1. Introduction
Hair follicles are specialized micro-organs with complex structures and distinct functions, extending from the follicular bulge to the basal region and consisting of integral components such as the hair germ and dermal papilla (DP) [
1]. The hair follicle passes through several cyclic phases, including the anagen, catagen, and telogen [
2], each characterized by distinct tissue and physiological features governed by extensive gene activation and silencing. The shift from telogen to anagen is characterized by the initiation of new hair shaft formations and the activation of many signaling pathways that regulate the expression of genes related to hair-specific keratin production, inner root sheath development, and pigmentation [
3].
MicroRNAs (miRNAs) are small, non-coding RNA molecules, generally around 24 nucleotides long, that are generated endogenously by cells. These molecules attach to complementary sequences in the 3′ untranslated region (3′UTR) of target mRNAs to inhibit translational expression, elucidating its regulatory role [
4]. Research has shown that microRNAs (miRNAs) are essential regulators of hair follicle growth and development.
miR-205 exhibits significant differences between growth and regression phases in mice, influencing hair follicle cycle transitions [
5]. Cao et al. posited that
miR-100 derived from exosome-mimetic nanovesicles (ReN-NVs) produced by neural progenitor cells enhances nuclear
β-
catenin expression, inhibits several Wnt negative regulators, and elevates
C-myc and
Cyclin D1 (
CCND1) levels, thereby facilitating the acceleration of hair follicle growth. Additionally,
miR-99/100 and let-7a/c have been identified as pivotal regulators for the regeneration of cardiomyocytes [
6]. Moreover, it has been established that
miR-218 regulates the Wnt signaling pathway by inhibiting the expression of the
SFRP2 gene, consequently affecting apoptosis and resulting in significant variations in rabbit hair length. This finding provides a foundation for future investigations on
miR-218′s function in hair follicle growth [
7].
In our prior research, we sequenced differentially expressed miRNAs from the skin tissues of Xinji fine-wool sheep and Small-tail Han sheep, each exhibiting unique wool characteristics, and revealed the differential expression of
miR-370-3p. Bioinformatic investigations indicated that SMAD family member 4 (
SMAD4) may be a target gene of
miR-370-3p. Moreover, a mechanism underlying the growth/rest switch in hair follicles has been proposed, which involves endothelial glycogen-dependent cross-signaling between the Wnt/β-catenin and BMP/Smad pathways [
8]. Studies have already shown that
SMAD4 and
SMAD7 are essential regulators of the formation and development of hair follicles. The bone morphogenetic protein (BMP) signaling pathway primarily mediates the effect of
SMAD4 on follicular development, whereas
SMAD7 disrupts TGF-β/activin/BMP signaling and promotes ubiquitin-mediated degradation of β-catenin, hence inhibiting the Wnt/β-catenin signaling cascade. These activities substantially influence follicular growth and differentiation, and their study demonstrated that
SMAD2 and
SMAD4 cooperatively regulate epidermal cell homeostasis via the TGF-β pathway [
9]. Owens et al. suggested that the loss of the
SMAD4 gene contributes to partial degeneration of hair follicles in the skin [
10], while Yang et al. highlighted the critical function of
SMAD4 in maintaining the shape of hair follicle stem cells [
11]. Yang et al. [
11] also highlighted the vital role of
SMAD4 in preserving the structural integrity of hair follicle stem cells, whereas Owens et al. [
10] proposed that the absence of the
SMAD4 gene leads to the deterioration of cutaneous hair follicles.
This study aimed to confirm the relationship between miR-370-3p and its target gene SMAD4 and to explore the specific mechanism by which they regulate hair follicle growth, development, and cycling, providing a theoretical basis for the miRNA regulation of dermal papilla cell (DPC) growth and development.
2. Materials and Methods
2.1. Ethical Statement
Three healthy Xinji fine-wool sheep and three healthy Small-tail Han sheep of the same age were selected from the Animal Husbandry Branch of Jilin Academy of Agricultural Sciences. Professional experimenters conducted regular checks on the health status of the experimental animals. To minimize environmental influences, both groups of sheep were kept in the same environment and fed with the same feed. Skin tissue samples were harvested during the anagen phase (October) and the telogen phase (January) of the hair follicle cycle for examination. The livestock collection was carried out following the regulations authorized by the Animal Welfare and Ethics Committee of Jilin Academy of Agricultural Sciences (JNK20210901001, 11 September 2021). Moreover, the international animal welfare guidelines were strictly followed, and euthanasia was performed on the experimental sheep. The drug used for euthanasia was sodium pentobarbital at a dosage of 120 mg/kg. All of the euthanasia procedures and sample collection were conducted by professionally trained and experienced experimental personnel to ensure the accuracy of the operation process and minimize pain for the animals.
2.2. Sample Collection
One-year-old female Small-tail Han and Xinji fine-wool sheep that were not in estrous were used in this study. They were chosen from the Jilin Academy of Agricultural Sciences’ Animal Husbandry Institute. After shearing the wool around the scapular region, the area was disinfected with 75% alcohol. Local anesthesia was administered by subcutaneous injection of lidocaine hydrochloride (Jianmin Pharmaceuticals, Wuhan, China). Under the guidance of professional experimenters, three 1-cm2 skin tissue samples were collected from each sheep. These three skin tissue samples were used for different experiments. The first skin tissue samples were placed in PBS containing 1% penicillin/streptomycin (Bioss, Beijing, China) and stored at 4 °C for DPC isolation. The second skin tissue samples were preserved in 4% paraformaldehyde (Biosharp, Hefei, China) and subsequently stored at room temperature for immunohistochemical analysis. In the third set of samples, the skin, heart, liver, spleen, lungs, kidneys, and muscle tissues were promptly snap-frozen in liquid nitrogen and stored at −80 °C for total RNA and miRNA isolation.
2.3. DPC Isolation and Culture
The skin tissues were subjected to washing with 75% ethanol, followed by three consecutive rinses with PBS containing 1% penicillin/streptomycin. This procedure was performed a total of three times. After washing, the hair shafts and subcutaneous fat layers were removed. DPCs from Small-tail Han sheep were isolated, cultured, and characterized in accordance with the protocols previously developed in our laboratory [
12]. The cells were kept in a CO
2 incubator (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C with 5% CO
2 in DMEM/F12 (BI, Shanghai, China) supplemented with 10% fetal bovine serum (Cell-Box, Hong Kong, China) and 1% penicillin/streptomycin solution.
2.4. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
The RNAeasyTM Animal RNA Extraction Kit (centrifugal column method) (Beyotime, Shanghai, China) was utilized for total RNA extraction. The concentration and purity of the RNA samples were evaluated using a spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA); and all samples tested had OD260/280 values of 1.8–2.0. Following this, genomic DNA was eliminated, and first-strand cDNA was produced with the HiFiScript Quick gDNA Removal cDNA Synthesis Kit (Cwbio, Taizhou, China), and the resultant cDNA was preserved at −20 °C. Gene expression measurement was conducted with the GoTaq® qPCR Master Mix (Promega, Beijing, China). The 20 μL reaction mixture comprised 10 μL of 2 × GoTaq® qPCR Master Mix, 0.4 μL of each forward and reverse primer, 2 μL of cDNA, and 7.2 μL of Nuclease-Free Water, Waltham, MA, USA. The PCR amplification was performed using the following parameters: an initial denaturation at 95 °C for 10 min, followed by 45 cycles of denaturation at 95 °C for 15 s, and annealing/extension at 60 °C for 1 min. β-actin was employed as the internal reference gene for normalization in the qPCR analysis.
Total miRNA was isolated using the miRcute miRNA Isolation Kit (TIANGEN, Beijing, China), according to the manufacturer’s instructions to ensure the efficient recovery of high-quality miRNA from tissue samples. First-strand cDNA was synthesized using the miRcute Enhanced miRNA cDNA First Strand Synthesis Kit (TIANGEN, Beijing, China), according to the manufacturer’s guidelines. The generated cDNA was subsequently preserved at −20 °C for future examination. The expression of miRNA was measured using the miRcute Enhanced miRNA Fluorescence Quantification Kit (TIANGEN, Beijing, China), adhering to the manufacturer’s guidelines, to precisely assess miRNA levels in the samples. The 20 μL reaction mixture consisted of 10 μL of 2 × PreMix (SYBR and ROX), 0.4 μL of forward primer (200 nM), 0.4 μL of reverse primer (200 nM), 1 μL of cDNA, and 8.2 μL of nuclease-free water. The PCR amplification method comprises an initial denaturation step at 95 °C for 15 min, followed by 5 cycles of denaturation at 94 °C for 20 s, annealing at 65 °C for 30 s, and elongation at 72 °C for 34 s. This was followed by 45 cycles of denaturation at 94 °C for 20 s and annealing at 60 °C for 34 s. U6 small nuclear RNA functioned as the internal control, utilizing the U6 forward primer sourced from Sangon Biotech (Shanghai, China) to normalize the miRNA expression data and reduce any variations in RNA input.
The expression levels of the target genes or miRNAs were quantified using the 2
−ΔΔCt method. All primers employed for qRT-PCR are listed in
Table 1.
2.5. 3′-UTR Luciferase Reporter Assay
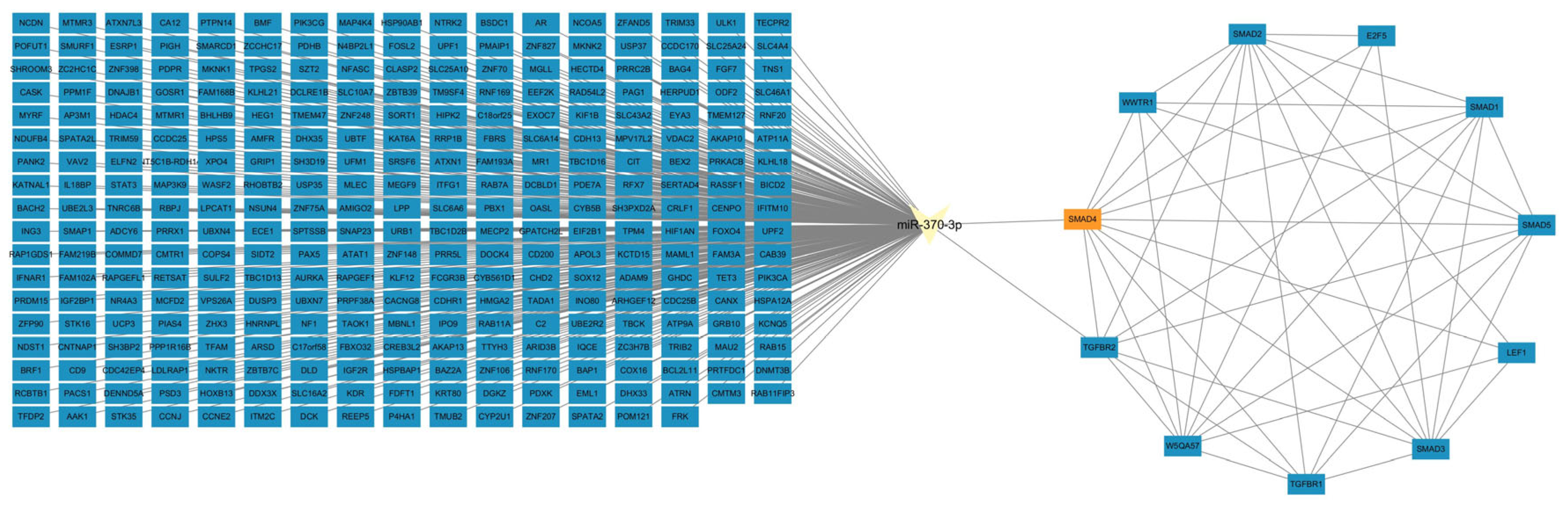
The target genes of miR-370-3p were identified utilizing four internet platforms: miRDB, miRWalk, TargetScan, and starBase. We selected the intersection of the prediction results from these four platforms, and among the genes in the intersection, SMAD4 was chosen for further investigation. The protein–protein interactions of SMAD4 were examined utilizing the web program STRING, and an miRNA–mRNA–mRNA interaction network was developed with Cytoscape 3.9.1.
The interaction sites between
miR-370-3p and
SMAD4 were predicted using RNAhybrid. According to the expected binding sites, plasmids were designed and constructed:
miR-370-3p-mimics, mimics negative control (NC), pmirglo-
miR-370-3p-
SMAD4-WT, and pmirglo-
miR-370-3p-
SMAD4-Mut. The constructs were co-transfected into HEK-293T cells using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). Samples were collected 48 h post-transfection. The assays employed a luciferase reporter gene kit (Promega, Beijing, China). The mutation sites are shown in
Table 2.
2.6. Cell Transfection
The miR-370-3p mimic, mimic negative control (miR-370-3p mimic-NC), miR-370-3p inhibitor, and inhibitor negative control (miR-370-3p inhibitor-NC) (GenePharma, Shanghai, China) were transfected into DPCs utilizing Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). DPCs were then uniformly seeded into a 6-well plate and allowed to proliferate until they reached 75% confluence with a complete growth medium. Transfection was subsequently executed utilizing Lipofectamine™ 2000 (all procedures were carried out under dim lighting). Subsequently, 3.75 μL of mimic or inhibitor was combined with 7.5 μL of Lipofectamine™ 2000 in 125 μL of Opti-MEM™ I reduced-serum medium and incubated at ambient temperature for 5 min. The Lipofectamine 2000 solution was thereafter mixed with the mimic or inhibitor combination and incubated at room temperature for 25 min. The incubated mixture was thereafter poured dropwise into each well of the 6-well plate, at a volume of 250 μL per well, and swirled gently. Subsequent to transfection, cells in the 6-well plates were cultured at 37 °C with 5% CO2 for 48 h before samples were collected for total RNA and total protein extraction.
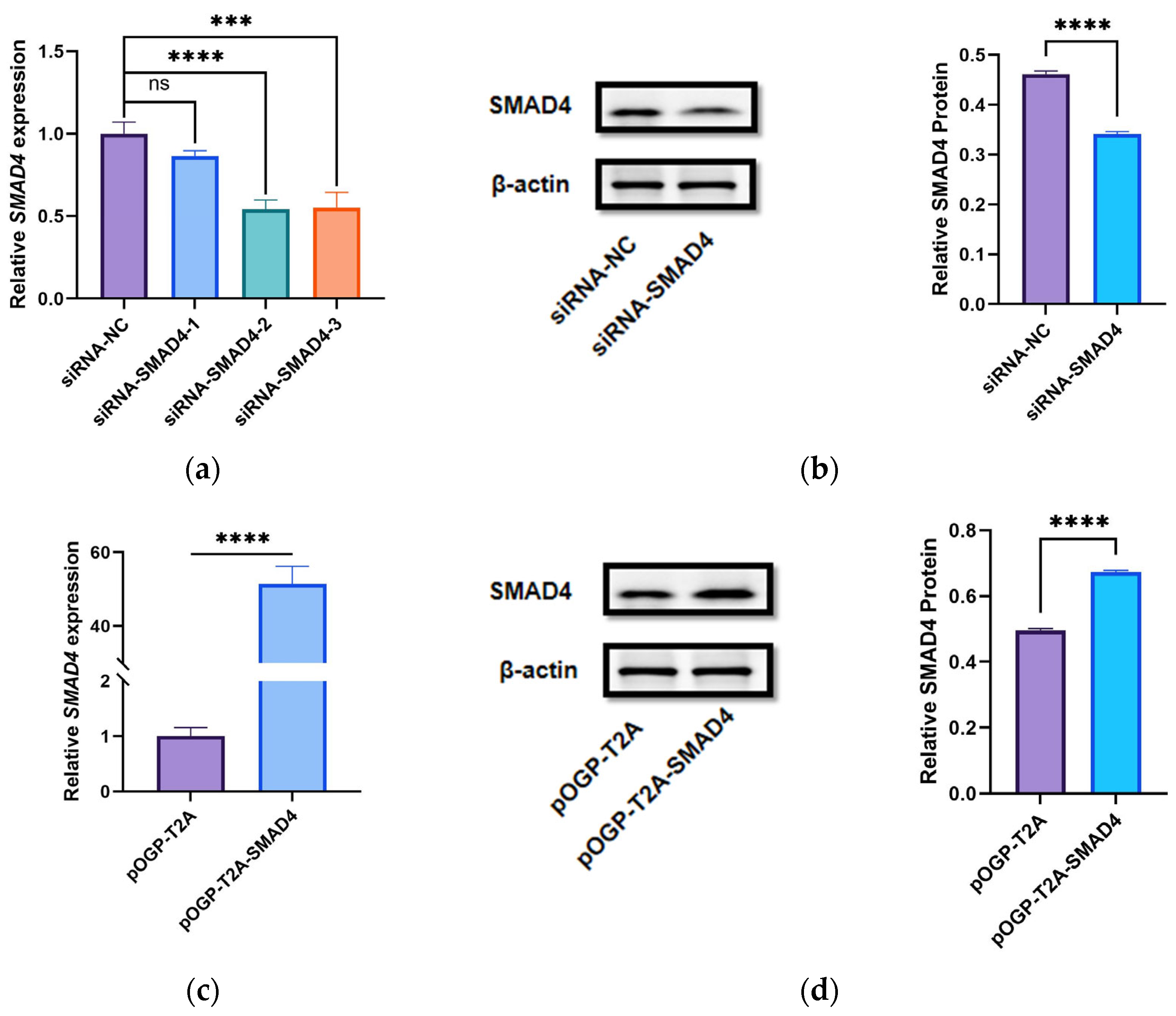
siRNA-
SMAD4, siRNA negative control (siRNA-NC), pOGP-T2A, and pOGP-T2A-
SMAD4 (pcDNA3.1, JST Scientific, Wuhan, China) were electroporated into DPCs using CUY21 EDIT II (BEX, Tokyo, Japan). Growth-arrested DPCs were seeded into culture bottles and cultured until reaching 90% confluence, with the complete medium being replaced by cell culture medium at 37 °C in a cell culture incubator with 5% CO
2 for 24 h. Following two PBS rinses, the cells were digested with 0.25% trypsin, centrifuged for five minutes at 1500×
g, and the supernatants removed. Cells were resuspended in 2 mL of Opti-MEM™ I reduced-serum medium, centrifuged at 1500×
g for 5 min, and the supernatants were then discarded. Cells were then resuspended in 2 mL of Opti-MEM™ (Invitrogen, Carlsbad, CA, USA) I reduced-serum medium, stained with trypan blue, counted using a cell counter, and 1 × 10
6 cells were removed and resuspended in 100 μL of Opti-MEM™ I reduced-serum medium. Subsequently, 1 μg of plasmid DNA was added, mixed thoroughly, and transferred into an electroporation cuvette. Electroporation parameters were set, and the cuvette was placed in the electroporator, and after verifying acceptable resistance, electroporation was performed. Post-electroporation, the cells were aspirated back into the culture bottle using a pipette and supplemented with cell culture medium. Fluorescence was detected using a fluorescence microscope after 24 h, and cells were collected 48 h post-electroporation for total RNA and total protein extraction. All sequences utilized are enumerated in
Table 3.
2.7. Western Blot
Skin tissue samples were collected and immersed in liquid nitrogen prior to being pulverized in a mortar. Following the lysis of the powder using RIPA tissue/cell lysis buffer (Solarbio, Beijing, China) and subsequent storage at 4 °C, the supernatant was obtained using centrifugation at 2000 rpm for five minutes. The culture media were withdrawn after 48 h of cell transfection, and the cells were lysed using a pre-cooled RIPA lysis solution containing 1% PMSF. The supernatants were subsequently collected. The protein content was determined using the BCA Protein Assay Kit (Cwbio, Taizhou, China).
Proteins were fractionated by SDS-PAGE (10%) and subsequently transferred to PVDF membranes (Sigma Aldrich, Shanghai, China). Blocking was performed using 5% Blotting Grade (Beyotime, Shanghai, China). The chemiluminescent reaction was conducted using Super ECL Plus (UElandy, Suzhou, China), prepared according to a specified ratio. The primary antibodies utilized were SMAD4 (1:1000, Proteintech, Wuhan, China), β-actin (1:2000, Proteintech, Wuhan, China), CCND1 (1:10,000, Proteintech, Wuhan, China), CCND2 (1:1000, Proteintech, Wuhan, China), c-MYC (1:10,000, Proteintech, Wuhan, China), and β-catenin (1:10,000, Proteintech, Wuhan, China).
2.8. Cell Counting Kit-8 (CCK-8) Assay
DPCs were uniformly seeded in a 96-well plate at a density of 1 × 103 cells per well. Cells underwent a 24 h incubation prior to transfection. Each well was administered 100 µL of fresh culture medium (DMEM/F12 + 10% FBS) supplemented with 10% CCK-8 (Beyotime, Shanghai, China) at 24, 48, 72, and 96 h post-transfection. The wells were subsequently incubated for two hours at 37 °C in darkness. A SpectraMax iD5 microplate reader (Molecular Devices, San Jose, CA, USA) was employed to measure absorbance at 450 nm to assess the reaction intensity and furnish quantitative data for further cellular response analysis.
2.9. Flow Cytometry Assay (FCM)
DPCs were inoculated in 6-well plates and grown until achieving 70–90% confluence prior to transfection. After 48 h of transfection, the progression of the cell cycle in DPCs was evaluated using a Cell Cycle and Apoptosis Detection Kit (Beyotime, Shanghai, China), following the manufacturer’s guidelines. Additionally, apoptosis in the DPCs was assessed using an Annexin V-FITC Apoptosis Detection Kit (Beyotime, Shanghai, China), following the provided protocol. Flow cytometry (Becton, Dickinson, and Company, Queensbury, NY, USA) was employed for comprehensive analysis of cell cycle distribution and apoptosis in DPCs following transfection, allowing for precise quantification and characterization of cellular responses.
2.10. Immunohistochemistry
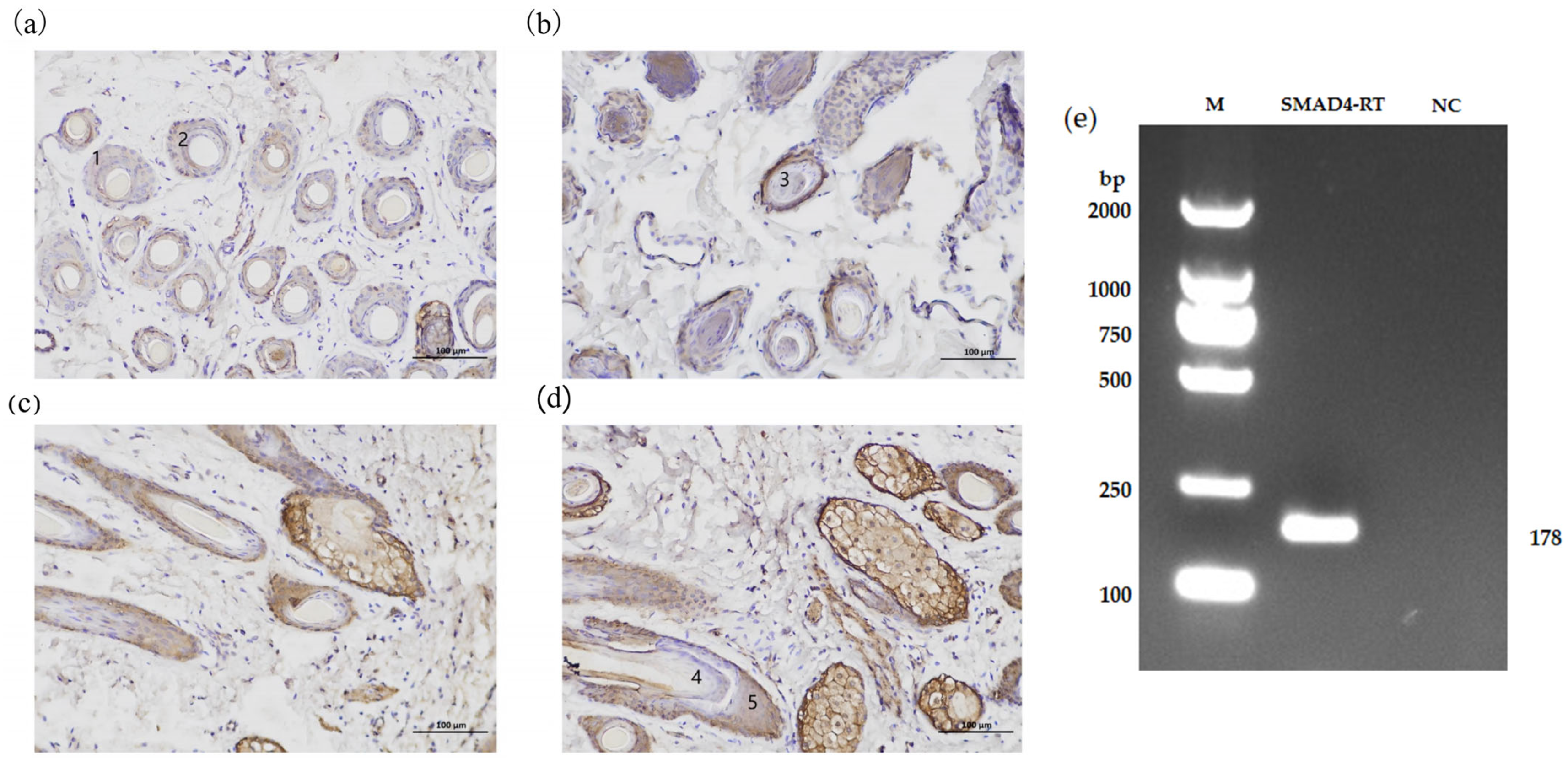
Skin tissue samples from Xinji fine-wool sheep and Small-tail Han sheep were harvested during both the growth and resting phases and thereafter preserved in 4% paraformaldehyde (Biosharp, Hefei, China) for 48 h to preserve tissue morphology for subsequent analysis. After trimming to the appropriate size, the skin tissues were washed in embedding cassettes for 24 h. Following dehydration and paraffin embedding, sections were taken at ambient temperature. The slides were incubated in a 3% hydrogen peroxide solution (ANNJET, Dezhou, China) in the dark at ambient temperature for 25 min to suppress endogenous peroxidase activity. Thereafter, non-specific binding sites were blocked by incubating the slides with 3% BSA (Servicebio, Wuhan, China) at ambient temperature for 30 min. The SMAD4 basic antibody (1:100, Proteintech, Wuhan, China) was diluted in PBS (Servicebio, Wuhan, China) and administered to the slides, followed by overnight incubation at 4 °C. Following PBS washing, the slides were incubated at room temperature for 50 min with an HRP-conjugated goat anti-mouse IgG secondary antibody (1:200, Servicebio, Wuhan, China). The reaction was visualized using DAB chromogenic reagent (Servicebio, Wuhan, China) under a microscope (E100, Nikon, Japan).
2.11. Statistical Analyses
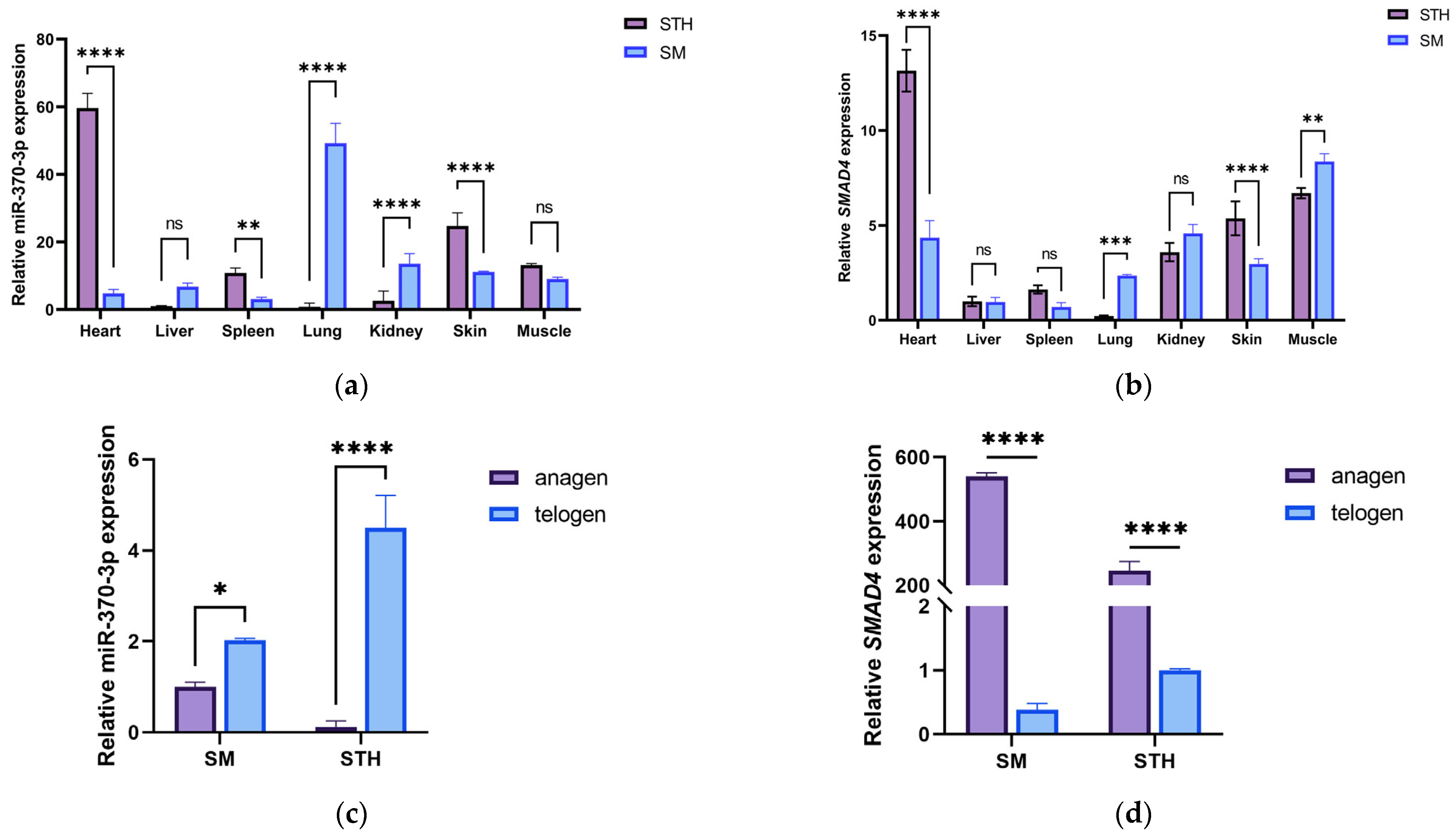
All data are presented as mean ± standard deviation (SD). Statistical analyses were conducted using SPSS 26 software (IBM, Chicago, IL, USA), with significance evaluated by t-tests or one-way ANOVA, subsequently followed by LSD and Duncan’s post hoc tests. Graphical representations and data analysis were performed using GraphPad Prism 9, the latest version (GraphPad Software, San Diego, CA, USA). Differences were considered nonsignificant (ns) when p > 0.05, with * p < 0.05 indicating significance, ** p < 0.01 indicating high significance, *** p < 0.001 indicating very high significance, and **** p < 0.0001 indicating extreme significance.
4. Discussion
Numerous cell types, including DPCs and hair follicle stem cells (HFSCs), carefully regulate the growth and development of hair follicles. HFSCs are vital for sustaining the long-term regeneration potential of hair follicles, whereas DPCs deliver crucial signals that direct follicular morphogenesis, cycling, and differentiation. Together, these cell populations interact within a highly coordinated microenvironment to ensure proper follicular function and hair production. Wnt/
β-catenin, Notch, Sonic Hedgehog (Shh), and BMP are among the several signaling pathways that control the growth and differentiation of hair follicles. These pathways control key processes like cell proliferation and differentiation, ensuring proper hair follicle function [
13]. The genetic regulatory network governing hair follicle development is highly complex, encompassing a range of signaling pathways, including Wnt, BMP, EDAR, and Shh. These pathways function interactively within the follicular microenvironment, coordinating cellular processes that regulate follicle morphogenesis and cycling [
2,
14]. MicroRNAs influence the biological processes of cancer, including invasion, metastasis, apoptosis, and proliferation, by controlling significant signaling pathways such as PI3K/Akt, MAPK, and Wnt [
15].
MicroRNAs (miRNAs) are diminutive, non-coding RNA molecules, typically around 24 nucleotides in length, that regulate gene expression post-transcriptionally. They affect outcomes by binding to specific areas in the 3′UTR of target mRNAs, leading to either mRNA destruction or translational repression. MiRNAs have a vital role in regulating multiple biological processes, including cell differentiation, proliferation, and apoptosis, through these pathways [
16]. They provide crucial regulatory functions in organismal growth and development, and research has underscored the important functions of
miR-370-3p in disorders like myocardial infarction, heart failure, lung cancer of non-small cells, and pneumonia [
17,
18,
19]. Moreover,
miR-370-3p has been linked to the regulation of tumors and malignancies. Jia et al. demonstrated that
CircCCNB1 suppresses cervical cancer proliferation through the
miR-370-3p/
SOX4 pathway, emphasizing the significance of circular RNAs (circRNAs) in the regulation of cancer-related signaling pathways [
20]. This study highlights the crucial function of
miR-370-3p in facilitating the regulatory impact of
circCCNB1 on carcinogenic mechanisms. Additionally, it has been suggested that
circ_0025033 facilitates the advancement of ovarian cancer by modulating the has-
miR-370-3p/
SLC1A5 pathway. This underscores the regulatory role of circular RNAs (circRNAs) in influencing miRNA activity, which in turn affects critical cellular processes such as amino acid transport. Such regulation may be integral to tumor growth and metabolism, thus highlighting the complex molecular interactions contributing to ovarian cancer progression [
21]. Experimental evidence by Li et al. confirmed that
circARID1A regulated glioblastoma migration and invasion via the
miR-370-3p/
TGFBR2 axis [
22]. Additionally,
miR-370-3p is implicated in the control of several physiological processes in rats, and studies have indicated its role in the regulation of goat hair follicle development. Considering the notable disparities in wool fiber diameter between Xinjiang fine-wool sheep and Tan sheep, together with prior research on
miR-370-3p in goats conducted by Haierhan, it has been deduced that
miR-370-3p may be linked to follicles for hair growth and development. This study gathered skin samples from sheep in October (hair follicle growth phase) and January (hair follicle resting phase) for fluorescence quantitative PCR, revealing significant differences in spatiotemporal expression of
miR-370-3p in skin tissues. This suggests that
miR-370-3p may have a regulatory role in the growth, cycle, and phenotype of sheep hair follicles. The analysis of
miR-370-3p tissue expression demonstrated significant variation in expression patterns between the two sheep breeds. Target gene validation using dual-luciferase reporter assays and detection at the gene and protein levels confirmed
SMAD4 as a target for
miR-370-3p, consistent with previous studies. Subsequent transient transfection assays on DPCs confirmed the targeting efficacy of
miR-370-3p on
SMAD4, leading to the downregulation of
SMAD4 expression.
Prior studies have highlighted the pivotal function of the SMAD family in regulating hair follicle cycling and the differentiation of associated cells, hence indicating its significance in the molecular pathways governing hair follicle growth and homeostasis.
SMAD2 has been suggested to modulate hair follicle development and growth in Angora rabbit dermal tissues through the TGF-
β signaling pathway, underscoring the critical role of SMAD family members in hair follicle dynamics [
23]. Research demonstrates that
miR-203a-3p inhibits
SMAD1 expression, hence facilitating the process of differentiation of HFSCs induced by loureirin A. This shows that
miR-203a-3p is pivotal in regulating HFSC differentiation by modulating the
SMAD1 signaling pathway [
12]. Nan et al. [
24] revealed that All-trans-retinoic acid (
ATRA) reduces DPC proliferation and promotes apoptosis via the TGF-β2/Smad2/3 signaling pathway, therefore decreasing hair follicle development. This finding underscores the regulatory influence of
ATRA on hair follicle dynamics via modulation of essential biological mechanisms, including growth and apoptosis.
SMAD7 is acknowledged as an antagonist of Wnt/β-catenin signaling, and its activation can alter the differentiation of skin cells from hair follicle production to sebaceous gland development. This indicates that
SMAD7 is essential in regulating the equilibrium between hair follicle and sebaceous gland development by opposing the Wnt/β-catenin pathway, therefore affecting skin appendage differentiation [
25]. Li et al. proposed that downregulation of the transcription factor
SMAD3 inhibits TGF-β signaling, inducing premature entry and prolongation of dormant hair follicles into the growth phase [
26]. These findings imply that the
SMAD4 protein may influence the hair follicle cycle and associated cellular phenotypes, thereby contributing to variations in wool traits. This suggests that
SMAD4 may regulate critical processes, including follicular differentiation, growth, and development, which are vital for defining wool characteristics. Additional study is necessary to clarify the specific molecular pathways by which
SMAD4 influences hair follicle dynamics and its possible impact on wool phenotypes. Research has demonstrated that the ablation of the
SMAD4 gene leads to the deterioration of hair follicles in the epidermis, highlighting its essential role in regulating hair follicle growth and development [
10,
27].
SMAD4 is essential for the proper development of hair follicles, and its functional deletion in mice disrupts the normal hair follicle cycle. This disturbance can result in abnormal skin development, thereby elevating the risk of skin cancers or carcinomas [
27,
28].
Park et al. suggested that the
Wip1 gene inhibits TGF-β signaling by regulating
SMAD4 phosphorylation [
29]. Wang et al. demonstrated that the inhibition of
TGF-β1 gene expression leads to reduced
SMAD4 phosphorylation, hence promoting apoptosis and impeding proliferation of cells, assault, and the transition from epithelial to mesenchymal in gallbladder cancer. The results highlight the crucial role of the TGF-β/
SMAD4 signaling pathway in regulating vital cellular processes in cancer growth and suggest potential therapeutic options for targeting this pathway in gallbladder cancer [
30]. Structural analyses reveal that the MH1 and MH2 modules of
SMAD4 are linked by a flexible region, enabling dynamic conformational changes. This flexibility is crucial for the protein’s function, as it facilitates transient interactions with other SMAD proteins and transcriptional regulators, thereby modulating its role in TGF-β signaling and cellular responses [
31]. These results indicate that
SMAD4 may modulate cell phenotypes by affecting cellular processes vital for the organism’s physiological functioning, such as proliferation, differentiation, and death. This regulation likely contributes to tissue development and overall homeostasis.
To elucidate the regulatory function of the SMAD4 gene in sheep, real-time quantitative PCR (qPCR) was utilized to assess the expression levels of SMAD4 across different tissues in Small-tail Han sheep and Xinji fine-wool sheep. We found that there were significant differences in heart, spleen, skin, lung, and muscle tissues between the two breeds. Considering the substantial disparities in wool fiber diameter and hair follicle density between Small-tail Han sheep and Xinji fine-wool sheep, together with the pronounced variations in SMAD4 gene expression in the skin tissues of both breeds in January (hair follicle growth phase), We hypothesize that the SMAD4 gene may be involved in the regulation of hair follicle growth and development, aligning with predictions derived from protein interaction analysis. Immunohistochemistry results further validated the expression of the SMAD4 protein in multiple parts of the hair follicle, confirming our hypothesis. This study validated the expression of the SMAD4 gene in several sheep tissues, underscoring its essential role in hair follicle growth and development. Future research will employ DPCs from this study to analyze the regulatory effects of miR-370-3p and SMAD4 genes on cell phenotypes and the mechanisms governing hair follicle cell cycles in Tan sheep.
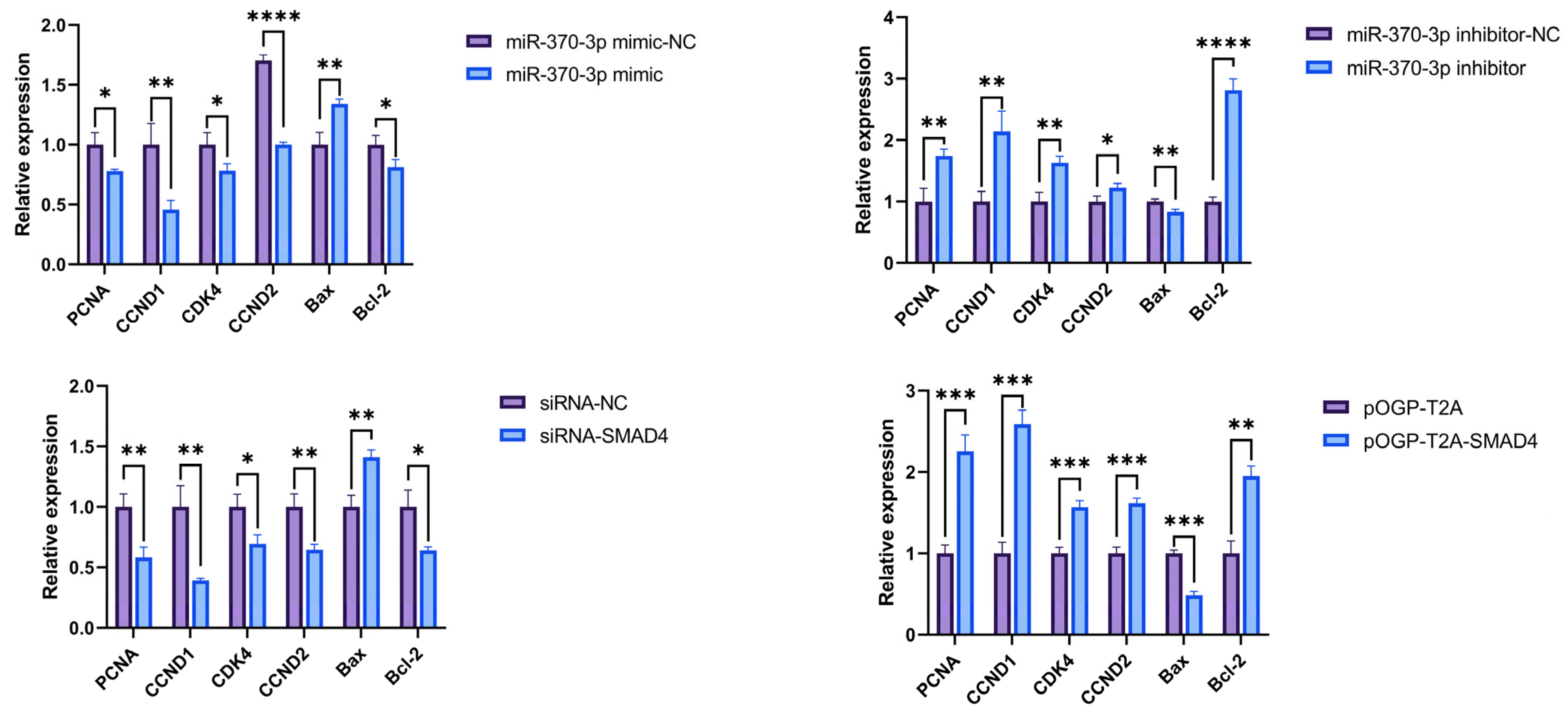
Through RT-qPCR, cell phenotype-related genes (
PCNA,
CCND1,
CDK4,
CCND2,
Bax,
Bcl-2) were preliminarily determined to be affected by
miR-370-3p in DPCs, influencing cell proliferation, cycle, and apoptosis, possibly through targeting
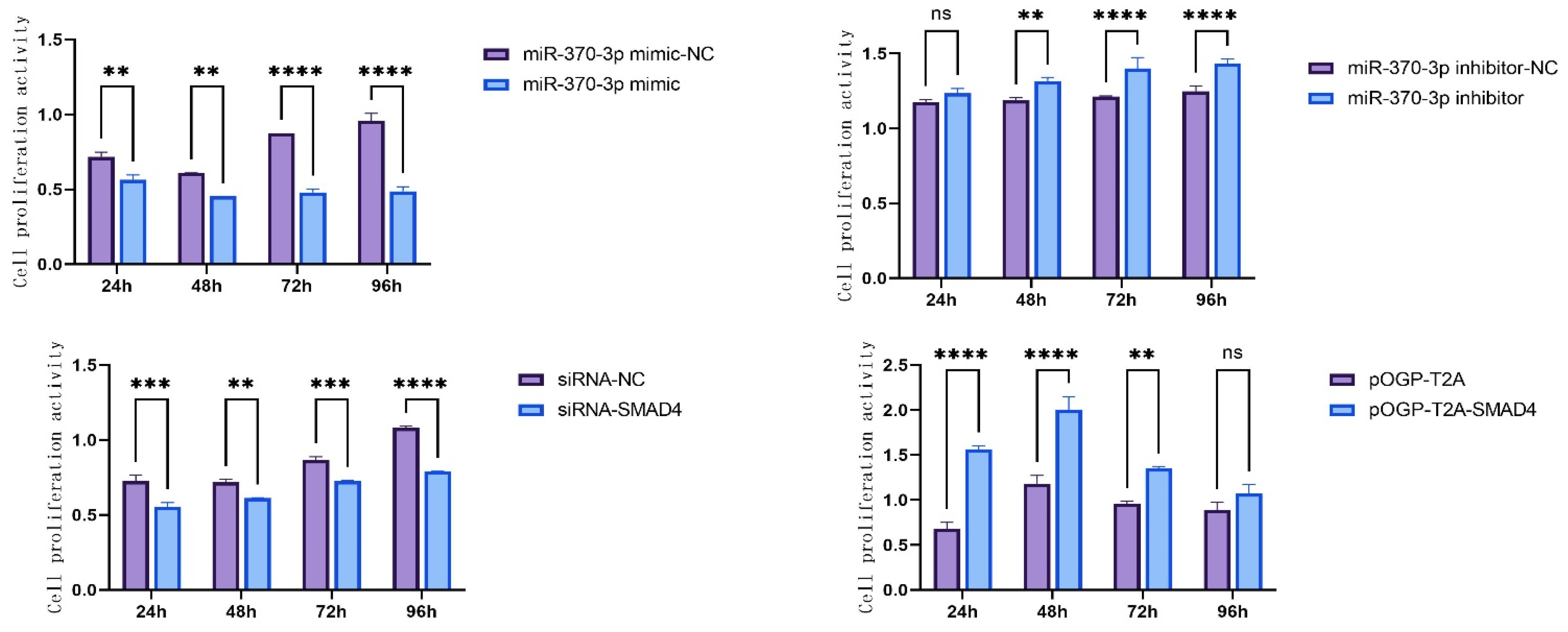
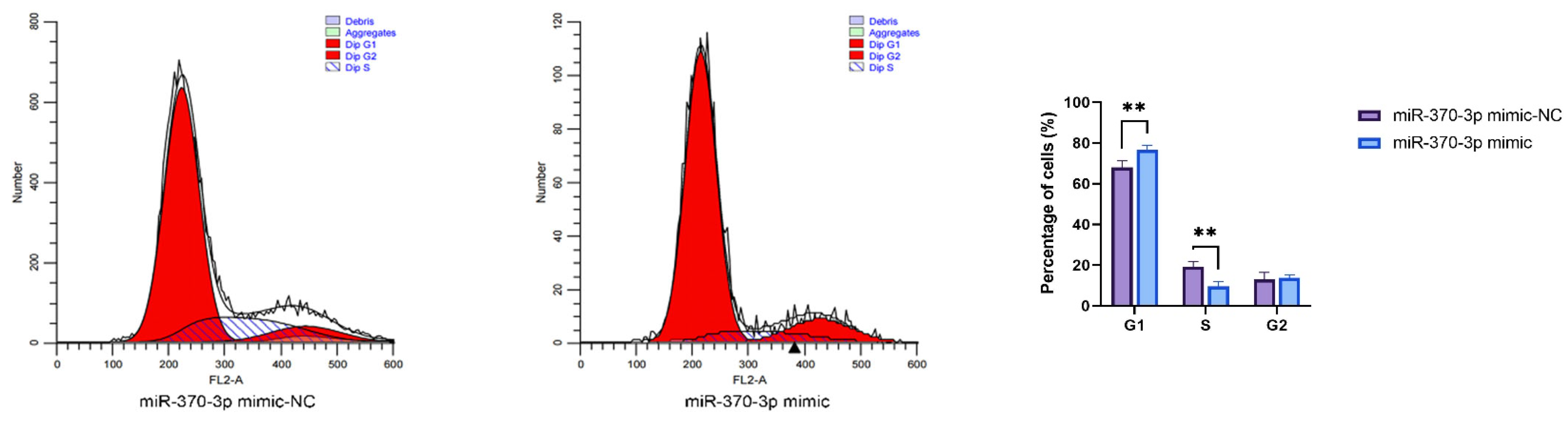
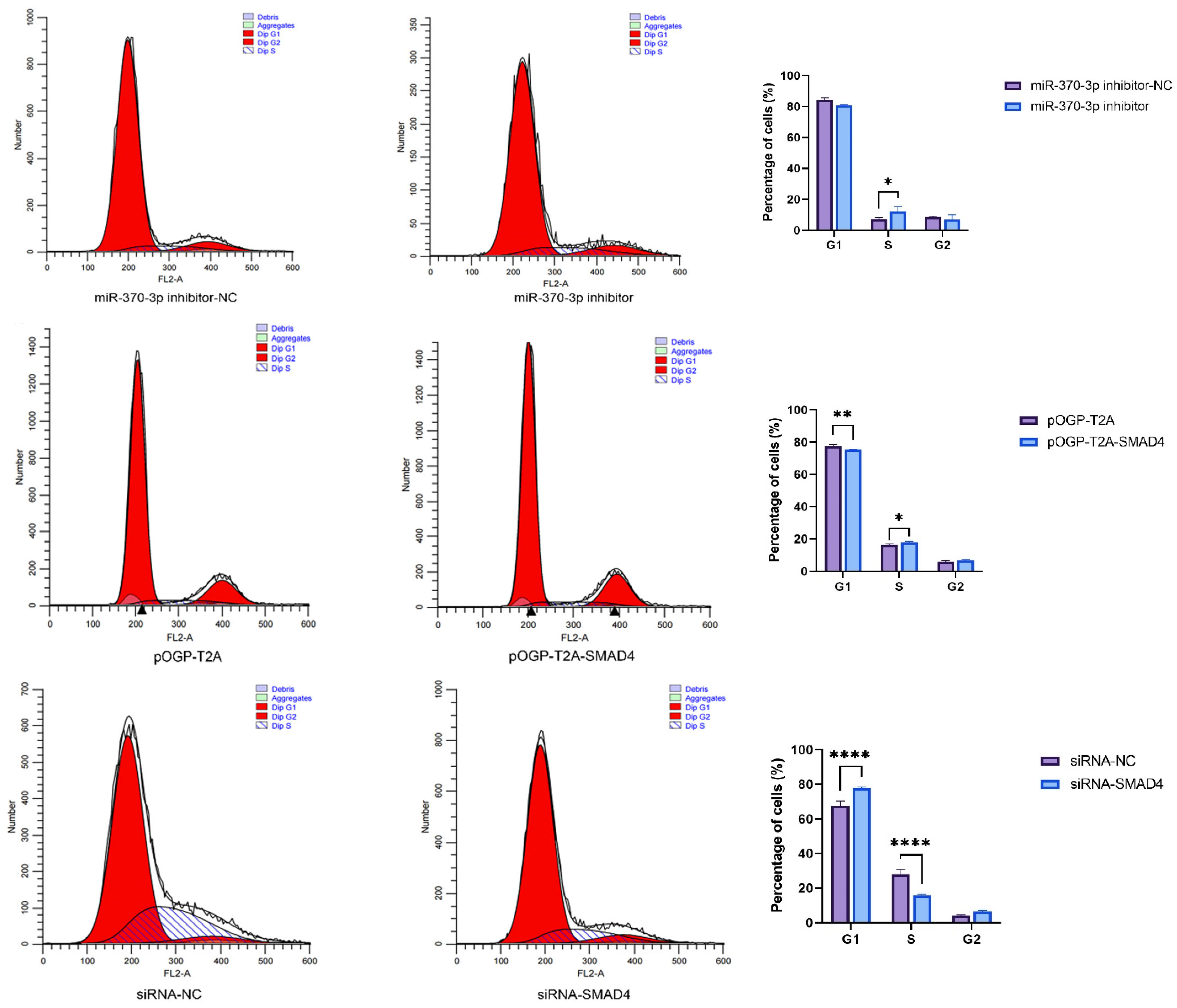
SMAD4. This study utilized CCK-8 assays and flow cytometry to substantiate these hypotheses, demonstrating that
miR-370-3p inhibits DPC proliferation and affects the cell cycle, perhaps by regulating related genes, consistent with previous research [
32]. Research using Annexin V-FITC and PI dual staining demonstrated that
miR-370-3p promotes cellular death. In contrast,
SMAD4 affects DPC phenotype differently; it promotes DPC proliferation, inhibits apoptosis, and has a certain effect on the cell cycle. Overall, it is inferred that
miR-370-3p participates in regulating DPC cell phenotypes by targeting
SMAD4.
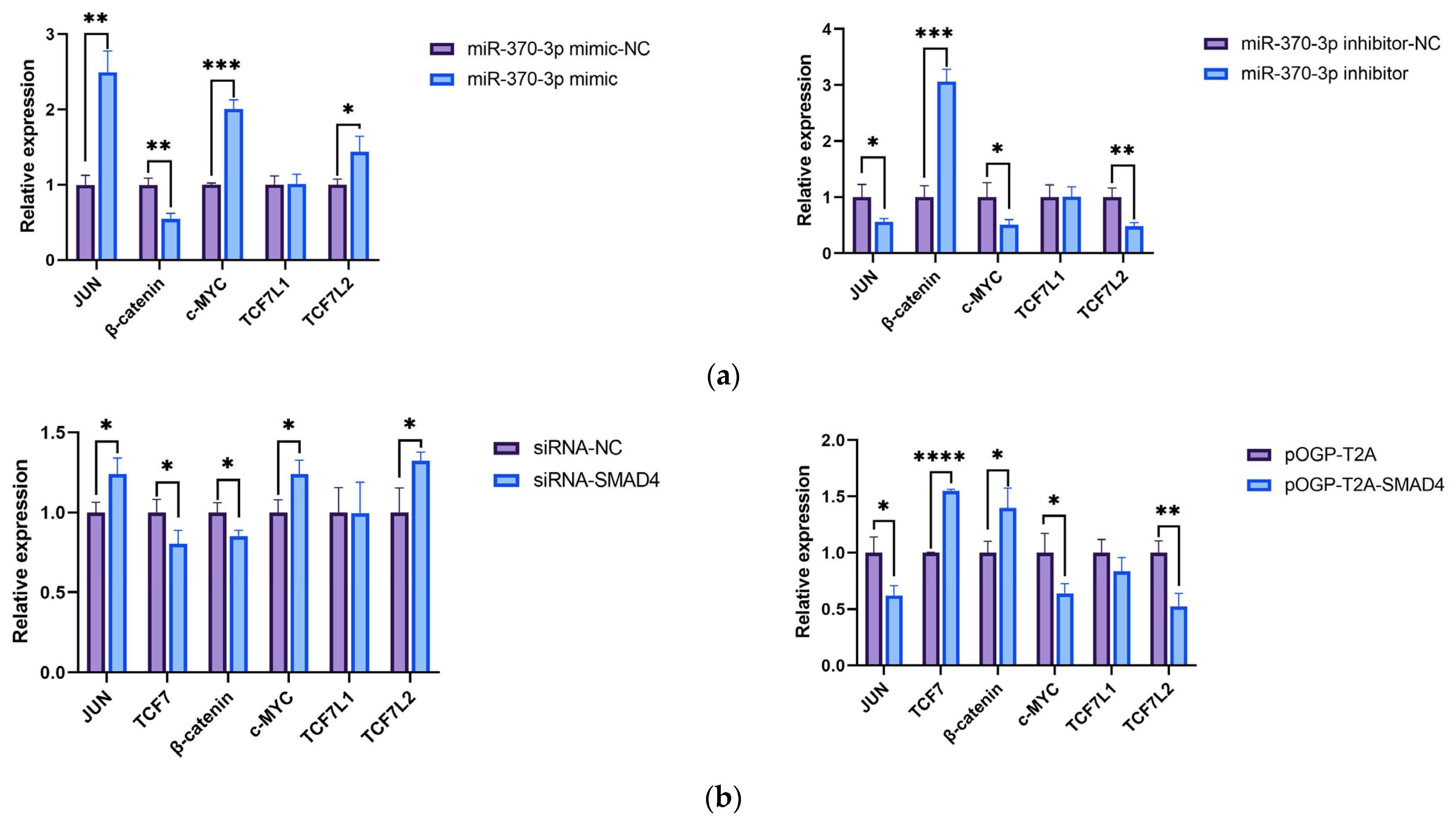
Currently, there is no relevant research on the effects of
miR-370-3p and
SMAD4 on sheep dermal papilla cells (DPCs). In this study, to explore the molecular mechanism by which
miR-370-3p regulates
SMAD4 and affects the development and growth of DPCs, the expression levels of relevant genes in DPCs were detected. The results showed that the targeting of
SMAD4 by
miR-370-3p could affect the expression of relevant genes such as
JUN,
c-MYC, and
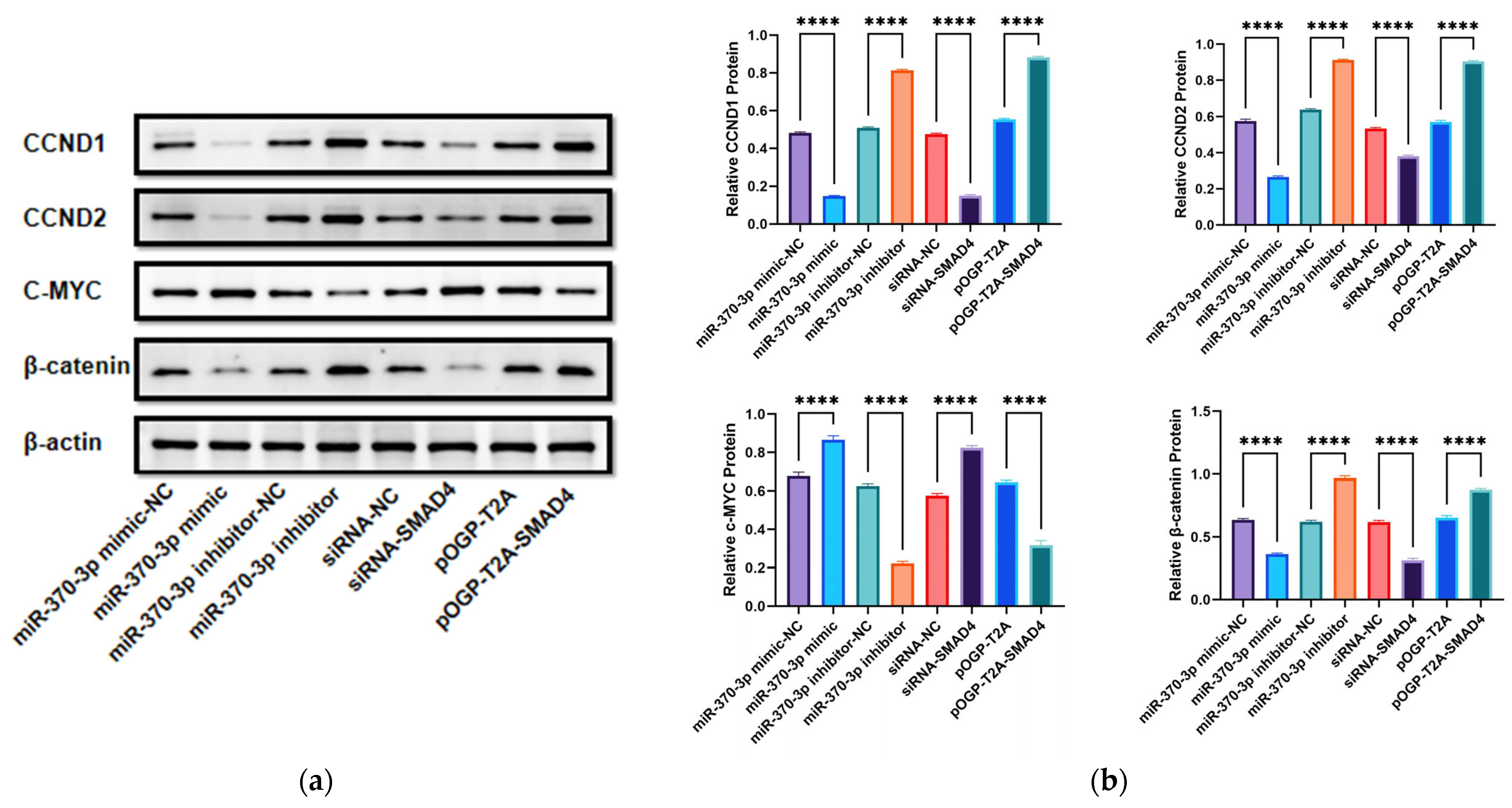
β-catenin. To further validate the results of real-time quantitative polymerase chain reaction (RT-qPCR), we selected several key proteins (cyclin D1 (CCND1), cyclin D2 (CCND2), β-catenin, and c-MYC) for Western blot analysis. The results were consistent with those of RT-qPCR. I would argue that
SMAD4 participates in several biological processes in cross-talk with the Wnt/β-catenin pathway [
33,
34,
35,
36]. Regarding the phenomenon that
SMAD4 affects the expression of the upstream gene β-catenin, we speculated that there might be a negative regulatory mechanism, which needs to be verified in the next stage of our research.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}