
Progranulin’s Protective Mechanisms and Therapeutic Potential in Cardiovascular Disease

 ,
, 
Abstract

1. Introduction
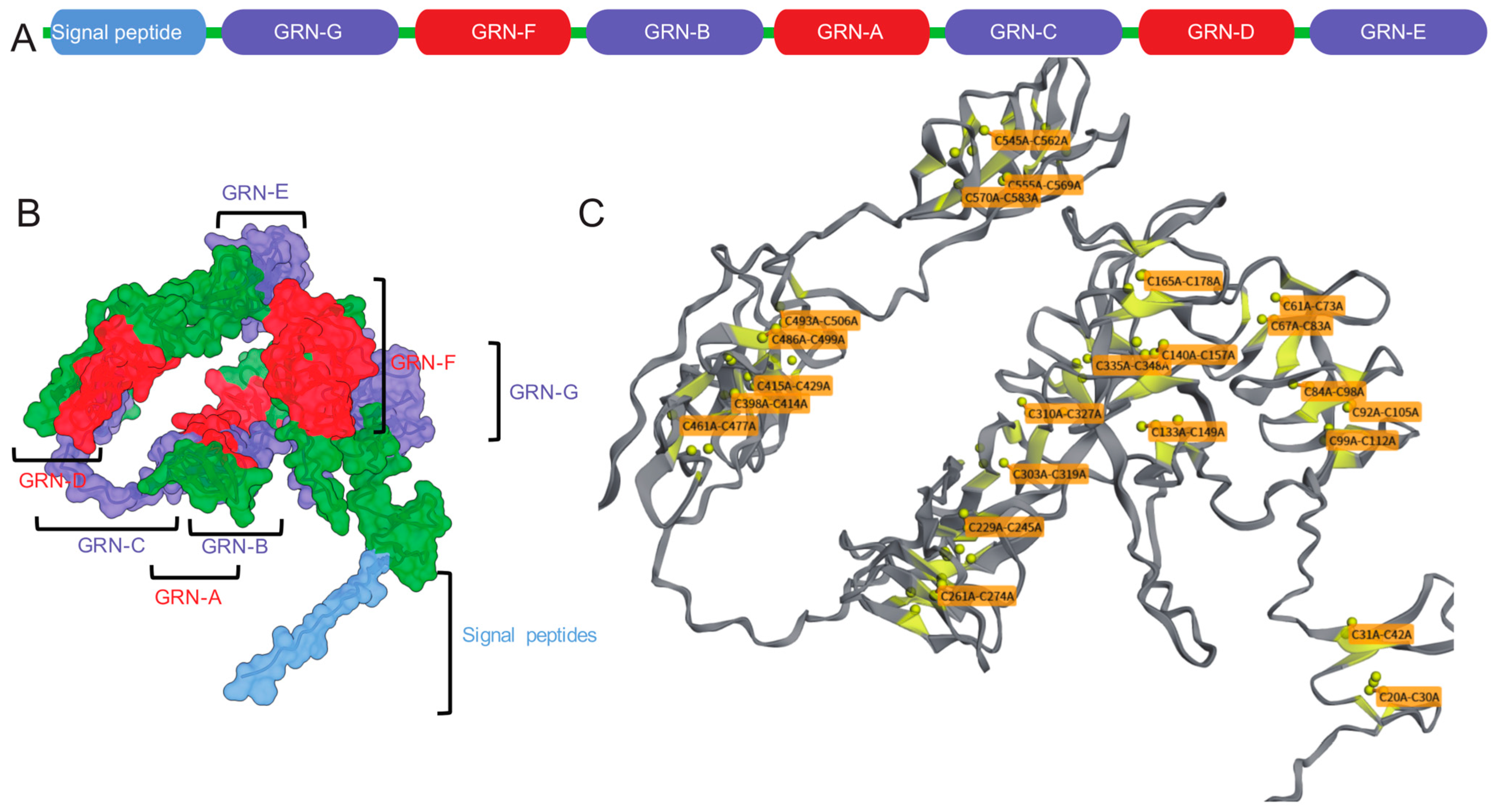
2. The Structure and Function of Progranulin
3. Progranulin and Cardiovascular Disease
3.1. Mechanisms of Progranulin’s Impact on Cardiac Pathologies
3.1.1. Myocardial Infarction and Ischemia-Reperfusion Injury
3.1.2. Sepsis-Induced and Diabetes-Induced Cardiomyopathy
3.1.3. Cardiac Aging and Hypertrophy
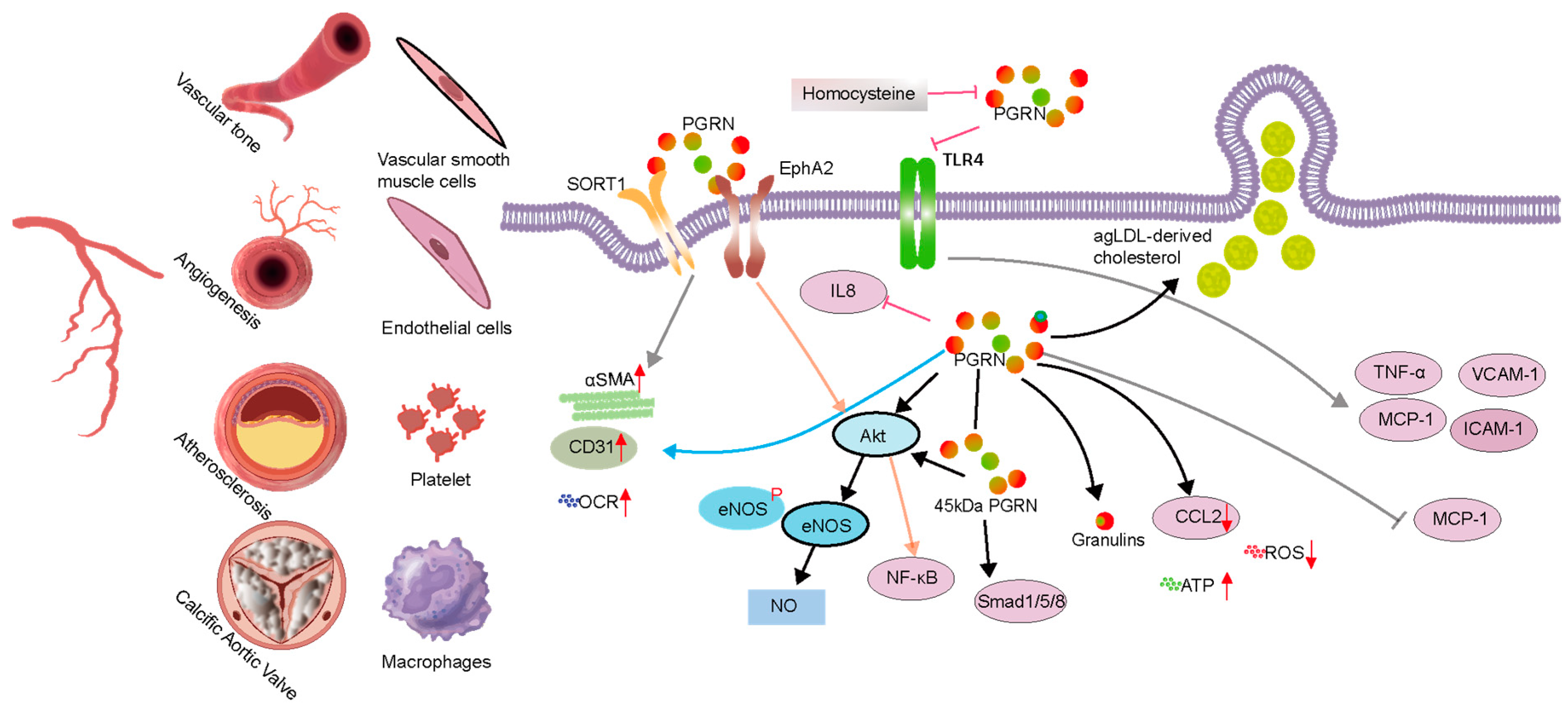
3.2. Mechanisms of Progranulin’s Impact on Vascular Diseases
3.2.1. Abnormal Angiogenesis or Vascular Injury
3.2.2. Atherosclerosis
3.3. Calcific Aortic Valve Disease
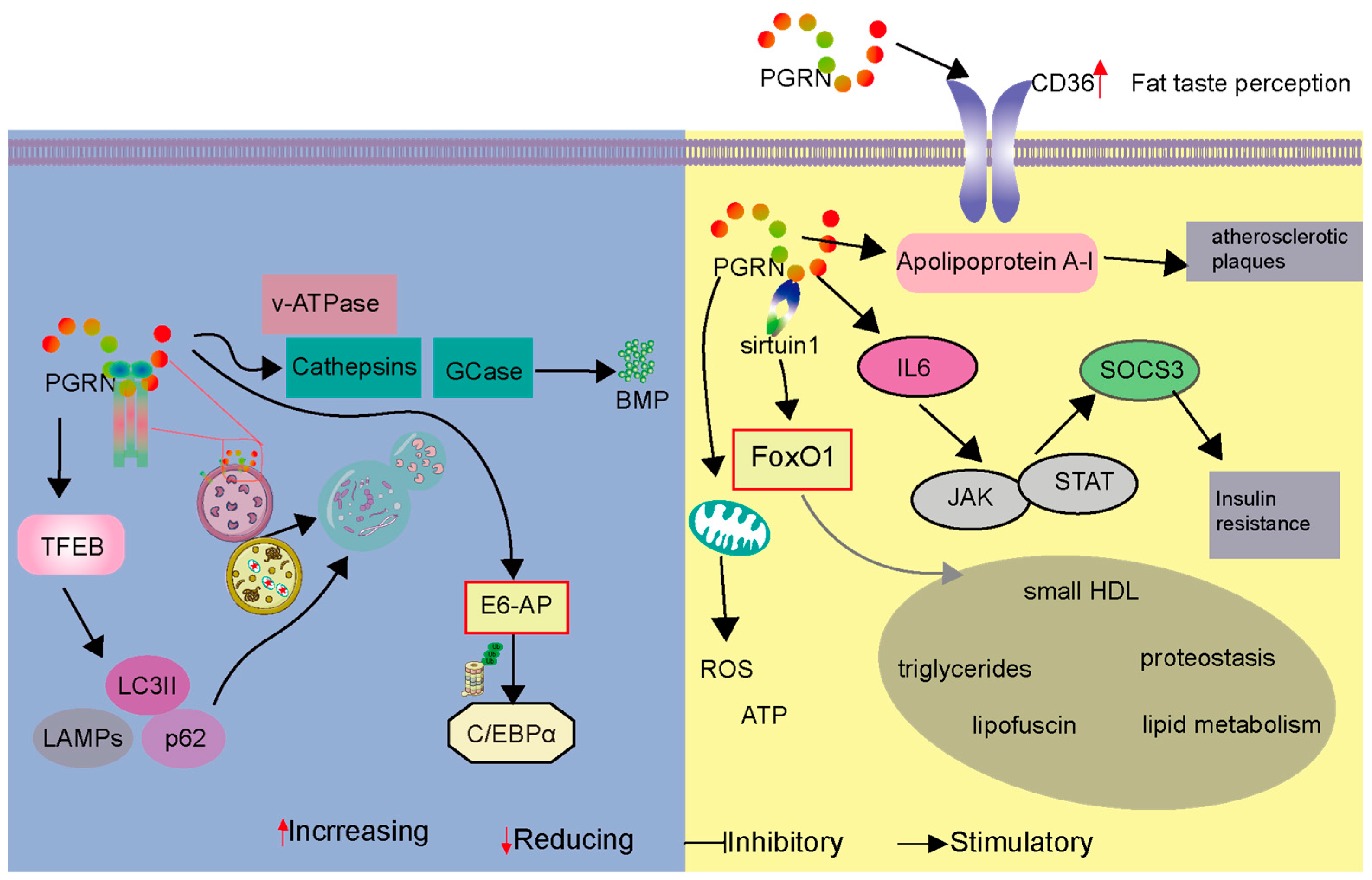
3.4. Progranulin’s Role in Cardiovascular Metabolism and Metabolic Disorders
3.5. PGRN as an Autophagosome and Lysosome Regulator
4. Therapeutic Approach Targeting PGRN
4.1. Protein Replacement
4.2. SORT1-Axis Modulation
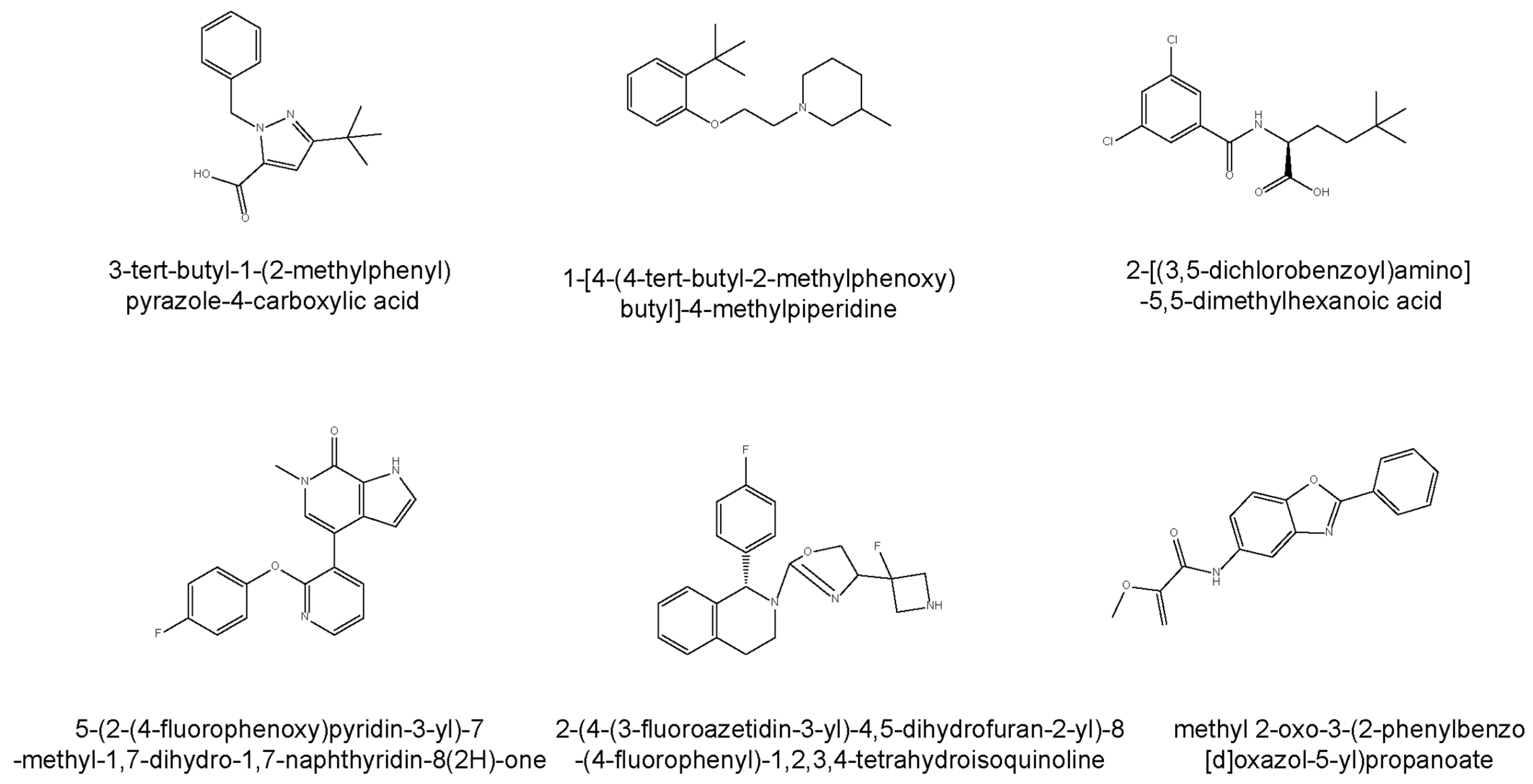
4.3. Allosteric Activators
5. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Form |
| PGRN | Progranulin |
| CVD | Cardiovascular Disease |
| MI | Myocardial Infarction |
| I/R | Ischemia-Reperfusion |
| PCI | Percutaneous Coronary Intervention |
| TNF | Tumor Necrosis Factor |
| TNFR | Tumor Necrosis Factor Receptor |
| PI3K | Phosphoinositide 3-Kinase |
| Akt | Protein Kinase B |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| VSMC | Vascular Smooth Muscle Cell |
| eNOS | Endothelial Nitric Oxide Synthase |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| IL-10 | Interleukin-10 |
| HDL | High-Density Lipoprotein |
| LDL | Low-Density Lipoprotein |
| agLDL | Aggregated Low-Density Lipoprotein |
| SIC | Sepsis-Induced Cardiomyopathy |
| CAVD | Calcific Aortic Valve Disease |
| hVICs | Human Valve Interstitial Cells |
| BMP | Bis(mono-acyl-glycero)phosphate |
| GCase | Glucocerebrosidase |
| TFEB | Transcription Factor EB |
| LAMP-1 | Lysosomal-Associated Membrane Protein 1 |
| OCR | Oxygen Consumption Rate |
| ROS | Reactive Oxygen Species |
| AAV | Adeno-Associated Virus |
| FTD | Frontotemporal Dementia |
| CSF | Cerebrospinal Fluid |
| SORT1 | Sortilin |
| PSAP | Prosaposin |
| BET | Bromodomain and Extra-terminal Domain |
| hERG | Human Ether-à-go-go-Related Gene |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy Type | Therapeutic Candidate | Mechanism of Action | Clinical Development |
|---|---|---|---|
| Direct Protein Replacement | AZP2006 | Binds to the PGRN/PSAP complex in lysosomes, promoting PGRN release and neurite outgrowth. | Completed Phase 1-2 trials. |
| Allosteric Activators | AZP2006 | Promotes PGRN release by binding to the PGRN/PSAP complex within lysosomal compartments. | Completed Phase 1-2 trials. |
| Gene Therapy | AAV-delivered GRN | Delivers functional GRN genes or promotes PGRN secretion through compounds like 5 and 6. | Preclinical and early clinical stages. |
| Targeting SORT1 Axis | AL001 | Monoclonal antibody that blocks the interaction between SORT1 and PGRN, increasing extracellular PGRN levels. | Increased PGRN in plasma/CSF; studies in mutation carriers. |
| Inhibitors of SORT1-PGRN Interaction | Compounds 1–3 | Small molecule inhibitors disrupting the SORT1-PGRN complex to increase extracellular PGRN. | Compound 3 shows low IC50, preclinical stage. |
| Modulators of Expression/Secretion | Compounds 4–6 | Modulate pathways like BET proteins or directly enhance PGRN secretion. | Preclinical; some in early clinical trials. |
References
- Vaduganathan, M.; Mensah, G.A.; Turco, J.V.; Fuster, V.; Roth, G.A. The Global Burden of Cardiovascular Diseases and Risk. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, H.; Tatton, N.; McCaughey, S.; Kurnellas, M.; Rosenthal, A. Progranulin as a Therapeutic Target in Neurodegenerative Diseases. Trends Pharmacol. Sci. 2022, 43, 641–652. [Google Scholar] [CrossRef]
- Sasaki, T.; Kuse, Y.; Nakamura, S.; Shimazawa, M.; Hara, H. Progranulin Deficiency Exacerbates Cardiac Remodeling after Myocardial Infarction. FASEB BioAdvances 2023, 5, 395–411. [Google Scholar] [CrossRef]
- Saeedi-Boroujeni, A.; Purrahman, D.; Shojaeian, A.; Poniatowski, Ł.A.; Rafiee, F.; Mahmoudian-Sani, M.-R. Progranulin (PGRN) as a Regulator of Inflammation and a Critical Factor in the Immunopathogenesis of Cardiovascular Diseases. J. Inflamm. 2023, 20, 1. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, X.; Li, B.; Tonetti, M.S.; Yang, Y.; Li, Y.; Liu, B.; Qian, S.; Gu, Y.; Wang, Q.; et al. Progranulin-Dependent Repair Function of Regulatory T Cells Drive Bone Fracture Healing. J. Clin. Investig. 2025, 135, e180679. [Google Scholar] [CrossRef]
- Pogonowska, M.; Poniatowski, Ł.A.; Wawrzyniak, A.; Królikowska, K.; Kalicki, B. The Role of Progranulin (PGRN) in the Modulation of Anti-Inflammatory Response in Asthma. Cent. Eur. J. Immunol. 2019, 44, 97–101. [Google Scholar] [CrossRef]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN Acts as a Novel Regulator of Mitochondrial Homeostasis by Facilitating Mitophagy and Mitochondrial Biogenesis to Prevent Podocyte Injury in Diabetic Nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef]
- Sieben, A.; Van Langenhove, T.; Engelborghs, S.; Martin, J.-J.; Boon, P.; Cras, P.; De Deyn, P.-P.; Santens, P.; Van Broeckhoven, C.; Cruts, M. The Genetics and Neuropathology of Frontotemporal Lobar Degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef]
- Bateman, A.; Bennett, H.P. Granulins: The Structure and Function of an Emerging Family of Growth Factors. Endocrinology 1998, 158, 145–151. [Google Scholar] [CrossRef]
- Palfree, R.G.E.; Bennett, H.P.J.; Bateman, A. The Evolution of the Secreted Regulatory Protein Progranulin. PLoS ONE 2015, 10, e0133749. [Google Scholar] [CrossRef]
- Songsrirote, K.; Li, Z.; Ashford, D.; Bateman, A.; Thomas-Oates, J. Development and Application of Mass Spectrometric Methods for the Analysis of Progranulin N-Glycosylation. J. Proteom. 2010, 73, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Sampognaro, P.J.; Argouarch, A.R.; Maynard, J.C.; Welch, M.; Patwardhan, A.; Courtney, E.C.; Zhang, J.; Mason, A.; Li, K.H.; et al. Processing of Progranulin into Granulins Involves Multiple Lysosomal Proteases and Is Affected in Frontotemporal Lobar Degeneration. Mol. Neurodegener. 2021, 16, 51. [Google Scholar] [CrossRef]
- Cui, Y.; Hettinghouse, A.; Liu, C. Progranulin: A Conductor of a Receptors Orchestra and a Therapeutic Target for Multiple Diseases. Cytokine Growth Factor. Rev. 2019, 45, 53–64. [Google Scholar] [CrossRef]
- Lee, W.C.; Almeida, S.; Prudencio, M.; Caulfield, T.R.; Zhang, Y.-J.; Tay, W.M.; Bauer, P.O.; Chew, J.; Sasaguri, H.; Jansen-West, K.R.; et al. Targeted Manipulation of the Sortilin–Progranulin Axis Rescues Progranulin Haploinsufficiency. Hum. Mol. Genet. 2014, 23, 1467–1478. [Google Scholar] [CrossRef]
- Jian, J.; Tian, Q.-Y.; Hettinghouse, A.; Zhao, S.; Liu, H.; Wei, J.; Grunig, G.; Zhang, W.; Setchell, K.D.R.; Sun, Y.; et al. Progranulin Recruits HSP70 to β-Glucocerebrosidase and Is Therapeutic Against Gaucher Disease. EBioMedicine 2016, 13, 212–224. [Google Scholar] [CrossRef]
- Zhao, X.; Liberti, R.; Jian, J.; Fu, W.; Hettinghouse, A.; Sun, Y.; Liu, C. Progranulin Associates with Rab2 and Is Involved in Autophagosome-Lysosome Fusion in Gaucher Disease. J. Mol. Med. 2021, 99, 1639–1654. [Google Scholar] [CrossRef]
- Liu, C.; Bosch, X. Progranulin: A Growth Factor, a Novel TNFR Ligand and a Drug Target. Pharmacol. Ther. 2012, 133, 124–132. [Google Scholar] [CrossRef]
- Ding, Y.; Wei, J.; Hettinghouse, A.; Li, G.; Li, X.; Einhorn, T.A.; Liu, C. Progranulin Promotes Bone Fracture Healing via TNFR Pathways in Mice with Type 2 Diabetes Mellitus. Ann. N. Y. Acad. Sci. 2021, 1490, 77–89. [Google Scholar] [CrossRef]
- Mundra, J.J.; Jian, J.; Bhagat, P.; Liu, C. Progranulin Inhibits Expression and Release of Chemokines CXCL9 and CXCL10 in a TNFR1 Dependent Manner. Sci. Rep. 2016, 6, 21115. [Google Scholar] [CrossRef]
- Tian, Q.; Zhao, Y.; Mundra, J.J.; Gonzalez-Gugel, E.; Jian, J.; Uddin, S.M.; Liu, C. Three TNFR-Binding Domains of PGRN Act Independently in Inhibition of TNFα Binding and Activity. Front. Biosci. (Landmark Ed.) 2014, 19, 1176–1185. [Google Scholar] [CrossRef]
- Russillo, M.C.; Sorrentino, C.; Scarpa, A.; Vinciguerra, C.; Cicarelli, G.; Cuoco, S.; Gagliardi, M.; Talarico, M.; Procopio, R.; Quattrone, A.; et al. A Novel Phenotype in an Italian Family with a Rare Progranulin Mutation. J. Neurol. 2022, 269, 6170–6177. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury Following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediators Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. The Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction Fourth Universal Definition of Myocardial Infarction (2018). Circulation 2018, 138, e618–e651. [Google Scholar] [CrossRef]
- Adabag, A.S.; Therneau, T.M.; Gersh, B.J.; Weston, S.A.; Roger, V.L. Sudden Death After Myocardial Infarction. JAMA 2008, 300, 2022–2029. [Google Scholar] [CrossRef]
- Tong, C.; Zhou, B. Cardioprotective Strategies in Myocardial Ischemia-Reperfusion Injury: Implications for Improving Clinical Translation. J. Mol. Cell. Cardiol. Plus 2024, 11, 100278. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, Y.; Zhang, S.; Li, M.; Wang, J. Serum Progranulin As a Risk Predictor in Patients with Acute Myocardial Infarction. Med. Sci. Monit. 2021, 27, e928864-1–e928864-6. [Google Scholar] [CrossRef]
- Sasaki, T.; Shimazawa, M.; Kanamori, H.; Yamada, Y.; Nishinaka, A.; Kuse, Y.; Suzuki, G.; Masuda, T.; Nakamura, S.; Hosokawa, M.; et al. Effects of Progranulin on the Pathological Conditions in Experimental Myocardial Infarction Model. Sci. Rep. 2020, 10, 11842. [Google Scholar] [CrossRef]
- Alyahya, A.M.; Al-Masri, A.; Hersi, A.; El Eter, E.; Husain, S.; Lateef, R.; Mawlana, O.H. The Effects of Progranulin in a Rat Model of Acute Myocardial Ischemia/Reperfusion Are Mediated by Activation of the P13K/Akt Signaling Pathway. Med. Sci. Monit. Basic. Res. 2019, 25, 229–237. [Google Scholar] [CrossRef]
- Jackman, K.; Kahles, T.; Lane, D.; Garcia-Bonilla, L.; Abe, T.; Capone, C.; Hochrainer, K.; Voss, H.; Zhou, P.; Ding, A.; et al. Progranulin Deficiency Promotes Post-Ischemic Blood-Brain Barrier Disruption. J. Neurosci. 2013, 33, 19579–19589. [Google Scholar] [CrossRef]
- de la Encarnación, A.; Alquézar, C.; Martín-Requero, Á. Increased Wnt Signaling and Reduced Viability in a Neuronal Model of Progranulin-Deficient Frontotemporal Lobar Degeneration. Mol. Neurobiol. 2015, 53, 7107–7118. [Google Scholar] [CrossRef]
- Tian, R.; Li, Y.; Yao, X. PGRN Suppresses Inflammation and Promotes Autophagy in Keratinocytes Through the Wnt/β-Catenin Signaling Pathway. Inflammation 2016, 39, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, Y.; Zhou, M.; Wang, X.; Wang, Z.; Wei, X.; Zhang, Y.; Su, Z.; Liang, K.; Tang, W.; et al. Therapeutic Potential of Progranulin in Hyperhomocysteinemia-Induced Cardiorenal Dysfunction. Hypertension 2017, 69, 259–266. [Google Scholar] [CrossRef]
- Raitano, S.; Ordovàs, L.; De Muynck, L.; Guo, W.; Espuny-Camacho, I.; Geraerts, M.; Khurana, S.; Vanuytsel, K.; Tóth, B.I.; Voets, T.; et al. Restoration of Progranulin Expression Rescues Cortical Neuron Generation in an Induced Pluripotent Stem Cell Model of Frontotemporal Dementia. Stem Cell Rep. 2015, 4, 16–24. [Google Scholar] [CrossRef]
- Xu, X.; Gou, L.; Zhou, M.; Yang, F.; Zhao, Y.; Feng, T.; Shi, P.; Ghavamian, A.; Zhao, W.; Yu, Y.; et al. Progranulin Protects against Endotoxin-Induced Acute Kidney Injury by Downregulating Renal Cell Death and Inflammatory Responses in Mice. Int. Immunopharmacol. 2016, 38, 409–419. [Google Scholar] [CrossRef]
- Egashira, Y.; Suzuki, Y.; Azuma, Y.; Takagi, T.; Mishiro, K.; Sugitani, S.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Kashimata, M.; et al. The Growth Factor Progranulin Attenuates Neuronal Injury Induced by Cerebral Ischemia-Reperfusion through the Suppression of Neutrophil Recruitment. J. Neuroinflammation 2013, 10, 105. [Google Scholar] [CrossRef]
- Sato, R.; Nasu, M. A Review of Sepsis-Induced Cardiomyopathy. J. Intensive Care 2015, 3, 48. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Yu, M.-M.; Shou, S.-T.; Chai, Y.-F. Sepsis-Induced Cardiomyopathy: Mechanisms and Treatments. Front. Immunol. 2017, 8, 1021. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, X.; Zhang, L.; Xu, F.; Tao, X.; Zhang, H.; Lin, X.; Kang, L.; Xiang, Y.; Lai, X.; et al. Progranulin Plays a Central Role in Host Defense during Sepsis by Promoting Macrophage Recruitment. Am. J. Respir. Crit. Care Med. 2016, 194, 1219–1232. [Google Scholar] [CrossRef]
- Yoo, W.; Lee, J.; Noh, K.H.; Lee, S.; Jung, D.; Kabir, M.H.; Park, D.; Lee, C.; Kwon, K.-S.; Kim, J.-S.; et al. Progranulin Attenuates Liver Fibrosis by Downregulating the Inflammatory Response. Cell Death Dis. 2019, 10, 758. [Google Scholar] [CrossRef]
- Shan, Y.; Zhang, X.; Zhou, G.; Ji, X.; Gu, Y. Increased Progranulin as an Independent Predictive Biomarker for Poor Prognosis in Sepsis. Cytokine 2022, 155, 155911. [Google Scholar] [CrossRef]
- Luo, Q.; He, X.; Zheng, Y.; Ning, P.; Xu, Y.; Yang, D.; Shang, Y.; Gao, Z. Elevated Progranulin as a Novel Biomarker to Predict Poor Prognosis in Community-Acquired Pneumonia. J. Infect. 2020, 80, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kuse, Y.; Nakamura, S.; Shimazawa, M. Progranulin-Deficient Macrophages Cause Cardiotoxicity under Hypoxic Conditions. Biochem. Biophys. Res. Commun. 2024, 691, 149341. [Google Scholar] [CrossRef] [PubMed]
- Milajerdi, A.; Maghbooli, Z.; Mohammadi, F.; Hosseini, B.; Mirzaei, K. Progranulin Concentration in Relation to Bone Mineral Density among Obese Individuals. Arch. Endocrinol. Metab. 2018, 62, 179–186. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Zhou, B.; Xu, L.; Kang, X.; Yang, W.; Wu, S.; Sun, H. PGRN Induces Impaired Insulin Sensitivity and Defective Autophagy in Hepatic Insulin Resistance. Mol. Endocrinol. 2015, 29, 528–541. [Google Scholar] [CrossRef]
- Wang, K.; Wei, J.; Liu, C. Progranulin Derivative Atsttrin Promotes Chondrogenesis and Cartilage Regeneration. Osteoarthr. Cartil. 2021, 29, S39–S40. [Google Scholar] [CrossRef]
- Kazama, K.; Hoshino, K.; Kodama, T.; Okada, M.; Yamawaki, H. Adipocytokine, Progranulin, Augments Acetylcholine-induced Nitric Oxide-mediated Relaxation through the Increases of cGMP Production in Rat Isolated Mesenteric Artery. Acta Physiol. 2017, 219, 781–789. [Google Scholar] [CrossRef]
- Yan, W.; Ding, A.; Kim, H.-J.; Zheng, H.; Wei, F.; Ma, X. Progranulin Controls Sepsis via C/EBPα-Regulated Il10 Transcription and Ubiquitin Ligase/Proteasome-Mediated Protein Degradation. J. Immunol. 2016, 197, 3393–3405. [Google Scholar] [CrossRef]
- Nádró, B.; Lőrincz, H.; Molnár, Á.; Szentpéteri, A.; Zöld, E.; Seres, I.; Páll, D.; Paragh, G.; Kempler, P.; Harangi, M.; et al. Effects of Alpha-Lipoic Acid Treatment on Serum Progranulin Levels and Inflammatory Markers in Diabetic Neuropathy. J. Int. Med. Res. 2021, 49, 03000605211012213. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Li, H.; Hastings, M.H.; Rhee, J.; Trager, L.E.; Roh, J.D.; Rosenzweig, A. Targeting Age-Related Pathways in Heart Failure. Circ. Res. 2020, 126, 533–551. [Google Scholar] [CrossRef]
- Root, J.; Mendsaikhan, A.; Nandy, S.; Taylor, G.; Wang, M.; Araujo, L.T.; Merino, P.; Ryu, D.; Holler, C.; Thompson, B.M.; et al. Granulins Rescue Inflammation, Lysosome Dysfunction, and Neuropathology in a Mouse Model of Progranulin Deficiency. bioRxiv 2023. [Google Scholar] [CrossRef]
- Yin, F.; Banerjee, R.; Thomas, B.; Zhou, P.; Qian, L.; Jia, T.; Ma, X.; Ma, Y.; Iadecola, C.; Beal, M.F.; et al. Exaggerated Inflammation, Impaired Host Defense, and Neuropathology in Progranulin-Deficient Mice. J. Exp. Med. 2010, 207, 117–128. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Malmstrom, T.K.; Niehoff, M.L.; Aziz, A.; Miller, D.K.; Morley, J.E. Serum Progranulin Levels Are Associated with Frailty in Middle-Aged Individuals. PLoS ONE 2020, 15, e0238877. [Google Scholar] [CrossRef]
- Zhu, Y.; Ohama, T.; Kawase, R.; Chang, J.; Inui, H.; Kanno, K.; Okada, T.; Masuda, D.; Koseki, M.; Nishida, M.; et al. Progranulin Deficiency Leads to Enhanced Age-Related Cardiac Hypertrophy through Complement C1q-Induced β-Catenin Activation. J. Mol. Cell Cardiol. 2020, 138, 197–211. [Google Scholar] [CrossRef]
- Tanaka, Y.; Ito, S.; Honma, Y.; Hasegawa, M.; Kametani, F.; Suzuki, G.; Kozuma, L.; Takeya, K.; Eto, M. Dysregulation of the Progranulin-Driven Autophagy-Lysosomal Pathway Mediates Secretion of the Nuclear Protein TDP-43. J. Biol. Chem. 2023, 299, 105272. [Google Scholar] [CrossRef]
- Zhang, L.; Saito, H.; Higashimoto, T.; Kaji, T.; Nakamura, A.; Iwamori, K.; Nagano, R.; Motooka, D.; Okuzaki, D.; Uezumi, A.; et al. Regulation of Muscle Hypertrophy through Granulin: Relayed Communication among Mesenchymal Progenitors, Macrophages, and Satellite Cells. Cell Rep. 2024, 43, 114052. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Nguyen, T.A.; Singh, R.K.; Eberle, D.; Zhang, J.; Abate, J.P.; Robles, A.; Koliwad, S.; Huang, E.J.; Maxfield, F.R.; et al. Progranulin in the Hematopoietic Compartment Protects Mice from Atherosclerosis. Atherosclerosis 2018, 277, 145–154. [Google Scholar] [CrossRef]
- Hu, S.-Y.; Tai, C.-C.; Li, Y.-H.; Wu, J.-L. Progranulin Compensates for Blocked IGF-1 Signaling to Promote Myotube Hypertrophy in C2C12 Myoblasts via the PI3K/Akt/mTOR Pathway. FEBS Lett. 2012, 586, 3485–3492. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Chen, H.-Y.; Li, Y.-W.; Wu, S.-Y.; Liu, W.-T.; Lin, G.-H.; Hu, S.-Y.; Chang, Z.-K.; Gong, H.-Y.; Liao, C.-H.; et al. Progranulin Regulates Zebrafish Muscle Growth and Regeneration through Maintaining the Pool of Myogenic Progenitor Cells. Sci. Rep. 2013, 3, 1176. [Google Scholar] [CrossRef]
- Singh, S.; Bruder-Nascimento, A.; Costa, R.M.; Alves, J.V.; Bharathi, S.; Goetzman, E.S.; Bruder-Nascimento, T. Adjusted Vascular Contractility Relies on Integrity of Progranulin Pathway: Insights into Mitochondrial Function. bioRxiv 2023. [Google Scholar]
- Singh, S.; Bruder do Nascimento, A.; Costa, R.M.; Bharathi, S.S.; Goetzman, E.; Bruder, T. Abstract 016: GRN Gene Mutation Leads To Vascular Abnormalities Via Mitochondrial Dysfunction And Oxidative Stress. Hypertension 2023, 80, A016. [Google Scholar] [CrossRef]
- Xu, B.; Chen, X.; Ding, Y.; Chen, C.; Liu, T.; Zhang, H. Abnormal Angiogenesis of Placenta in Progranulin-deficient Mice. Mol. Med. Rep. 2020, 22, 3482–3492. [Google Scholar] [CrossRef]
- Tian, D.; Qin, Q.; Li, M.; Li, X.; Xu, Q.; Lv, Q. Homocysteine Impairs Endothelial Cell Barrier Function and Angiogenic Potential via the Progranulin/EphA2 Pathway. Front. Pharmacol. 2021, 11, 614760. [Google Scholar] [CrossRef]
- Tian, D.; Qin, Q.; Liu, R.; Wang, Z.; Li, X.; Xu, Q.; Lv, Q. Diagnostic Value of Circulating Progranulin and Its Receptor EphA2 in Predicting the Atheroma Burden in Patients with Coronary Artery Disease. Dis. Markers 2021, 2021, 6653501. [Google Scholar] [CrossRef]
- Bruder-Nascimento, A.; Awata, W.M.C.; Alves, J.V.; Singh, S.; Costa, R.M.; Bruder-Nascimento, T. Progranulin Maintains Blood Pressure and Vascular Tone Dependent on EphrinA2 and Sortilin1 Receptors and Endothelial Nitric Oxide Synthase Activation. JAHA 2023, 12, e030353. [Google Scholar] [CrossRef]
- Kaur, J.; Mukheja, S.; Varma, S.; Kalra, H.S.; Khosa, B.S.; Vohra, K. Serum Progranulin/Tumor Necrosis Factor-α Ratio as Independent Predictor of Systolic Blood Pressure in Overweight Hypertensive Patients: A Cross-Sectional Study. Egypt. Heart J. 2020, 72, 25. [Google Scholar] [CrossRef]
- Singh, S.; Bruder, A.; Costa, R.M.; Alves, J.V.; Bharathi, S.; Goetzman, E.S.; Bruder-Nascimento, T. Vascular Contractility Relies on Integrity of Progranulin Pathway: Insights Into Mitochondrial Function. J. Am. Heart Assoc. 2025, 14, e037640. [Google Scholar] [CrossRef]
- Hwang, H.-J.; Jung, T.W.; Hong, H.C.; Choi, H.Y.; Seo, J.-A.; Kim, S.G.; Kim, N.H.; Choi, K.M.; Choi, D.S.; Baik, S.H.; et al. Progranulin Protects Vascular Endothelium against Atherosclerotic Inflammatory Reaction via Akt/eNOS and Nuclear Factor-κB Pathways. PLoS ONE 2013, 8, e76679. [Google Scholar] [CrossRef]
- Kawase, R.; Ohama, T.; Matsuyama, A.; Matsuwaki, T.; Okada, T.; Yamashita, T.; Yuasa-Kawase, M.; Nakaoka, H.; Nakatani, K.; Inagaki, M.; et al. Deletion of Progranulin Exacerbates Atherosclerosis in ApoE Knockout Mice. Cardiovasc. Res. 2013, 100, 125–133. [Google Scholar] [CrossRef]
- Kojima, Y.; Ono, K.; Inoue, K.; Takagi, Y.; Kikuta, K.; Nishimura, M.; Yoshida, Y.; Nakashima, Y.; Matsumae, H.; Furukawa, Y.; et al. Progranulin Expression in Advanced Human Atherosclerotic Plaque. Atherosclerosis 2009, 206, 102–108. [Google Scholar] [CrossRef]
- Yin, L.; Wang, L.; Shi, Z.; Ji, X.; Liu, L. The Role of Peroxisome Proliferator-Activated Receptor Gamma and Atherosclerosis: Post-Translational Modification and Selective Modulators. Front. Physiol. 2022, 13, 826811. [Google Scholar] [CrossRef]
- Campbell, C.A.; Fursova, O.; Cheng, X.; Snella, E.; McCune, A.; Li, L.; Solchenberger, B.; Schmid, B.; Sahoo, D.; Morton, M.; et al. A Zebrafish Model of Granulin Deficiency Reveals Essential Roles in Myeloid Cell Differentiation. Blood Adv. 2021, 5, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.H.P.; He, Z.; Kriazhev, L.; Shan, X.; Palfree, R.G.E.; Bateman, A. Regulation of Progranulin Expression in Myeloid Cells. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2006, 291, R1602–R1612. [Google Scholar] [CrossRef] [PubMed]
- Al-Yahya, A.M.; Al-Masri, A.A.; El Eter, E.A.; Hersi, A.; Lateef, R.; Mawlana, O. Progranulin Inhibits Platelet Aggregation and Prolongs Bleeding Time in Rats. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3240–3248. [Google Scholar]
- Huang, G.; An, L.; Fan, M.; Zhang, M.; Chen, B.; Zhu, M.; Wu, J.; Liu, Y.; Wang, Y.; Huang, Q.; et al. Potential Role of Full-Length and Nonfull-Length Progranulin in Affecting Aortic Valve Calcification. J. Mol. Cell. Cardiol. 2020, 141, 93–104. [Google Scholar] [CrossRef]
- Powell-Wiley, T.M.; Poirier, C.P.; Burke, V.C.L.E.; Després, J.-P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef]
- Nicoletto, B.B.; Canani, L.H. The Role of Progranulin in Diabetes and Kidney Disease. Diabetol. Metab. Syndr. 2015, 7, 117. [Google Scholar] [CrossRef]
- Schumann, L.; Wilken-Schmitz, A.; Trautmann, S.; Vogel, A.; Schreiber, Y.; Hahnefeld, L.; Gurke, R.; Geisslinger, G.; Tegeder, I. Increased Fat Taste Preference in Progranulin-Deficient Mice. Nutrients 2021, 13, 4125. [Google Scholar] [CrossRef]
- Nádró, B.; Lőrincz, H.; Juhász, L.; Szentpéteri, A.; Sztanek, F.; Varga, É.; Páll, D.; Paragh, G.; Harangi, M. Determination of Serum Progranulin in Patients with Untreated Familial Hypercholesterolemia. Biomedicines 2022, 10, 771. [Google Scholar] [CrossRef]
- Okura, H.; Yamashita, S.; Ohama, T.; Saga, A.; Yamamoto-Kakuta, A.; Hamada, Y.; Sougawa, N.; Ohyama, R.; Sawa, Y.; Matsuyama, A. HDL/Apolipoprotein A-I Binds to Macrophage-Derived Progranulin and Suppresses Its Conversion into Proinflammatory Granulins. J. Atheroscler. Thromb. 2010, 17, 568–577. [Google Scholar] [CrossRef]
- Boland, S.; Swarup, S.; Ambaw, Y.A.; Malia, P.C.; Richards, R.C.; Fischer, A.W.; Singh, S.; Aggarwal, G.; Spina, S.; Nana, A.L.; et al. Deficiency of the Frontotemporal Dementia Gene GRN Results in Gangliosidosis. Nat. Commun. 2022, 13, 5924. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Sayana, R.; Suarez-Nieto, M.V.; Sarno, J.; Nyame, K.; Xiong, J.; Pimentel Vera, L.N.; Arozqueta Basurto, J.; Corbo, M.; Limaye, A.; et al. CNS-Wide Repopulation by Hematopoietic-Derived Microglia-like Cells Corrects Progranulin Deficiency in Mice. Nat. Commun. 2024, 15, 5654. [Google Scholar] [CrossRef] [PubMed]
- Wesolek, A.; Skoracka, K.; Skrypnik, K.; Suliburska, J.; Bogdanski, P.; Szulinska, M.; Skrypnik, D. Assessment of Progranulin and Fam19a5 Protein Blood Levels in Patients with Metabolic Syndrome. J. Physiol. Pharmacol. 2022, 73, 13. [Google Scholar] [CrossRef]
- Rodríguez-Periñán, G.; de la Encarnación, A.; Moreno, F.; López de Munain, A.; Martínez, A.; Martín-Requero, Á.; Alquézar, C.; Bartolomé, F. Progranulin Deficiency Induces Mitochondrial Dysfunction in Frontotemporal Lobar Degeneration with TDP-43 Inclusions. Antioxidants 2023, 12, 581. [Google Scholar] [CrossRef]
- Orogo, A.M.; Gustafsson, Å.B. Therapeutic Targeting of Autophagy: Potential and Concerns in Treating Cardiovascular Disease. Circ. Res. 2015, 116, 489–503. [Google Scholar] [CrossRef]
- Davis, S.E.; Roth, J.R.; Aljabi, Q.; Hakim, A.R.; Savell, K.E.; Day, J.J.; Arrant, A.E. Delivering Progranulin to Neuronal Lysosomes Protects against Excitotoxicity. J. Biol. Chem. 2021, 297, 100993. [Google Scholar] [CrossRef]
- Tanaka, Y.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M. Increased Lysosomal Biogenesis in Activated Microglia and Exacerbated Neuronal Damage After Traumatic Brain Injury in Progranulin-Deficient Mice. Neuroscience 2013, 250, 8–19. [Google Scholar] [CrossRef]
- Hasan, S.; Fernandopulle, M.S.; Humble, S.W.; Frankenfield, A.M.; Li, H.; Prestil, R.; Johnson, K.R.; Ryan, B.J.; Wade-Martins, R.; Ward, M.E.; et al. Multi-Modal Proteomic Characterization of Lysosomal Function and Proteostasis in Progranulin-Deficient Neurons. bioRxiv 2023. [Google Scholar] [CrossRef]
- Doyle, J.J.; Maios, C.; Vrancx, C.; Duhaime, S.; Chitramuthu, B.; Bennett, H.P.J.; Bateman, A.; Parker, J.A. Chemical and Genetic Rescue of in Vivo Progranulin-Deficient Lysosomal and Autophagic Defects. Proc. Natl. Acad. Sci. USA 2021, 118, e2022115118. [Google Scholar] [CrossRef]
- Chang, M.C.; Srinivasan, K.; Friedman, B.A.; Suto, E.; Modrusan, Z.; Lee, W.P.; Kaminker, J.S.; Hansen, D.V.; Sheng, M. Progranulin Deficiency Causes Impairment of Autophagy and TDP-43 Accumulation. J. Exp. Med. 2017, 214, 2611–2628. [Google Scholar] [CrossRef]
- Zhou, X.; Paushter, D.H.; Pagan, M.D.; Kim, D.; Nunez Santos, M.; Lieberman, R.L.; Overkleeft, H.S.; Sun, Y.; Smolka, M.B.; Hu, F. Progranulin Deficiency Leads to Reduced Glucocerebrosidase Activity. PLoS ONE 2019, 14, e0212382. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, N.; An, Z. Engineering Antibody and Protein Therapeutics to Cross the Blood–Brain Barrier. Antib. Ther. 2022, 5, 311–331. [Google Scholar] [CrossRef]
- Logan, T.; Simon, M.J.; Rana, A.; Cherf, G.M.; Srivastava, A.; Davis, S.S.; Low, R.L.Y.; Chiu, C.-L.; Fang, M.; Huang, F.; et al. Rescue of a Lysosomal Storage Disorder Caused by Grn Loss of Function with a Brain Penetrant Progranulin Biologic. Cell 2021, 184, 4651–4668.e25. [Google Scholar] [CrossRef]
- Berger, A.C.; Cohen, I.; Jafarnejad, M.; Chiu, C.; Bhalla, A.; Damo, L.; Shanbhag, N.M.; Simen, A.; Lu, H.; Zicha, S.; et al. Safety and Pharmacokinetics of Single Ascending Doses of TAK-594/DNL593, a Brain-penetrant Progranulin Replacement Therapy, in Healthy Volunteers: Interim Results from Part A of a Phase 1/2 Clinical Trial. Alzheimer’s Dement. 2023, 19, e075068. [Google Scholar] [CrossRef]
- Sevigny, J.; Uspenskaya, O.; Heckman, L.D.; Wong, L.C.; Hatch, D.A.; Tewari, A.; Vandenberghe, R.; Irwin, D.J.; Saracino, D.; Le Ber, I.; et al. Progranulin AAV Gene Therapy for Frontotemporal Dementia: Translational Studies and Phase 1/2 Trial Interim Results. Nat. Med. 2024, 30, 1406–1415. [Google Scholar] [CrossRef]
- Kashyap, S.N.; WIlson, K.I.; Arrant, A.E.; Roberson, E.D. Neuronal Transduction of AAV-Progranulin Corrects Microglial Lipofuscinosis and Pro-Inflammatory Microglial Morphology in a Mouse Model of Progranulin Deficiency. Mol. Ther. 2023, 31, 585. [Google Scholar]
- Terryn, J.; Verfaillie, C.M.; Van Damme, P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front. Mol. Neurosci. 2021, 14, 713031. [Google Scholar] [CrossRef]
- Yoon, T.; Lee, L.E.; Ahn, S.S.; Pyo, J.Y.; Song, J.J.; Park, Y.-B.; Lee, S.-W. Serum Progranulin as a Predictive Marker for High Activity of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. J. Clin. Lab. Anal. 2021, 35, e24048. [Google Scholar] [CrossRef]
- Buccellato, F.R.; D’Anca, M.; Tartaglia, G.M.; Fabbro, M.D.; Galimberti, D. Frontotemporal Dementia: From Genetics to Therapeutic Approaches. Expert Opin. Investig. Drugs 2024, 33, 561–573. [Google Scholar] [CrossRef]
- Alberici, A.; Archetti, S.; Pilotto, A.; Premi, E.; Cosseddu, M.; Bianchetti, A.; Semeraro, F.; Salvetti, M.; Muiesan, M.L.; Padovani, A.; et al. Results from a Pilot Study on Amiodarone Administration in Monogenic Frontotemporal Dementia with Granulin Mutation. Neurol. Sci. 2014, 35, 1215–1219. [Google Scholar] [CrossRef]
- Boylan, M.A.; Pincetic, A.; Romano, G.; Tatton, N.; Kenkare-Mitra, S.; Rosenthal, A. Targeting Progranulin as an Immuno-Neurology Therapeutic Approach. Int. J. Mol. Sci. 2023, 24, 15946. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; O’Donnell, R.; Drew, Y.; Curtin, N.J.; Sharma Saha, S. Characterisation of Ovarian Cancer Cell Line NIH-OVCAR3 and Implications of Genomic, Transcriptomic, Proteomic and Functional DNA Damage Response Biomarkers for Therapeutic Targeting. Cancers 2020, 12, 1939. [Google Scholar] [CrossRef]
- Reich, M.; Simon, M.J.; Polke, B.; Paris, I.; Werner, G.; Schrader, C.; Spieth, L.; Davis, S.S.; Robinson, S.; de Melo, G.L.; et al. Peripheral Expression of Brain-Penetrant Progranulin Rescues Pathologies in Mouse Models of Frontotemporal Lobar Degeneration. Sci. Transl. Med. 2024, 16, eadj7308. [Google Scholar] [CrossRef]
- Almeida, M.R.; Tábuas-Pereira, M.; Baldeiras, I.; Lima, M.; Durães, J.; Massano, J.; Pinto, M.; Cruto, C.; Santana, I. Characterization of Progranulin Gene Mutations in Portuguese Patients with Frontotemporal Dementia. Int. J. Mol. Sci. 2023, 25, 511. [Google Scholar] [CrossRef]
- Kurnellas, M.; Mitra, A.; Schwabe, T.; Paul, R.; Arrant, A.E.; Roberson, E.D.; Ward, M.; Yeh, F.; Long, H.; Rosenthal, A. Latozinemab, a Novel Progranulin-Elevating Therapy for Frontotemporal Dementia. J. Transl. Med. 2023, 21, 387. [Google Scholar] [CrossRef]
- Stachel, S.J.; Ginnetti, A.T.; Johnson, S.A.; Cramer, P.; Wang, Y.; Bukhtiyarova, M.; Krosky, D.; Stump, C.; Hurzy, D.M.; Schlegel, K.-A.; et al. Identification of Potent Inhibitors of the Sortilin-Progranulin Interaction. Bioorg. Med. Chem. Lett. 2020, 30, 127403. [Google Scholar] [CrossRef]
- Rosenthal, Z.C.; Fass, D.M.; Payne, N.C.; She, A.; Patnaik, D.; Hennig, K.M.; Tesla, R.; Werthmann, G.C.; Guhl, C.; Reis, S.A.; et al. Epigenetic Modulation through BET Bromodomain Inhibitors as a Novel Therapeutic Strategy for Progranulin-Deficient Frontotemporal Dementia. Sci. Rep. 2024, 14, 9064. [Google Scholar] [CrossRef]
- Peng, X.; Lanter, J.C.; Chen, A.Y.-P.; Brand, M.A.; Wozniak, M.K.; Hoekman, S.; Longin, O.; Regeling, H.; Zonneveld, W.; Bell, R.P.L.; et al. Discovery of Oxazoline Enhancers of Cellular Progranulin Release. Bioorg. Med. Chem. Lett. 2023, 80, 129048. [Google Scholar] [CrossRef]
- Tesla, R.; Guhl, C.; Werthmann, G.C.; Dixon, D.; Cenik, B.; Addepalli, Y.; Liang, J.; Fass, D.M.; Rosenthal, Z.; Haggarty, S.J.; et al. Benzoxazole-Derivatives Enhance Progranulin Expression and Reverse the Aberrant Lysosomal Proteome Caused by GRN Haploinsufficiency. Nat. Commun. 2024, 15, 6125. [Google Scholar] [CrossRef]
- Callizot, N.; Estrella, C.; Burlet, S.; Henriques, A.; Brantis, C.; Barrier, M.; Campanari, M.L.; Verwaerde, P. AZP2006, a New Promising Treatment for Alzheimer’s and Related Diseases. Sci. Rep. 2021, 11, 16806. [Google Scholar] [CrossRef]
- Verwaerde, P.; Estrella, C.; Burlet, S.; Barrier, M.; Marotte, A.-A.; Clincke, G. First-In-Human Safety, Tolerability, and Pharmacokinetics of Single and Multiple Doses of AZP2006, A Synthetic Compound for the Treatment of Alzheimer’s Disease and Related Diseases. J. Alzheimers Dis. 2024, 98, 715–727. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, G.; Lu, Y.; Wu, J.; Ren, C.; Lin, R.; Zhang, C. Progranulin’s Protective Mechanisms and Therapeutic Potential in Cardiovascular Disease. Cells 2025, 14, 762. https://doi.org/10.3390/cells14110762
Qiao G, Lu Y, Wu J, Ren C, Lin R, Zhang C. Progranulin’s Protective Mechanisms and Therapeutic Potential in Cardiovascular Disease. Cells. 2025; 14(11):762. https://doi.org/10.3390/cells14110762
Chicago/Turabian StyleQiao, Gan, Yongxiang Lu, Jianping Wu, Chunyang Ren, Roudian Lin, and Chunxiang Zhang. 2025. "Progranulin’s Protective Mechanisms and Therapeutic Potential in Cardiovascular Disease" Cells 14, no. 11: 762. https://doi.org/10.3390/cells14110762
APA StyleQiao, G., Lu, Y., Wu, J., Ren, C., Lin, R., & Zhang, C. (2025). Progranulin’s Protective Mechanisms and Therapeutic Potential in Cardiovascular Disease. Cells, 14(11), 762. https://doi.org/10.3390/cells14110762