Blood Adv. 2023, 7, 1692–1696. [Google Scholar] [CrossRef]
- Brysland, S.A.; Maqbool, M.G.; Talaulikar, D.; Gardiner, E.E. Bleeding propensity in Waldenström macroglobulinemia: Potential causes and evaluation. Thromb. Haemost. 2022, 122, 1843–1857. [Google Scholar] [CrossRef]
- Elvers, M.; Pozgaj, R.; Pleines, I.; May, F.; Kuijpers, M.J.; Heemskerk, J.M.; Yu, P.; Nieswandt, B. Platelet hyperreactivity and a prothrombotic phenotype in mice with a gain-of-function mutation in phospholipase Cgamma2. J. Thromb. Haemost. 2010, 8, 1353–1363. [Google Scholar] [CrossRef]
- Quinton, T.M.; Ozdener, F.; Dangelmaier, C.; Daniel, J.L.; Kunapuli, S.P. Glycoprotein VI–mediated platelet fibrinogen receptor activation occurs through calcium-sensitive and PKC-sensitive pathways without a requirement for secreted ADP. Blood 2002, 99, 3228–3234. [Google Scholar] [CrossRef]
- Ohlmann, P.; Eckly, A.; Freund, M.; Cazenave, J.-P.; Offermanns, S.; Gachet, C. ADP induces partial platelet aggregation without shape change and potentiates collagen-induced aggregation in the absence of Gαq. Blood 2000, 96, 2134–2139. [Google Scholar] [CrossRef]
- Martin, A.-C.; Zlotnik, D.; Bonete, G.P.; Baron, E.; Decouture, B.; Belleville-Rolland, T.; Le Bonniec, B.; Poirault-Chassac, S.; Alessi, M.-C.; Gaussem, P.; et al. Epinephrine restores platelet functions inhibited by ticagrelor: A mechanistic approach. Eur. J. Pharmacol. 2020, 866, 172798. [Google Scholar] [CrossRef]
- Smolenski, A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 2012, 10, 167–176. [Google Scholar] [CrossRef]
- Vanags, D.M.; Rodgers, S.E.; Duncan, E.M.; Lloyd, J.V.; Bochner, F. Potentiation of ADP-induced aggregation in human platelet-rich plasma by 5-hydroxytryptamine and adrenaline. Br. J. Pharmacol. 1992, 106, 917–923. [Google Scholar] [CrossRef]
- Singh, S.; Malm, C.J.; Ramström, S.; Hesse, C.; Jeppsson, A. Adrenaline enhances in vitro platelet activation and aggregation in blood samples from ticagrelor-treated patients. Res. Pract. Thromb. Haemost. 2018, 2, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; de Frutos, P.; Aparicio, C.; Melis, E.; Savi, P.; Lupu, F.; Arnout, J.; Dewerchin, M.; Hoylaerts, M.; Herbert, J.; et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat. Med. 2001, 7, 215–221. [Google Scholar] [CrossRef]
- Hers, I. Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kalpha pathway. Blood 2007, 110, 4243–4252. [Google Scholar] [CrossRef]
- Blair, T.A.; Moore, S.F.; Williams, C.M.; Poole, A.W.; Vanhaesebroeck, B.; Hers, I. Phosphoinositide 3-kinases p110α and p110β have differential roles in insulin-like growth factor-1-mediated Akt phosphorylation and platelet priming. Arter. Thromb. Vasc. Biol. 2014, 34, 1681–1688. [Google Scholar] [CrossRef]
- Falcinelli, E.; Guglielmini, G.; Torti, M.; Gresele, P. Intraplatelet signaling mechanisms of the priming effect of matrix metalloproteinase-2 on platelet aggregation. J. Thromb. Haemost. 2005, 3, 2526–2535. [Google Scholar] [CrossRef]
- Sawicki, G.; Salas, E.; Murat, J.; Miszta-Lane, H.; Radomski, M.W. Release of gelatinase A during platelet activation mediates aggregation. Nature 1997, 386, 616–619. [Google Scholar] [CrossRef]
- Sebastiano, M.; Momi, S.; Falcinelli, E.; Bury, L.; Hoylaerts, M.F.; Gresele, P. A novel mechanism regulating human platelet activation by MMP-2-mediated PAR1 biased signaling. Blood 2017, 129, 883–895. [Google Scholar] [CrossRef]
- Valiyaveettil, M.; Kar, N.; Ashraf, M.Z.; Byzova, T.V.; Febbraio, M.; Podrez, E.A. Oxidized high-density lipoprotein inhibits platelet activation and aggregation via scavenger receptor BI. Blood 2008, 111, 1962–1971. [Google Scholar] [CrossRef]
- Maaninka, K.; Neuvonen, M.; Kerkelä, E.; Hyvärinen, K.; Palviainen, M.; Kamali-Moghaddam, M.; Federico, A.; Greco, D.; Laitinen, S.; Öörni, K.; et al. OxLDL sensitizes platelets for increased formation of extracellular vesicles capable of finetuning macrophage gene expression. Eur. J. Cell Biol. 2023, 102, 151311. [Google Scholar] [CrossRef]
- Korporaal, S.J.; Van Eck, M.; Adelmeijer, J.; Ijsseldijk, M.; Out, R.; Lisman, T.; Lenting, P.J.; Van Berkel, T.J.; Akkerman, J.W. Platelet activation by oxidized low density lipoprotein is mediated by CD36 and scavenger receptor-A. Arter. Thromb. Vasc. Biol. 2007, 27, 2476–2483. [Google Scholar] [CrossRef] [PubMed]
- Wraith, K.S.; Magwenzi, S.; Aburima, A.; Wen, Y.; Leake, D.; Naseem, K.M. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase-signaling pathways. Blood 2013, 122, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.J.; Kohs, T.C.L.; Mueller, P.A.; Pang, J.; Reitsma, S.E.; Parra-Izquierdo, I.; Melrose, A.R.; Yang, L.; Choi, J.; Zientek, K.D.; et al. Effect of antiplatelet agents and tyrosine kinase inhibitors on oxLDL-mediated procoagulant platelet activity. Blood Adv. 2023, 7, 1366–1378. [Google Scholar] [CrossRef]
- Capitanio, A.M.; Niewiarowski, S.; Rucinski, B.; Tuszynski, G.P.; Cierniewski, C.S.; Hershock, D.; Kornecki, E. Interaction of platelet factor 4 with human platelets. Biochim. Biophys. Acta 1985, 839, 161–173. [Google Scholar] [CrossRef]
- Dickhout, A.; Tullemans, B.M.E.; Heemskerk, J.W.M.; Thijssen, V.; Kuijpers, M.J.E.; Koenen, R.R. Galectin-1 and platelet factor 4 (CXCL4) induce complementary platelet responses in vitro. PLoS ONE 2021, 16, e0244736. [Google Scholar] [CrossRef]
- Buka, R.J.; Montague, S.J.; Moran, L.A.; Martin, E.M.; Slater, A.; Watson, S.P.; Nicolson, P.L.R. PF4 activates the c-Mpl–Jak2 pathway in platelets. Blood 2024, 143, 64–69. [Google Scholar] [CrossRef]
- Vezza, R.; Roberti, R.; Nenci, G.G.; Gresele, P. Prostaglandin E2 potentiates platelet aggregation by priming protein kinase C. Blood 1993, 82, 2704–2713. [Google Scholar] [CrossRef]
- Schober, L.J.; Khandoga, A.L.; Dwivedi, S.; Penz, S.M.; Maruyama, T.; Brandl, R.; Siess, W. The role of PGE(2) in human atherosclerotic plaque on platelet EP(3) and EP(4) receptor activation and platelet function in whole blood. J. Thromb. Thrombolysis 2011, 32, 158–166. [Google Scholar] [CrossRef]
- Smith, J.P.; Haddad, E.V.; Downey, J.D.; Breyer, R.M.; Boutaud, O. PGE2 decreases reactivity of human platelets by activating EP2 and EP4. Thromb. Res. 2010, 126, e23–e29. [Google Scholar] [CrossRef]
- Braune, S.; Küpper, J.-H.; Jung, F. Effect of prostanoids on human platelet function: An overview. Int. J. Mol. Sci. 2020, 21, 9020. [Google Scholar] [CrossRef]
- Kojok, K.; Akoum, S.E.; Mohsen, M.; Mourad, W.; Merhi, Y. CD40L Priming of Platelets via NF-κB Activation is CD40- and TAK1-Dependent. J. Am. Heart Assoc. 2018, 7, e03677. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, D.; Hachem, A.; Théorêt, J.-F.; Gillis, M.-A.; Mourad, W.; Merhi, Y. Enhanced levels of soluble CD40 ligand exacerbate platelet aggregation and thrombus formation through a CD40-dependent tumor necrosis factor receptor & associated factor-2/Rac1/p38 Mitogen-Activated Protein Kinase signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2424–2433. [Google Scholar] [CrossRef]
- Kojok, K.; Mohsen, M.; El Kadiry, A.E.H.; Mourad, W.; Merhi, Y. Aspirin reduces the potentiating effect of CD40L on platelet aggregation via inhibition of Myosin Light Chain. J. Am. Heart Assoc. 2020, 9, e013396. [Google Scholar] [CrossRef]
- Yatomi, Y.; Yamamura, S.; Ruan, F.; Igarashi, Y. Sphingosine 1-phosphate induces platelet activation through an extracellular action and shares a platelet surface receptor with lysophosphatidic acid. J. Biol. Chem. 1997, 272, 5291–5297. [Google Scholar] [CrossRef]
- Nugent, D.; Xu, Y. Sphingosine-1-phosphate: Characterization of its inhibition of platelet aggregation. Platelets 2000, 11, 226–232. [Google Scholar] [CrossRef]
- Liu, H.; Jackson, M.L.; Goudswaard, L.J.; Moore, S.F.; Hutchinson, J.L.; Hers, I. Sphingosine-1-phosphate modulates PAR1-mediated human platelet activation in a concentration-dependent biphasic manner. Sci. Rep. 2021, 11, 15308. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Tijssen, M.R.; Thijssen-Timmer, D.C.; Gusnanto, A.; Steward, M.; Burns, P.; Langford, C.F.; Ellis, P.D.; Dudbridge, F.; Zwaginga, J.J.; et al. Comparative gene expression profiling of in vitro differentiated megakaryocytes and erythroblasts identifies novel activatory and inhibitory platelet membrane proteins. Blood 2007, 109, 3260–3269. [Google Scholar] [CrossRef]
- Veninga, A.; Baaten, C.; De Simone, I.; Tullemans, B.M.E.; Kuijpers, M.J.E.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Effects of Platelet Agonists and Priming on the Formation of Platelet Populations. Thromb. Haemost. 2022, 122, 726–738. [Google Scholar] [CrossRef]
- Moore, S.F.; Smith, N.R.; Blair, T.A.; Durrant, T.N.; Hers, I. Critical roles for the phosphatidylinositide 3-kinase isoforms p110β and p110γ in thrombopoietin-mediated priming of platelet function. Sci. Rep. 2019, 9, 1468. [Google Scholar] [CrossRef]
- Roell, K.R.; Reif, D.M.; Motsinger-Reif, A.A. An introduction to terminology and methodology of chemical synergy-perspectives from across disciplines. Front. Pharmacol. 2017, 8, 158. [Google Scholar] [CrossRef]
- Spalding, A.; Vaitkevicius, H.; Dill, S.; MacKenzie, S.; Schmaier, A.; Lockette, W. Mechanism of epinephrine-induced platelet aggregation. Hypertension 1998, 31, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.L.; Bergeron, A.L.; Sun, C.W.; Dong, J.F.; Bray, P.F. Platelet hyperreactivity generalizes to multiple forms of stimulation. J. Thromb. Haemost. 2006, 4, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Blair, T.A.; Moore, S.F.; Hers, I. Circulating primers enhance platelet function and induce resistance to antiplatelet therapy. J. Thromb. Haemost. 2015, 13, 1479–1493. [Google Scholar] [CrossRef]
- Gerotziafas, G.T.; Elalamy, I.; Lecrubier, C.; Lebrazi, J.; Mirshahi, M.; Potevin, F.; Lecompte, T.; Samama, M.M. The role of platelet factor 4 in platelet aggregation induced by the antibodies implicated in heparin-induced thrombocytopenia. Blood Coagul. Fibrinolysis 2001, 12, 511–520. [Google Scholar] [CrossRef]
- Cai, Z.; Greene, M.I.; Zhu, Z.; Zhang, H. Structural Features and PF4 Functions that Occur in Heparin-Induced Thrombocytopenia (HIT) Complicated by COVID-19. Antibodies 2020, 9, 52. [Google Scholar] [CrossRef]
- Tang, X.; Fuchs, D.; Tan, S.; Trauelsen, M.; Schwartz, T.W.; Wheelock, C.E.; Li, N.; Haeggström, J.Z. Activation of metabolite receptor GPR91 promotes platelet aggregation and transcellular biosynthesis of leukotriene C(4). J. Thromb. Haemost. 2020, 18, 976–984. [Google Scholar] [CrossRef]
- Spath, B.; Hansen, A.; Bokemeyer, C.; Langer, F. Succinate reverses in-vitro platelet inhibition by acetylsalicylic acid and P2Y receptor antagonists. Platelets 2012, 23, 60–68. [Google Scholar] [CrossRef]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef]
- Balogh, I.; Hafizi, S.; Stenhoff, J.; Hansson, K.; Dahlbäck, B. Analysis of Gas6 in human platelets and plasma. Arter. Thromb. Vasc. Biol. 2005, 25, 1280–1286. [Google Scholar] [CrossRef]
- Gresele, P.; Falcinelli, E.; Momi, S. Potentiation and priming of platelet activation: A potential target for antiplatelet therapy. Trends Pharmacol. Sci. 2008, 29, 352–360. [Google Scholar] [CrossRef]
- Marturano, A.; Hendrickx, M.L.V.; Falcinelli, E.; Sebastiano, M.; Guglielmini, G.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S.; Declerck, P.J.; Gresele, P. Development of anti-matrix metalloproteinase-2 (MMP-2) nanobodies as potential therapeutic and diagnostic tools. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102103. [Google Scholar] [CrossRef] [PubMed]
- Bocancia-Mateescu, L.A.; Stan, D.; Mirica, A.C.; Ghita, M.G.; Stan, D.; Ruta, L.L. Nanobodies as diagnostic and therapeutic tools for cardiovascular diseases (CVDs). Pharmaceuticals 2023, 16, 863. [Google Scholar] [CrossRef]
- Koride, S.; Nayak, S.; Banfield, C.; Peterson, M.C. Evaluating the role of Janus Kinase pathways in platelet homeostasis using a systems modeling approach. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 478–488. [Google Scholar] [CrossRef]
- Rossi, J.F.; Lu, Z.Y.; Jourdan, M.; Klein, B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 2015, 21, 1248–1257. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Rose-John, S. The soluble interleukin 6 receptor: Advanced therapeutic options in inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef]
- Smolen, J.S.; Schoels, M.M.; Nishimoto, N.; Breedveld, F.C.; Burmester, G.R.; Dougados, M.; Emery, P.; Ferraccioli, G.; Gabay, C.; Gibofsky, A.; et al. Consensus statement on blocking the effects of interleukin-6 and in particular by interleukin-6 receptor inhibition in rheumatoid arthritis and other inflammatory conditions. Ann. Rheum. Dis. 2013, 72, 482–492. [Google Scholar] [CrossRef]
- Hartman, J.; Frishman, W.H. Inflammation and atherosclerosis: A review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol. Rev. 2014, 22, 147–151. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef]
- Sehgal, P.B.; Grieninger, G.; Tosato, G. Regulation of the acute phase and immune responses: Interleukin-6. Ann. N. Y. Acad. Sci. 1989, 557, 583. [Google Scholar]
- Yamasaki, K.; Taga, T.; Hirata, Y.; Yawata, H.; Kawanishi, Y.; Seed, B.; Taniguchi, T.; Hirano, T.; Kishimoto, T. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science 1988, 241, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Bazan, J.F. Haemopoietic receptors and helical cytokines. Immunol. Today 1990, 11, 350–354. [Google Scholar] [CrossRef]
- Metcalfe, R.D.; Putoczki, T.L.; Griffin, M.D.W. Structural understanding of interleukin 6 family cytokine signaling and targeted therapies: Focus on interleukin 11. Front. Immunol. 2020, 11, 1424. [Google Scholar] [CrossRef]
- Keystone, E.; Omair, M.A. Interleukin-6 inhibition. In Rheumatology, 6th ed.; Hochberg, M.C., Silman, A.J., Smolen, J.S., Weinblatt, M.E., Weisman, M.H., Eds.; Mosby: Philadelphia, PA, USA, 2015; pp. 485–491. [Google Scholar]
- Marta, R.F.; Goette, N.P.; Lev, P.R.; Chazarreta, C.D.; Pirola, C.J.; Molinas, F.C. Normal platelets possess the soluble form of IL-6 receptor. Cytokine 2005, 29, 13–17. [Google Scholar] [CrossRef]
- Tajima, S.; Tsuji, K.; Ebihara, Y.; Sui, X.; Tanaka, R.; Muraoka, K.; Yoshida, M.; Yamada, K.; Yasukawa, K.; Taga, T.; et al. Analysis of interleukin 6 receptor and gp130 expressions and proliferative capability of human CD34+ cells. J. Exp. Med. 1996, 184, 1357–1364. [Google Scholar] [CrossRef]
- Weich, N.S.; Wang, A.; Fitzgerald, M.; Neben, T.Y.; Donaldson, D.; Giannotti, J.; Yetz-Aldape, J.; Leven, R.M.; Turner, K.J. Recombinant human interleukin-11 directly promotes megakaryocytopoiesis in vitro. Blood 1997, 90, 3893–3902. [Google Scholar] [CrossRef]
- Schumacher, N.; Meyer, D.; Mauermann, A.; von der Heyde, J.; Wolf, J.; Schwarz, J.; Knittler, K.; Murphy, G.; Michalek, M.; Garbers, C.; et al. Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by A Disintegrin and Metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. J. Biol. Chem. 2015, 290, 26059–26071. [Google Scholar] [CrossRef]
- Jones, S.A.; Horiuchi, S.; Topley, N.; Yamamoto, N.; Fuller, G.M. The soluble interleukin 6 receptor: Mechanisms of production and implications in disease. FASEB J. 2001, 15, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, M.J.; Chow, D.-c.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 α-Receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef]
- Mackiewicz, A.; Schooltink, H.; Heinrich, P.C.; Rose-John, S. Complex of soluble human IL-6-receptor/IL-6 up-regulates expression of acute-phase proteins. J. Immunol. 1992, 149, 2021–2027. [Google Scholar] [CrossRef]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: Generation and biological function. Biochem. J. 1994, 300 Pt 2, 281–290. [Google Scholar] [CrossRef]
- Stoyan, T.; Michaelis, U.; Schooltink, H.; van Dam, M.; Rudolph, R.; Heinrich, P.C.; Rose-John, S. Recombinant soluble human interleukin-6 receptor. Eur. J. Biochem. 1993, 216, 239–245. [Google Scholar] [CrossRef]
- Schumacher, N.; Yan, K.; Gandraß, M.; Müller, M.; Krisp, C.; Häsler, R.; Carambia, A.; Nofer, J.-R.; Bernardes, J.P.; Khouja, M.; et al. Cell-autonomous hepatocyte-specific GP130 signaling is sufficient to trigger a robust innate immune response in mice. J. Hepatol. 2021, 74, 407–418. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Möller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T(H)17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef]
- Xu, S.; Deng, K.-Q.; Lu, C.; Fu, X.; Zhu, Q.; Wan, S.; Zhang, L.; Huang, Y.; Nie, L.; Cai, H.; et al. Interleukin-6 classic and trans-signaling utilize glucose metabolism reprogramming to achieve anti- or pro-inflammatory effects. Metabolism 2024, 155, 155832. [Google Scholar] [CrossRef]
- Barnes, T.C.; Anderson, M.E.; Moots, R.J. The many faces of interleukin-6: The role of IL-6 in inflammation, vasculopathy, and fibrosis in systemic sclerosis. Int. J. Rheumatol. 2011, 2011, 721608. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef]
- Goldstein, I.; Paakinaho, V.; Baek, S.; Sung, M.-H.; Hager, G.L. Synergistic gene expression during the acute phase response is characterized by transcription factor assisted loading. Nat. Commun. 2017, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Schur, P.H. Laboratory evaluation of patients with systemic lupus erythematosus. In Systemic Lupus Erythematosus, 4th ed.; Lahita, R.G., Ed.; Academic Press: San Diego, CA, USA, 2004; pp. 633–657. [Google Scholar]
- Cahill, C.M.; Rogers, J.T. Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1. J. Biol. Chem. 2008, 283, 25900–25912. [Google Scholar] [CrossRef] [PubMed]
- Castell, J.V.; Gómez-Lechón, M.J.; David, M.; Andus, T.; Geiger, T.; Trullenque, R.; Fabra, R.; Heinrich, P.C. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989, 242, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef]
- Tanaka, T.; Kishimoto, T. The biology and medical implications of interleukin-6. Cancer Immunol. Res. 2014, 2, 288–294. [Google Scholar] [CrossRef]
- Tylutka, A.; Walas, Ł.; Zembron-Lacny, A. Level of IL-6, TNF, and IL-1β and age-related diseases: A systematic review and meta-analysis. Front. Immunol. 2024, 15, 1330386. [Google Scholar] [CrossRef]
- Müller-Newen, G.; Küster, A.; Hemmann, U.; Keul, R.; Horsten, U.; Martens, A.; Graeve, L.; Wijdenes, J.; Heinrich, P.C. Soluble IL-6 receptor potentiates the antagonistic activity of soluble gp130 on IL-6 responses. J. Immunol. 1998, 161, 6347–6355. [Google Scholar] [CrossRef]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef]
- Garbers, C.; Aparicio-Siegmund, S.; Rose-John, S. The IL-6/gp130/STAT3 signaling axis: Recent advances towards specific inhibition. Curr. Opin. Immunol. 2015, 34, 75–82. [Google Scholar] [CrossRef]
- Taga, T.; Hibi, M.; Hirata, Y.; Yamasaki, K.; Yasukawa, K.; Matsuda, T.; Hirano, T.; Kishimoto, T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 1989, 58, 573–581. [Google Scholar] [CrossRef]
- Huang, M.; Wang, L.; Zhang, Q.; Zhou, L.; Liao, R.; Wu, A.; Wang, X.; Luo, J.; Huang, F.; Zou, W.; et al. Interleukins in platelet biology: Unraveling the complex regulatory network. Pharmaceuticals 2024, 17, 109. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Maekawa, T.; Watanabe, S.; Tsuji, K.; Nakahata, T. Erythroid progenitors differentiate and mature in response to endogenous erythropoietin. J. Clin. Investig. 2000, 106, 263–270. [Google Scholar] [CrossRef]
- Rennick, D.; Jackson, J.; Yang, G.; Wideman, J.; Lee, F.; Hudak, S. Interleukin-6 interacts with interleukin-4 and other hematopoietic growth factors to selectively enhance the growth of megakaryocytic, erythroid, myeloid, and multipotential progenitor cells. Blood 1989, 73, 1828–1835. [Google Scholar] [CrossRef]
- Bruno, E.; Hoffman, R. Effect of interleukin 6 on in vitro human megakaryocytopoiesis: Its interaction with other cytokines. Exp. Hematol. 1989, 17, 1038–1043. [Google Scholar]
- Navarro, S.; Mitjavila, M.T.; Katz, A.; Doly, J.; Vainchenker, W. Expression of interleukin 6 and its specific receptor by untreated and PMA-stimulated human erythroid and megakaryocytic cell lines. Exp. Hematol. 1991, 19, 11–17. [Google Scholar]
- Imai, T.; Koike, K.; Kubo, T.; Kikuchi, T.; Amano, Y.; Takagi, M.; Okumura, N.; Nakahata, T. Interleukin-6 supports human megakaryocytic proliferation and differentiation in vitro. Blood 1991, 78, 1969–1974. [Google Scholar] [CrossRef]
- Kimura, H.; Ishibashi, T.; Uchida, T.; Maruyama, Y.; Friese, P.; Burstein, S.A. Interleukin 6 is a differentiation factor for human megakaryocytes in vitro. Eur. J. Immunol. 1990, 20, 1927–1931. [Google Scholar] [CrossRef]
- Sui, X.; Tsuji, K.; Ebihara, Y.; Tanaka, R.; Muraoka, K.; Yoshida, M.; Yamada, K.; Yasukawa, K.; Taga, T.; Kishimoto, T.; et al. Soluble interleukin-6 (IL-6) receptor with IL-6 stimulates megakaryopoiesis from human CD34(+) cells through glycoprotein (gp)130 signaling. Blood 1999, 93, 2525–2532. [Google Scholar] [CrossRef]
- Tajika, K.; Ikebuchi, K.; Inokuchi, K.; Hasegawa, S.; Dan, K.; Sekiguchi, S.; Nakahata, T.; Asano, S. IL-6 and SCF exert different effects on megakaryocyte maturation. Br. J. Haematol. 1998, 100, 105–111. [Google Scholar] [CrossRef]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef]
- Geddis, A.E. Megakaryopoiesis. Semin. Hematol. 2010, 47, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Thattaliyath, B.; Cykowski, M.; Jagadeeswaran, P. Young thrombocytes initiate the formation of arterial thrombi in zebrafish. Blood 2005, 106, 118–124. [Google Scholar] [CrossRef]
- Angénieux, C.; Couvidou, A.; Brouard, N.; Eckly, A.; Dupuis, A.; Mangin, P.H.; Maître, B. Discriminating young platelets on human leukocyte antigen-I expression highlights their extremely high reactivity potential. Res. Pract. Thromb. Haemost. 2023, 7, 100006. [Google Scholar] [CrossRef]
- Peng, J.; Friese, P.; George, J.N.; Dale, G.L.; Burstein, S.A. Alteration of platelet function in dogs mediated by interleukin-6. Blood 1994, 83, 398–403. [Google Scholar] [CrossRef]
- Burstein, S.A.; Peng, J.; Friese, P.; Wolf, R.F.; Harrison, P.; Downs, T.; Hamilton, K.; Comp, P.; Dale, G.L. Cytokine-induced alteration of platelet and hemostatic function. Stem Cells 1996, 14 (Suppl. S1), 154–162. [Google Scholar] [CrossRef]
- Oleksowicz, L.; Puszkin, E.; Mrowiec, Z.; Isaacs, R.; Dutcher, J.P. Alterations in platelet function in patients receiving interleukin-6 as cytokine therapy. Cancer Investig. 1996, 14, 307–316. [Google Scholar] [CrossRef]
- Lupia, E.; Bosco, O.; Bergerone, S.; Dondi, A.E.; Goffi, A.; Oliaro, E.; Cordero, M.; Del Sorbo, L.; Trevi, G.; Montrucchio, G. Thrombopoietin contributes to enhanced platelet activation in patients with unstable angina. J. Am. Coll. Cardiol. 2006, 48, 2195–2203. [Google Scholar] [CrossRef]
- Pasquet, J.M.; Gross, B.S.; Gratacap, M.P.; Quek, L.; Pasquet, S.; Payrastre, B.; van Willigen, G.; Mountford, J.C.; Watson, S.P. Thrombopoietin potentiates collagen receptor signaling in platelets through a phosphatidylinositol 3-kinase-dependent pathway. Blood 2000, 95, 3429–3434. [Google Scholar] [CrossRef]
- Rodríguez-Liñares, B.; Watson, S.P. Thrombopoietin potentiates activation of human platelets in association with JAK2 and TYK2 phosphorylation. Biochem. J. 1996, 316 Pt 1, 93–98. [Google Scholar] [CrossRef]
- Majka, M.; Ratajczak, J.; Villaire, G.; Kubiczek, K.; Marquez, L.A.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Thrombopoietin, but not cytokines binding to gp130 protein-coupled receptors, activates MAPKp42/44, AKT, and STAT proteins in normal human CD34+ cells, megakaryocytes, and platelets. Exp. Hematol. 2002, 30, 751–760. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Yildirim, A.; Granger, D.N.; Gavins, F.N.E. Novel role of T Cells and IL-6 (Interleukin-6) in angiotensin II-induced microvascular dysfunction. Hypertension 2019, 73, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Oleksowicz, L.; Mrowiec, Z.; Isaacs, R.; Dutcher, J.P.; Puszkin, E. Morphologic and ultrastructural evidence of interleukin-6 induced platelet activation. Am. J. Hematol. 1995, 48, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Mauro, D.; Iacono, D.; Pantano, I.; Raimondi, M.; Marchesano, M.L.; Riccio, F.; Pellegrino, A.; Liakouli, V.; Ciccia, F. AB0470 Effect of tofacitinib in modulating platelet function in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2023, 82, 1428. [Google Scholar] [CrossRef]
- Lu, W.-J.; Lin, K.-C.; Huang, S.-Y.; Thomas, P.A.; Wu, Y.-H.; Wu, H.-C.; Lin, K.-H.; Sheu, J.-R. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb. Res. 2014, 133, 1088–1096. [Google Scholar] [CrossRef]
- Aleva, F.E.; van de Veerdonk, F.L.; Li, Y.; Tunjungputri, R.N.; Simons, S.; De Groot, P.G.; Netea, M.M.; Heijdra, Y.F.; de Mast, Q.; van der Ven, A. The effects of signal transducer and activator of transcription three mutations on human platelets. Platelets 2018, 29, 602–609. [Google Scholar] [CrossRef]
- Eaton, N.; Subramaniam, S.; Schulte, M.L.; Drew, C.; Jakab, D.; Haberichter, S.L.; Weiler, H.; Falet, H. Bleeding diathesis in mice lacking JAK2 in platelets. Blood Adv. 2021, 5, 2969–2981. [Google Scholar] [CrossRef]
- Rodríguez-Liñares, B.; Watson, S.P. Phosphorylation of JAK2 in thrombin-stimulated human platelets. FEBS Lett. 1994, 352, 335–338. [Google Scholar] [CrossRef]
- Randi, M.L.; Brunati, A.M.; Scapin, M.; Frasson, M.; Deana, R.; Magrin, E.; Fabris, F.; Donella-Deana, A. Src tyrosine kinase preactivation is associated with platelet hypersensitivity in essential thrombocythemia and polycythemia vera. Blood 2010, 115, 667–676. [Google Scholar] [CrossRef]
- Houck, K.L.; Yuan, H.; Tian, Y.; Solomon, M.; Cramer, D.; Liu, K.; Zhou, Z.; Wu, X.; Zhang, J.; Oehler, V.; et al. Physical proximity and functional cooperation of glycoprotein 130 and glycoprotein VI in platelet membrane lipid rafts. J. Thromb. Haemost. 2019, 17, 1500–1510. [Google Scholar] [CrossRef]
- Zegeye, M.M.; Lindkvist, M.; Fälker, K.; Kumawat, A.K.; Paramel, G.; Grenegård, M.; Sirsjö, A.; Ljungberg, L.U. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun. Signal. 2018, 16, 55. [Google Scholar] [CrossRef]
- Bongartz, H.; Gille, K.; Hessenkemper, W.; Mandel, K.; Lewitzky, M.; Feller, S.M.; Schaper, F. The multi-site docking protein Grb2-associated binder 1 (Gab1) enhances interleukin-6-induced MAPK-pathway activation in an SHP2-, Grb2-, and time-dependent manner. Cell Commun. Signal. 2019, 17, 135. [Google Scholar] [CrossRef]
- Moshapa, F.T.; Riches-Suman, K.; Palmer, T.M. Therapeutic targeting of the proinflammatory IL-6-JAK/STAT signalling pathways responsible for vascular restenosis in Type 2 diabetes mellitus. Cardiol. Res. Pract. 2019, 2019, 9846312. [Google Scholar] [CrossRef] [PubMed]
- Horsten, U.; Schmitz-Van de Leur, H.; Müllberg, J.; Heinrich, P.C.; Rose-John, S. The membrane distal half of gp130 is responsible for the formation of a ternary complex with IL-6 and the IL-6 receptor. FEBS Lett. 1995, 360, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- van Willigen, G.; Gorter, G.; Akkerman, J.W. Thrombopoietin increases platelet sensitivity to alpha-thrombin via activation of the ERK2-cPLA2 pathway. Thromb. Haemost. 2000, 83, 610–616. [Google Scholar]
- Flevaris, P.; Li, Z.; Zhang, G.; Zheng, Y.; Liu, J.; Du, X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 2009, 113, 893–901. [Google Scholar] [CrossRef]
- Guidetti, G.F.; Canobbio, I.; Torti, M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv. Biol. Regul. 2015, 59, 36–52. [Google Scholar] [CrossRef]
- Moore, S.F.; Agbani, E.O.; Wersäll, A.; Poole, A.W.; Williams, C.M.; Zhao, X.; Li, Y.; Hutchinson, J.L.; Hunter, R.W.; Hers, I. Opposing roles of GSK3α and GSK3β phosphorylation in platelet function and thrombosis. Int. J. Mol. Sci. 2021, 22, 656. [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Tiwari, A.; Singh, N.; Gautam, D.; Sonkar, V.K.; Agarwal, V.; Dash, D. Aerobic glycolysis fuels platelet activation: Small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica 2019, 104, 806–818. [Google Scholar] [CrossRef]
- Laurent, P.A.; Séverin, S.; Hechler, B.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.P. Platelet PI3Kβ and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Grebenciucova, E.; VanHaerents, S. Interleukin 6: At the interface of human health and disease. Front. Immunol. 2023, 14, 1255533. [Google Scholar] [CrossRef]
- Akira, S.; Taga, T.; Kishimoto, T. Interleukin-6 in biology and medicine. Adv. Immunol. 1993, 54, 1–78. [Google Scholar] [CrossRef]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. S2), S3. [Google Scholar] [CrossRef]
- Braunwald, E. Shattuck lecture—Cardiovascular medicine at the turn of the millennium: Triumphs, concerns, and opportunities. N. Engl. J. Med. 1997, 337, 1360–1369. [Google Scholar] [CrossRef]
- Kannel, W.B.; Dawber, T.R.; Kagan, A.; Revotskie, N.; Stokes, J., III. Factors of risk in the development of coronary heart disease—Six year follow-up experience. The Framingham Study. Ann. Intern. Med. 1961, 55, 33–50. [Google Scholar] [CrossRef]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rane, M. Interleukin-6 signaling and anti-interleukin-6 therapeutics in cardiovascular disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D.; Frenette, P.S. The vessel wall and its interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D.; Burger, P.C. Platelets in inflammation and thrombosis. Arter. Thromb. Vasc. Biol. 2003, 23, 2131–2137. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Prapiadou, S.; Živković, L.; Thorand, B.; George, M.J.; van der Laan, S.W.; Malik, R.; Herder, C.; Koenig, W.; Ueland, T.; Kleveland, O.; et al. Proteogenomic data integration reveals CXCL10 as a potentially downstream causal mediator for IL-6 signaling on atherosclerosis. Circulation 2024, 149, 669–683. [Google Scholar] [CrossRef]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Katkenov, N.; Mukhatayev, Z.; Kozhakhmetov, S.; Sailybayeva, A.; Bekbossynova, M.; Kushugulova, A. Systematic review on the role of IL-6 and IL-1β in cardiovascular diseases. J. Cardiovasc. Dev. Dis. 2024, 11, 206. [Google Scholar] [CrossRef]
- Müller, K.; Aichele, S.; Herkommer, M.; Bigalke, B.; Stellos, K.; Htun, P.; Fateh-Moghadam, S.; May, A.E.; Flather, M.; Gawaz, M.; et al. Impact of inflammatory markers on platelet inhibition and cardiovascular outcome including stent thrombosis in patients with symptomatic coronary artery disease. Atherosclerosis 2010, 213, 256–262. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A.; Pfister, R.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [CrossRef]
- Hwang, S.J.; Park, K.W.; Kwon, D.A.; Kang, H.J.; Koo, B.K.; Chae, I.H.; Gwon, H.C.; Park, S.J.; Seung, K.B.; Ahn, T.; et al. High plasma interleukin-6 is associated with drug-eluting stent thrombosis: Possible role of inflammatory cytokines in the development of stent thrombosis from the Korea Stent Thrombosis Registry. Circ. J. 2011, 75, 1350–1357. [Google Scholar] [CrossRef]
- Gremmel, T.; Perkmann, T.; Kopp, C.W.; Seidinger, D.; Eichelberger, B.; Koppensteiner, R.; Steiner, S.; Panzer, S. Interleukin-6 and asymmetric dimethylarginine are associated with platelet activation after percutaneous angioplasty with stent implantation. PLoS ONE 2015, 10, e0122586. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.L.; Russell, J.; Harris, N.R.; Senchenkova, E.Y.; Yildirim, A.; Granger, D.N. Platelet abnormalities during colonic inflammation. Inflamm. Bowel Dis. 2013, 19, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; Mariette, X. Systemic and organ-specific immune-related manifestations of COVID-19. Nat. Rev. Rheumatol. 2021, 17, 315–332. [Google Scholar] [CrossRef]
- Henry, B.M.; Oliveira, M.H.S.d.; Benoit, S.; Plebani, M.; Lippi, G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): A meta-analysis. Clin. Chem. Lab. Med. 2020, 58, 1021–1028. [Google Scholar] [CrossRef]
- Ghasemzadeh, M.; Ahmadi, J.; Hosseini, E. Platelet-leukocyte crosstalk in COVID-19: How might the reciprocal links between thrombotic events and inflammatory state affect treatment strategies and disease prognosis? Thromb. Res. 2022, 213, 179–194. [Google Scholar] [CrossRef]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic. Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef]
- Lokau, J.; Garbers, Y.; Vicente, M.M.; Dittrich, A.; Meltendorf, S.; Lingel, H.; Münster-Kühnel, A.K.; Brunner-Weinzierl, M.; Garbers, C. Long-term increase in soluble interleukin-6 receptor levels in convalescents after mild COVID-19 infection. Front. Immunol. 2024, 15, 1488745. [Google Scholar] [CrossRef]
- Goudswaard, L.J.; Williams, C.M.; Khalil, J.; Burley, K.L.; Hamilton, F.; Arnold, D.; Milne, A.; Lewis, P.A.; Heesom, K.J.; Mundell, S.J.; et al. Alterations in platelet proteome signature and impaired platelet integrin α(IIb)β(3) activation in patients with COVID-19. J. Thromb. Haemost. 2023, 21, 1307–1321. [Google Scholar] [CrossRef]
- Le Joncour, A.; Biard, L.; Vautier, M.; Bugaut, H.; Mekinian, A.; Maalouf, G.; Vieira, M.; Marcelin, A.-G.; Rosenzwajg, M.; Klatzmann, D.; et al. Neutrophil–platelet and monocyte–platelet aggregates in COVID-19 patients. Thromb. Haemost. 2020, 120, 1733–1735. [Google Scholar] [CrossRef]
- Srzić, I.; Nesek Adam, V.; Tunjić Pejak, D. Sepsis definition: what’s new in the treatment guidelines. Acta Clin. Croat. 2022, 61, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Taeb, A.M.; Hooper, M.H.; Marik, P.E. Sepsis: Current definition, pathophysiology, diagnosis, and management. Nutr. Clin. Pract. 2017, 32, 296–308. [Google Scholar] [CrossRef]
- Esper, R.C.; Ramos, R.C. Sepsis: Un reto para el internista. Med. Int. Mex. 2005, 21, 206–222. [Google Scholar]
- Liu, Y.; Chen, L. Impact of interleukin 6 levels on acute lung injury risk and disease severity in critically ill sepsis patients. World J. Clin. Cases 2024, 12, 5374–5381. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, F.W.; Thomas, M.; Arnold, D.; Palmer, T.; Moran, E.; Mentzer, A.J.; Maskell, N.; Baillie, K.; Summers, C.; Hingorani, A.; et al. Therapeutic potential of IL6R blockade for the treatment of sepsis and sepsis-related death: A Mendelian randomisation study. PLoS Med. 2023, 20, e1004174. [Google Scholar] [CrossRef]
- Takahashi, W.; Nakada, T.-a.; Yazaki, M.; Oda, S. Interleukin-6 levels act as a diagnostic marker for infection and a prognostic marker in patients with organ dysfunction in intensive care Units. Shock 2016, 46, 254–260. [Google Scholar] [CrossRef]
- Behnes, M.; Bertsch, T.; Lepiorz, D.; Lang, S.; Trinkmann, F.; Brueckmann, M.; Borggrefe, M.; Hoffmann, U. Diagnostic and prognostic utility of soluble CD 14 subtype (presepsin) for severe sepsis and septic shock during the first week of intensive care treatment. Crit. Care 2014, 18, 507. [Google Scholar] [CrossRef]
- Hack, C.E.; De Groot, E.R.; Fert-Bersma, R.J.F.; Nuijens, J.H.; Strack Van Schijndel, R.J.M.; Eerenberg-Belmer, A.J.M.; Thijs, L.G.; Aarden, L.A. Increased Plasma Levels of Interleukin-6 in Sepsis. Blood 1989, 74, 1704–1710. [Google Scholar] [CrossRef]
- Venkata, C.; Kashyap, R.; Farmer, J.C.; Afessa, B. Thrombocytopenia in adult patients with sepsis: Incidence, risk factors, and its association with clinical outcome. J. Intensive Care 2013, 1, 9. [Google Scholar] [CrossRef]
- Vandijck, D.M.; Blot, S.I.; De Waele, J.J.; Hoste, E.A.; Vandewoude, K.H.; Decruyenaere, J.M. Thrombocytopenia and outcome in critically ill patients with bloodstream infection. Heart Lung 2010, 39, 21–26. [Google Scholar] [CrossRef]
- Shannon, O. The role of platelets in sepsis. Res. Pract. Thromb. Haemost. 2021, 5, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Scully, M. How I treat disseminated intravascular coagulation. Blood 2018, 131, 845–854. [Google Scholar] [CrossRef]
- Shimizu, M.; Konishi, A.; Nomura, S. Examination of biomarker expressions in sepsis-related DIC patients. Int. J. Gen. Med. 2018, 11, 353–361. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Activation Threshold | Receptor(s) Involved | Mechanism | References |
|---|---|---|---|---|
| Epinephrine | ↓ | α2A | Stimulates Gαi/z- and Gαq-signalling; activates PI3K/Akt | [50,51] |
| Gas6 | ↓ | Tyro3, UFO and MerTK | Activates PI3K/Akt | [52,53] |
| IGF-1 | ↓ | IGF-1R | Activates PI3K/Akt | [54,55] |
| MMP2 | ↓ | PAR-1 with αIibβ3 as a cofactor | Cleaves PAR-1 at a non-canonical site, stimulates Gαq, Gα12-13 and Gαi/z responses; activates PI3K/Akt | [56,57,58] |
| Ox-LDL | ↓ | CD36 (via ligation) | Induction of tyrosine phosphorylation and Rho kinase activation | [59,60,61,62,63] |
| PF4 | ↓ | c-Mpl | Activates JAK2/STAT3 and PI3K/Akt | [64,65,66] |
| PGE2 | ↑/↓ | Primarily EP3-4 | EP3 stimulates Gαi/z-signalling (at low PGE2 concentrations: priming); EP4 stimulates Gαs-signalling (at high PGE2 concentrations: inhibition) | [67,68,69,70] |
| sCD40L | ↓ | CD40 | Activates p38 MAPK and NF-κB | [71,72,73] |
| S1P | ↑/↓ | S1PR1,4-5 | S1PR1 stimulates Gαi/z-signalling (at low S1P concentrations: priming); S1PR4-5 stimulates Gα12-13-signalling, potentially impairing Gαq-signalling (at high concentrations: inhibitory) | [74,75,76] |
| Succinate | ↓ | SUCNR1 | Stimulates Gαi/z- and Gαq-signalling | [77,78] |
| TPO | ↓ | c-Mpl | Activates JAK2/STAT3, PI3K/Akt and MAPK | [16,79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webb, C.E.; Vautrinot, J.; Hers, I. IL-6 as a Mediator of Platelet Hyper-Responsiveness. Cells 2025, 14, 766. https://doi.org/10.3390/cells14110766
Webb CE, Vautrinot J, Hers I. IL-6 as a Mediator of Platelet Hyper-Responsiveness. Cells. 2025; 14(11):766. https://doi.org/10.3390/cells14110766
Chicago/Turabian StyleWebb, Connor Elliot, Jordan Vautrinot, and Ingeborg Hers. 2025. "IL-6 as a Mediator of Platelet Hyper-Responsiveness" Cells 14, no. 11: 766. https://doi.org/10.3390/cells14110766
APA StyleWebb, C. E., Vautrinot, J., & Hers, I. (2025). IL-6 as a Mediator of Platelet Hyper-Responsiveness. Cells, 14(11), 766. https://doi.org/10.3390/cells14110766