
The Competitive Interaction of Alveolar Wall Distention with Elastin Crosslinking: A Mechanistic Approach to Emergent Phenomena in Pulmonary Emphysema
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Emergent Phenomena in the Pathogenesis of Pulmonary Emphysema
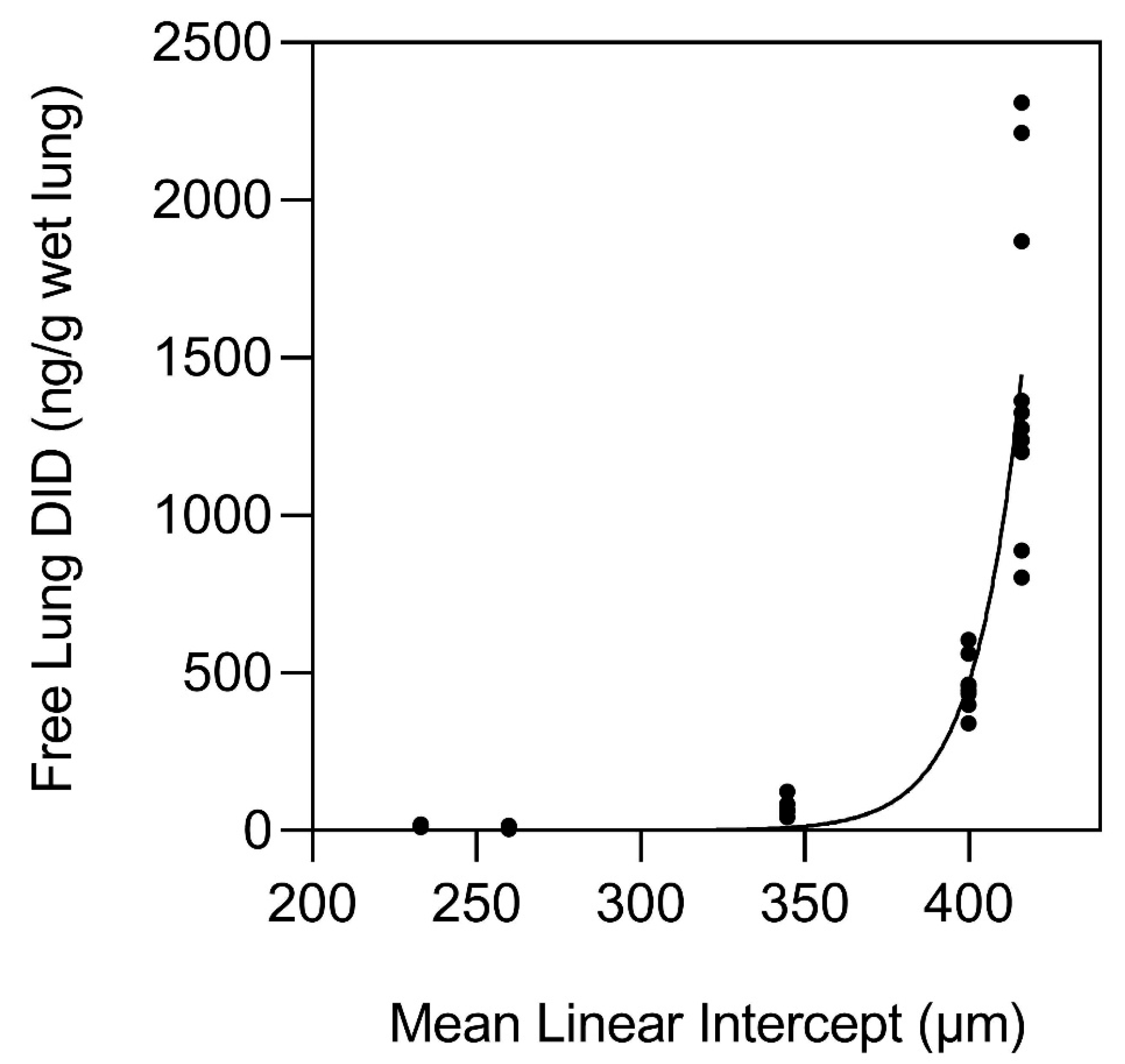
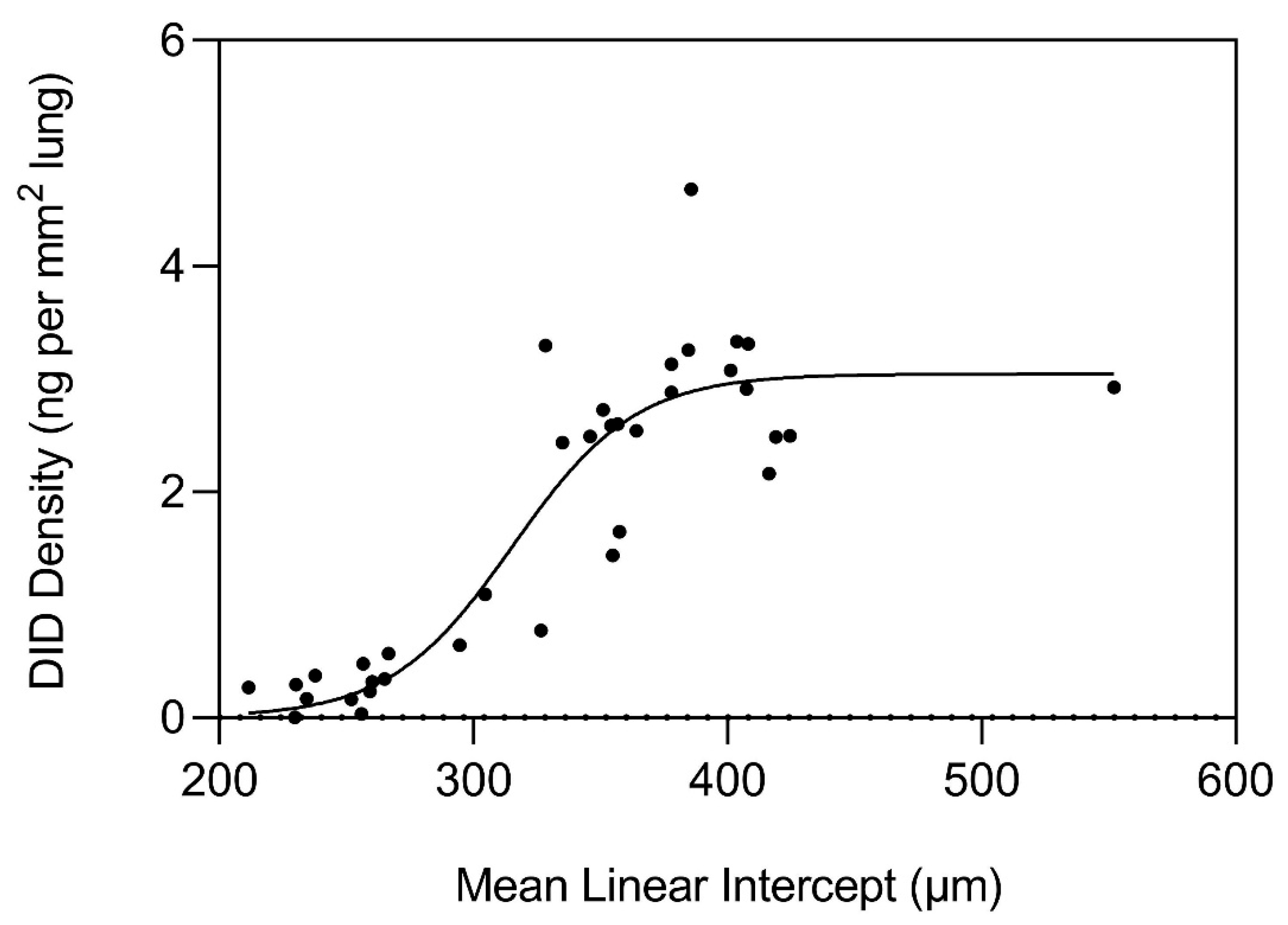
3. The Nonlinear Relationship Between Elastin Crosslinking and Airspace Enlargement
4. Modeling the Relationship Between Elastic Fibers and Mechanical Strain
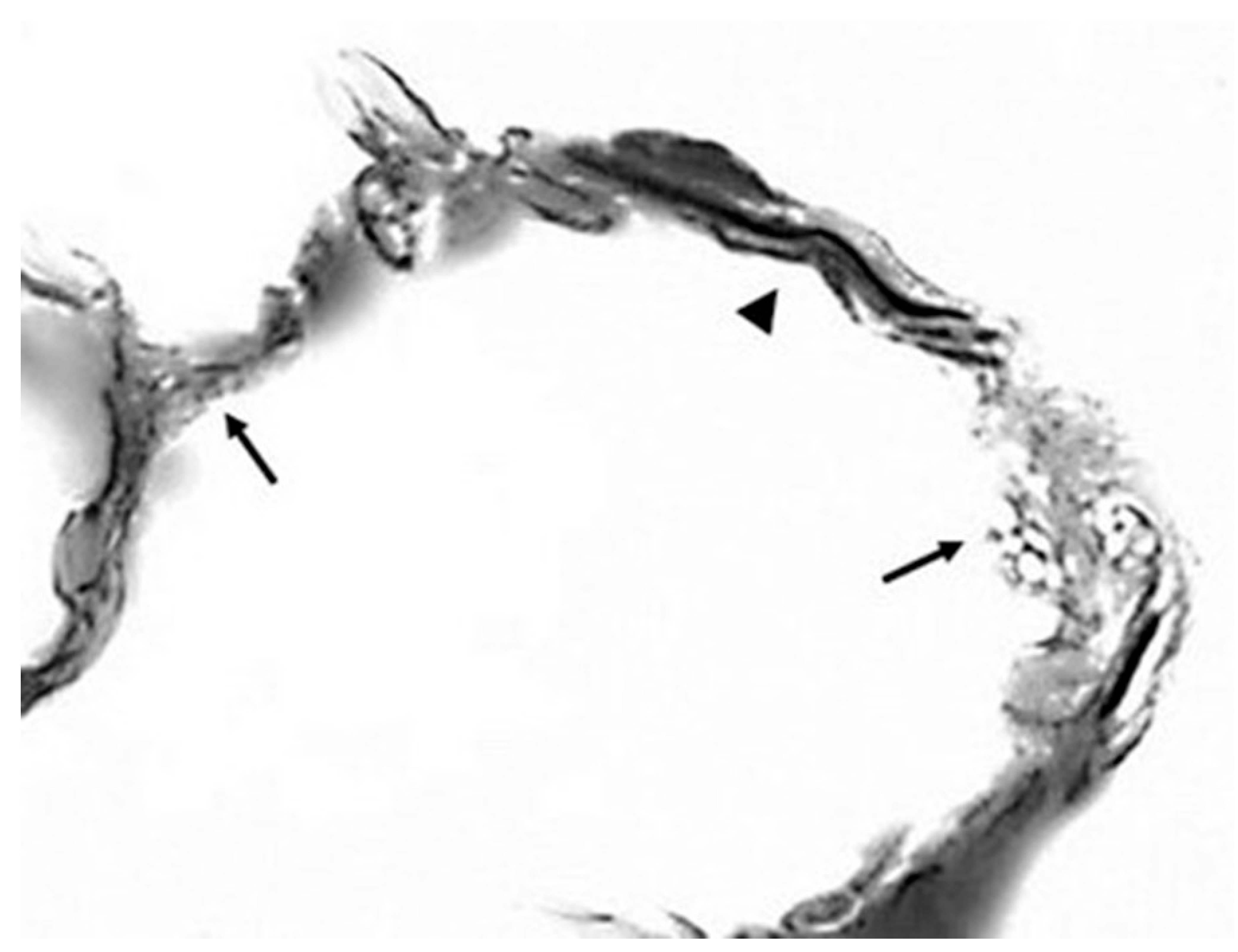
5. The Effect of Elastic Fiber Fragmentation on the Transmission of Mechanical Force
6. A Feedback Loop Involving Structurally Altered Elastic Fibers
7. The Effect of Increased Crosslink Density on the Structural Integrity of Elastic Fibers
8. The Role of Genetic Abnormalities
9. The Application of the Concept of Emergence to Therapeutic Intervention
10. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mecham, R.P. Elastin in lung development and disease pathogenesis. Matrix Biol. 2018, 73, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.R. Bench to Bedside and Back: The Evolving Story of Alpha-1 Antitrypsin Deficiency. Am. J. Respir. Cell Mol. Biol. 2020, 63, 403–404. [Google Scholar] [CrossRef] [PubMed]
- Sandhaus, R.A.; Turino, G. Neutrophil elastase-mediated lung disease. COPD J. Chronic Obstr. Pulm. Dis. 2013, 10 (Suppl. 1), 60–63. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Cao, J.; Ma, X.; Wang, X.; Guo, X.; Yan, N.; Fan, C.; Bao, S.; Fan, J. Animal models of chronic obstructive pulmonary disease: A systematic review. Front. Med. 2024, 11, 1474870. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burgess, J.K.; Gosens, R. Mechanotransduction and the extracellular matrix: Key drivers of lung pathologies and drug responsiveness. Biochem. Pharmacol. 2024, 228, 116255. [Google Scholar] [CrossRef]
- Geitner, C.M.; Becher, T.; Frerichs, I.; Weiler, N.; Bates, J.H.T.; Wall, W.A. An approach to studying recruitment/derecruitment dynamics in a patient-specific computational model of an injured human lung. Int. J. Numer. Methods Biomed. Eng. 2023, 39, e3745. [Google Scholar] [CrossRef]
- Suki, B.; Jesudason, R.; Sato, S.; Parameswaran, H.; Araujo, A.D.; Majumdar, A.; Allen, P.G.; Bartolák-Suki, E. Mechanical failure, stress redistribution, elastase activity, and binding site availability on elastin during the progression of emphysema. Pulm. Pharmacol. Ther. 2012, 25, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Szabari, M.V.; Parameswaran, H.; Sato, S.; Hantos, Z.; Bartolák-Suki, E.; Suki, B. Acute mechanical forces cause deterioration in lung structure and function in elastase-induced emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L567–L574. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, G.Q.; Jusup, M.; Jin, Z.; Wang, Y.; Wang, Z. Pattern transitions in spatial epidemics: Mechanisms and emergent properties. Phys. Life Rev. 2016, 19, 43–73. [Google Scholar] [CrossRef]
- Browne, C.A.; Amchin, D.B.; Schneider, J.; Datta, S.S. Infection Percolation: A Dynamic Network Model of Disease Spreading. Front. Phys. 2021, 9, 645954. [Google Scholar] [CrossRef]
- Artime, O.; De Domenico, M. From the origin of life to pandemics: Emergent phenomena in complex systems. Philos. Trans. A Math. Phys. Eng. Sci. 2022, 380, 20200410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Macklem, P.T. The molecular-clinical divorce. Am. J. Respir. Crit. Care Med. 2003, 168, 500. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Caves, J.M.; Ravi, S.; Li, W.; Chaikof, E.L. Effects of crosslinking on the mechanical properties, drug release and cytocompatibility of protein polymers. Acta Biomater. 2014, 10, 26–33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fagiola, M.; Reznik, S.; Riaz, M.; Qyang, Y.; Lee, S.; Avella, J.; Turino, G.; Cantor, J. The relationship between elastin cross linking and alveolar wall rupture in human pulmonary emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L747–L755. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, D.E.; Hoidal, J.R. Lung fibrosis and emphysema: Divergent responses to a common injury? Science 1982, 217, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Merrilees, M.J.; Ching, P.S.; Beaumont, B.; Hinek, A.; Wight, T.N.; Black, P.N. Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease. Respir. Res. 2008, 9, 41. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Black, P.N.; Ching, P.S.; Beaumont, B.; Ranasinghe, S.; Taylor, G.; Merrilees, M.J. Changes in elastic fibres in the small airways and alveoli in COPD. Eur. Respir. J. 2008, 31, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Deslee, G.; Woods, J.C.; Moore, C.M.; Liu, L.; Conradi, S.H.; Milne, M.; Gierada, D.S.; Pierce, J.; Patterson, A.; Lewit, R.A.; et al. Elastin expression in very severe human COPD. Eur. Respir. J. 2009, 34, 324–331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schräder, C.U.; Heinz, A.; Majovsky, P.; Karaman Mayack, B.; Brinckmann, J.; Sippl, W.; Schmelzer, C.E.H. Elastin is heterogeneously cross-linked. J. Biol. Chem. 2018, 293, 15107–15119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stone, P.J.; Morris, S.M.; Thomas, K.M.; Schuhwerk, K.; Mitchelson, A. Repair of elastase-digested elastic fibers in acellular matrices by replating with neonatal rat-lung lipid interstitial fibroblasts or other elastogenic cell types. Am. J. Respir. Cell Mol. Biol. 1997, 17, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.D.; Hunt, G.W.; Almond, D.P. Evidence of emergent scaling in mechanical systems. Philos. Mag. 2006, 86, 3325–3338. [Google Scholar] [CrossRef]
- Mehraban, S.; Gu, G.; Ma, S.; Liu, X.; Turino, G.; Cantor, J. The Proinflammatory Activity of Structurally Altered Elastic Fibers. Am. J. Respir. Cell Mol. Biol. 2020, 63, 699–706. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cantor, J. Desmosine as a biomarker for the emergent properties of pulmonary emphysema. Front. Med. 2023, 10, 1322283. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, P.; Dong, S. The Influence of Crosslink Density on the Failure Behavior in Amorphous Polymers by Molecular Dynamics Simulations. Materials 2016, 9, 234. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nietupski, C.A.; Moset Zupan, A.; Schutte, S.C. Impact of Cyclic Strain on Elastin Synthesis in a 3D Human Myometrial Culture Model. Tissue Eng. Part C Methods 2024, 30, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.H.; Balestrini, J.L.; Udelsman, B.V.; Zhou, K.C.; Zhao, L.; Ferruzzi, J.; Starcher, B.C.; Levene, M.J.; Humphrey, J.D.; Niklason, L.E. Biaxial Stretch Improves Elastic Fiber Maturation, Collagen Arrangement, and Mechanical Properties in Engineered Arteries. Tissue Eng. Part C Methods 2016, 22, 524–533. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Silverman, E.K. Genetics of COPD. Annu. Rev. Physiol. 2020, 82, 413–431. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Murphy-Ryan, M.; Psychogios, A.; Lindor, N.M. Hereditary disorders of connective tissue: A guide to the emerging differential diagnosis. Genet. Med. 2010, 12, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Tun, M.H.; Borg, B.; Godfrey, M.; Hadley-Miller, N.; Chan, E.D. Respiratory manifestations of Marfan syndrome: A narrative review. J. Thorac. Dis. 2021, 13, 6012–6025. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cooney, A.L.; Wambach, J.A.; Sinn, P.L.; McCray, P.B., Jr. Gene Therapy Potential for Genetic Disorders of Surfactant Dysfunction. Front. Genome Ed. 2022, 3, 785829. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bentley, A.R.; Kritchevsky, S.B.; Harris, T.B.; Newman, A.B.; Bauer, D.C.; Meibohm, B.; Clark, A.G.; Cassano, P.A. Health, Aging, Body Composition Study. Genetic variation in antioxidant enzymes and lung function. Free Radic. Biol. Med. 2012, 52, 1577–1583. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gordon, M.; Duffy, S.; Criner, G.J. Lung volume reduction surgery or bronchoscopic lung volume reduction: Is there an algorithm for allocation? J. Thorac. Dis. 2018, 10 (Suppl. 23), S2816–S2823. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rogliani, P.; Manzetti, G.M.; Gholamalishahi, S.; Bafadhel, M.; Calzetta, L. Inhaled corticosteroids in chronic obstructive pulmonary disease: A systematic review and meta-analysis on mortality protection-making a long story short. Expert. Rev. Respir. Med. 2025, 12, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.O.; Cerreta, J.M.; Keller, S.; Turino, G.M. Modulation of airspace enlargement in elastase-induced emphysema by intratracheal instillment of hyaluronidase and hyaluronic acid. Exp. Lung Res. 1995, 21, 423–436. [Google Scholar] [CrossRef]
- Cantor, J.O.; Shteyngart, B.; Cerreta, J.M.; Liu, M.; Armand, G.; Turino, G.M. The effect of hyaluronan on elastic fiber injury in vitro and elastase-induced airspace enlargement in vivo. Proc. Soc. Exp. Biol. Med. 2000, 225, 65–71. [Google Scholar]
- Cantor, J.O. Potential therapeutic applications of hyaluronan in the lung. Int. J. Chron. Obstruct Pulmon Dis. 2007, 2, 283–288. [Google Scholar] [PubMed] [PubMed Central]
- Cantor, J.; Armand, G.; Turino, G. Lung hyaluronan levels are decreased in alpha-1 antiprotease deficiency COPD. Respir. Med. 2015, 109, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.O.; Ma, S.; Liu, X.; Campos, M.A.; Strange, C.; Stocks, J.M.; Devine, M.S.; El Bayadi, S.G.; Lipchik, R.J.; Sandhaus, R.A.; et al. A 28-day clinical trial of aerosolized hyaluronan in alpha-1 antiprotease deficiency COPD using desmosine as a surrogate marker for drug efficacy. Respir. Med. 2021, 182, 106402. [Google Scholar] [CrossRef]
- Heinz, A. Elastases and elastokines: Elastin degradation and its significance in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 252–273. [Google Scholar] [CrossRef]
- Li, B.; Daggett, V. Molecular basis for the extensibility of elastin. J. Muscle Res. Cell Motil. 2002, 23, 561–573. [Google Scholar] [CrossRef]
- Depenveiller, C.; Baud, S.; Belloy, N.; Bochicchio, B.; Dandurand, J.; Dauchez, M.; Pepe, A.; Pomès, R.; Samouillan, V.; Debelle, L. Structural and physical basis for the elasticity of elastin. Q. Rev. Biophys. 2024, 57, e3. [Google Scholar] [CrossRef] [PubMed]
- Trębacz, H.; Barzycka, A. Mechanical Properties and Functions of Elastin: An Overview. Biomolecules 2023, 13, 574. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takahashi, A.; Majumdar, A.; Parameswaran, H.; Bartolák-Suki, E.; Suki, B. Proteoglycans maintain lung stability in an elastase-treated mouse model of emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 26–33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strange, C. Anti-Proteases and Alpha-1 Antitrypsin Augmentation Therapy. Respir. Care 2018, 63, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Kuna, P.; Jenkins, M.; O’Brien, C.D.; Fahy, W.A. AZD9668, a neutrophil elastase inhibitor, plus ongoing budesonide/formoterol in patients with COPD. Respir. Med. 2012, 106, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.D.; Costa, E.; Guedes, R.C.; Moreira, R. Targeting COPD: Advances on low-molecular-weight inhibitors of human neutrophil elastase. Med. Res. Rev. 2013, 33, E73–E101. [Google Scholar] [CrossRef]
- Groutas, W.C.; Dou, D.; Alliston, K.R. Neutrophil elastase inhibitors. Expert. Opin. Ther. Pat. 2011, 21, 339–354. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Clark, A.R.; Kumar, H.; Burrowes, K. Capturing complexity in pulmonary system modelling. Proc. Inst. Mech. Eng. H 2017, 231, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Sturmberg, J.P. Multimorbidity and chronic disease: An emergent perspective. J. Eval. Clin. Pract. 2014, 20, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.H.; Davis, G.S.; Majumdar, A.; Butnor, K.J.; Suki, B. Linking parenchymal disease progression to changes in lung mechanical function by percolation. Am. J. Respir. Crit. Care Med. 2007, 176, 617–623. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Winkler, T.; Suki, B. Emergent structure-function relations in emphysema and asthma. Crit. Rev. Biomed. Eng. 2011, 39, 263–280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cantor, J. Elastin Peptides as a Potential Disease Vector in the Pathogenesis of Pulmonary Emphysema: An Investigation of This Hypothesis. Life 2025, 15, 356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tembely, D.; Henry, A.; Vanalderwiert, L.; Toussaint, K.; Bennasroune, A.; Blaise, S.; Sartelet, H.; Jaisson, S.; Galés, C.; Martiny, L.; et al. The Elastin Receptor Complex: An Emerging Therapeutic Target Against Age-Related Vascular Diseases. Front. Endocrinol. 2022, 13, 815356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maldonado-Franco, A.; Giraldo-Cadavid, L.F.; Tuta-Quintero, E.; Bastidas Goyes, A.R.; Botero-Rosas, D.A. The Challenges of Spirometric Diagnosis of COPD. Can. Respir. J. 2023, 2023, 6991493. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhu, D.; Qiao, C.; Dai, H.; Hu, Y.; Xi, Q. Diagnostic efficacy of visual subtypes and low attenuation area based on HRCT in the diagnosis of COPD. BMC Pulm. Med. 2022, 22, 81. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stockley, R.A.; Halpin, D.M.G.; Celli, B.R.; Singh, D. Chronic Obstructive Pulmonary Disease Biomarkers and Their Interpretation. Am. J. Respir. Crit. Care Med. 2019, 199, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Pantazopoulos, I.; Magounaki, K.; Kotsiou, O.; Rouka, E.; Perlikos, F.; Kakavas, S.; Gourgoulianis, K. Incorporating Biomarkers in COPD Management: The Research Keeps Going. J. Pers. Med. 2022, 12, 379. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cantor, J. Desmosine: The Rationale for Its Use as a Biomarker of Therapeutic Efficacy in the Treatment of Pulmonary Emphysema. Diagnostics 2025, 15, 578. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantor, J. The Competitive Interaction of Alveolar Wall Distention with Elastin Crosslinking: A Mechanistic Approach to Emergent Phenomena in Pulmonary Emphysema. Cells 2025, 14, 702. https://doi.org/10.3390/cells14100702
Cantor J. The Competitive Interaction of Alveolar Wall Distention with Elastin Crosslinking: A Mechanistic Approach to Emergent Phenomena in Pulmonary Emphysema. Cells. 2025; 14(10):702. https://doi.org/10.3390/cells14100702
Chicago/Turabian StyleCantor, Jerome. 2025. "The Competitive Interaction of Alveolar Wall Distention with Elastin Crosslinking: A Mechanistic Approach to Emergent Phenomena in Pulmonary Emphysema" Cells 14, no. 10: 702. https://doi.org/10.3390/cells14100702
APA StyleCantor, J. (2025). The Competitive Interaction of Alveolar Wall Distention with Elastin Crosslinking: A Mechanistic Approach to Emergent Phenomena in Pulmonary Emphysema. Cells, 14(10), 702. https://doi.org/10.3390/cells14100702