

Retinoid X Receptor as a Therapeutic Target to Treat Neurological Disorders Associated with α-Synucleinopathy

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. AAV Vectors
2.2. Primary Mouse Cortical Neuron Culture
2.3. Intracerebral Injection of PFFs and AAV Vectors
2.4. Behavioral Test
2.5. Isolation and Processing of Tissues
2.6. Immunohistochemistry
2.7. Quantitation of Phospho-Synuclein Aggregates
2.8. Unbiased Stereology
2.9. DA Measurements
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
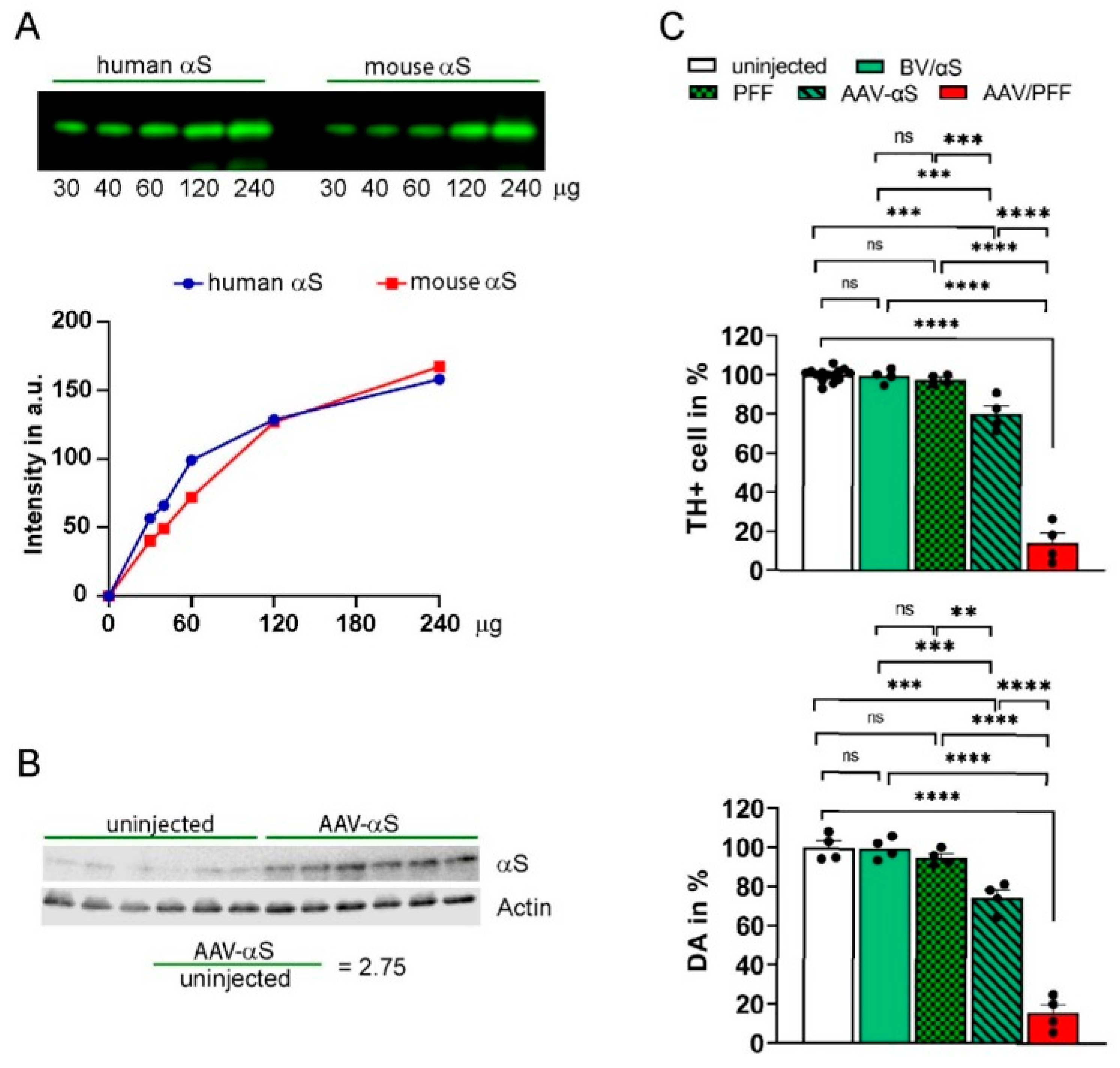
3.1. Development of PD-like Mouse Model Associated with αS-Induced Neurodegeneration
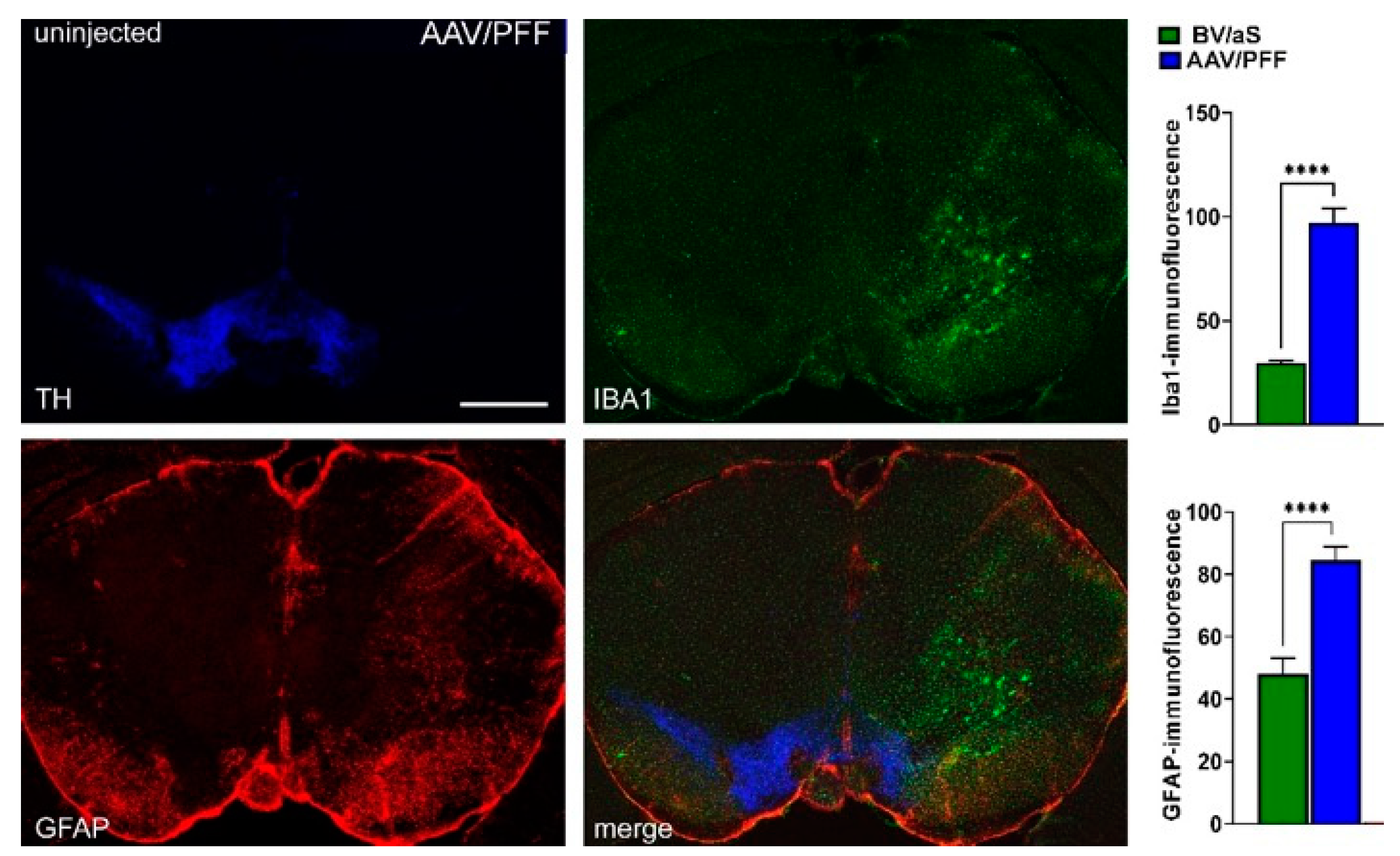
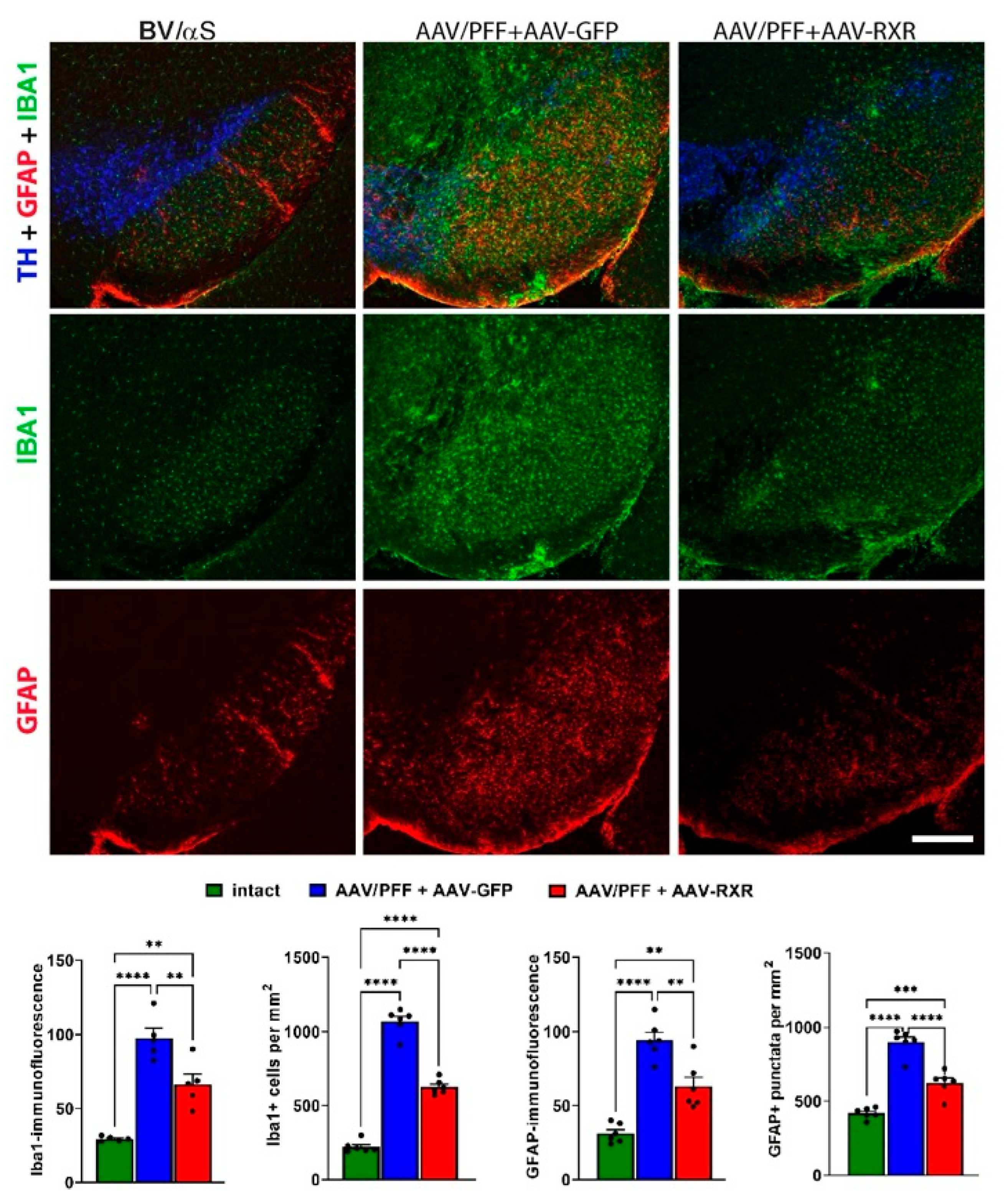
3.2. Combined AAV/PFF Injection Induce Severe Neurodegeneration Accompanied by Inflammatory Response and Gliosis
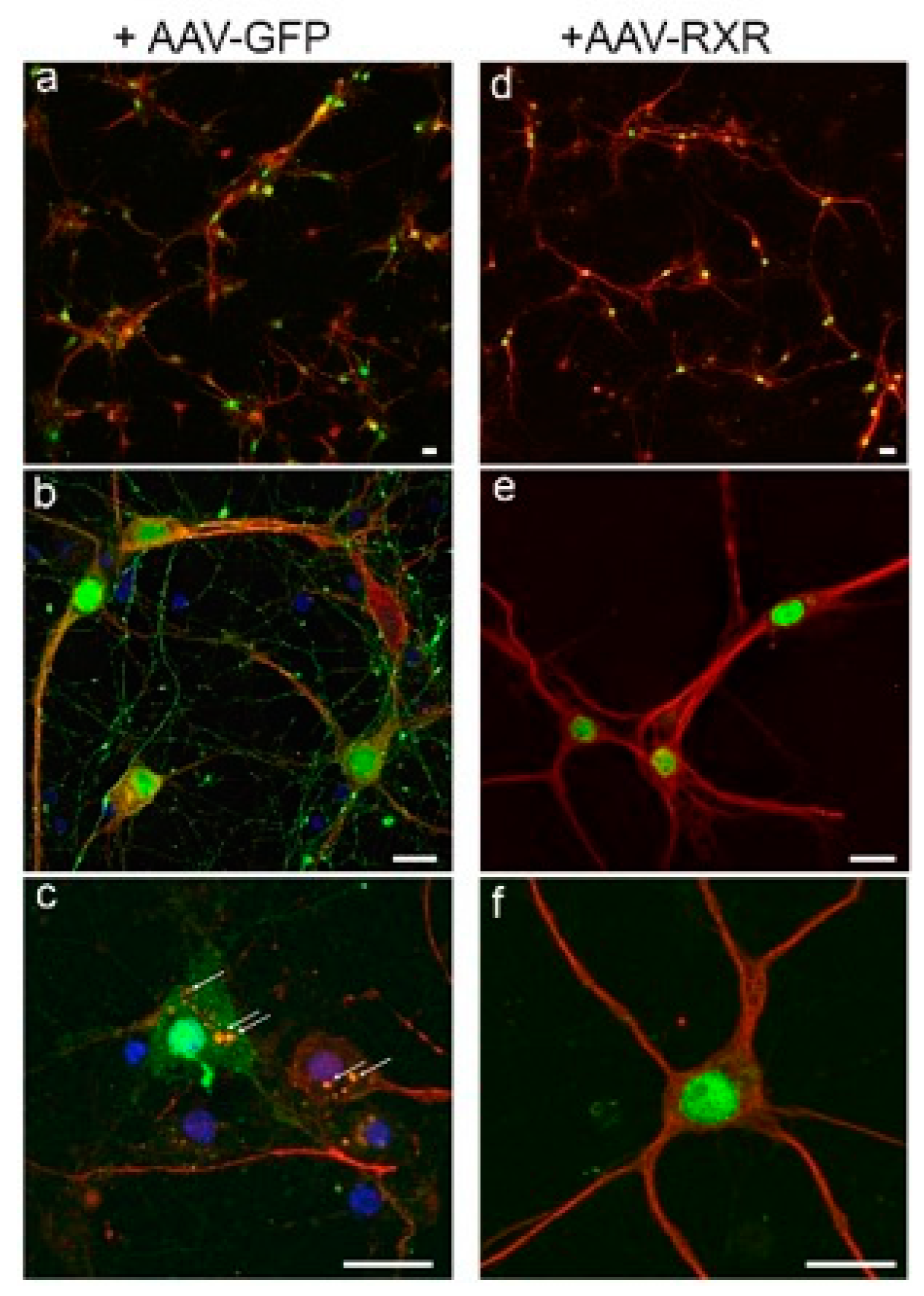
3.3. RXR Diminishes LB-like Formation in Primary Mouse Cortical Neurons
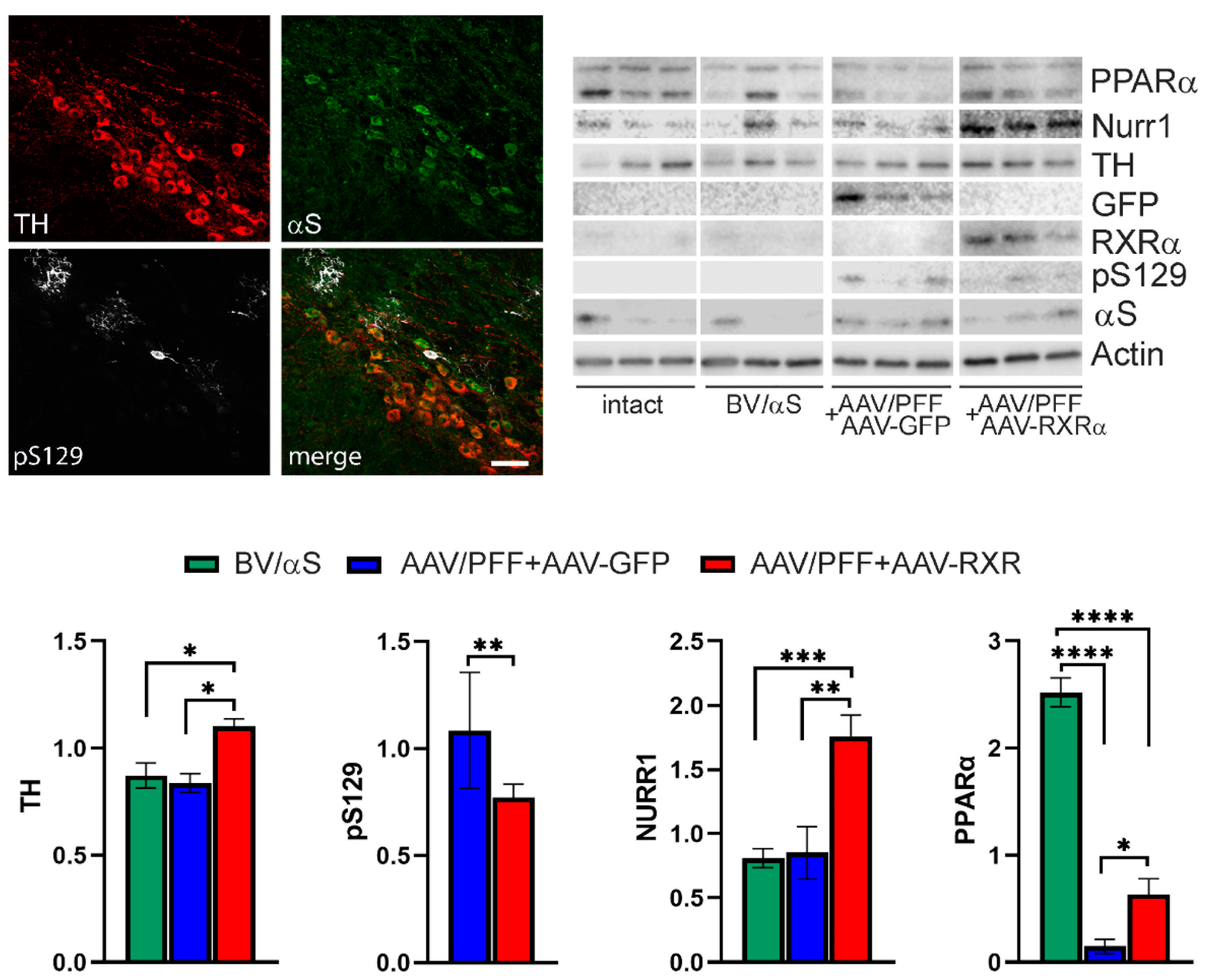
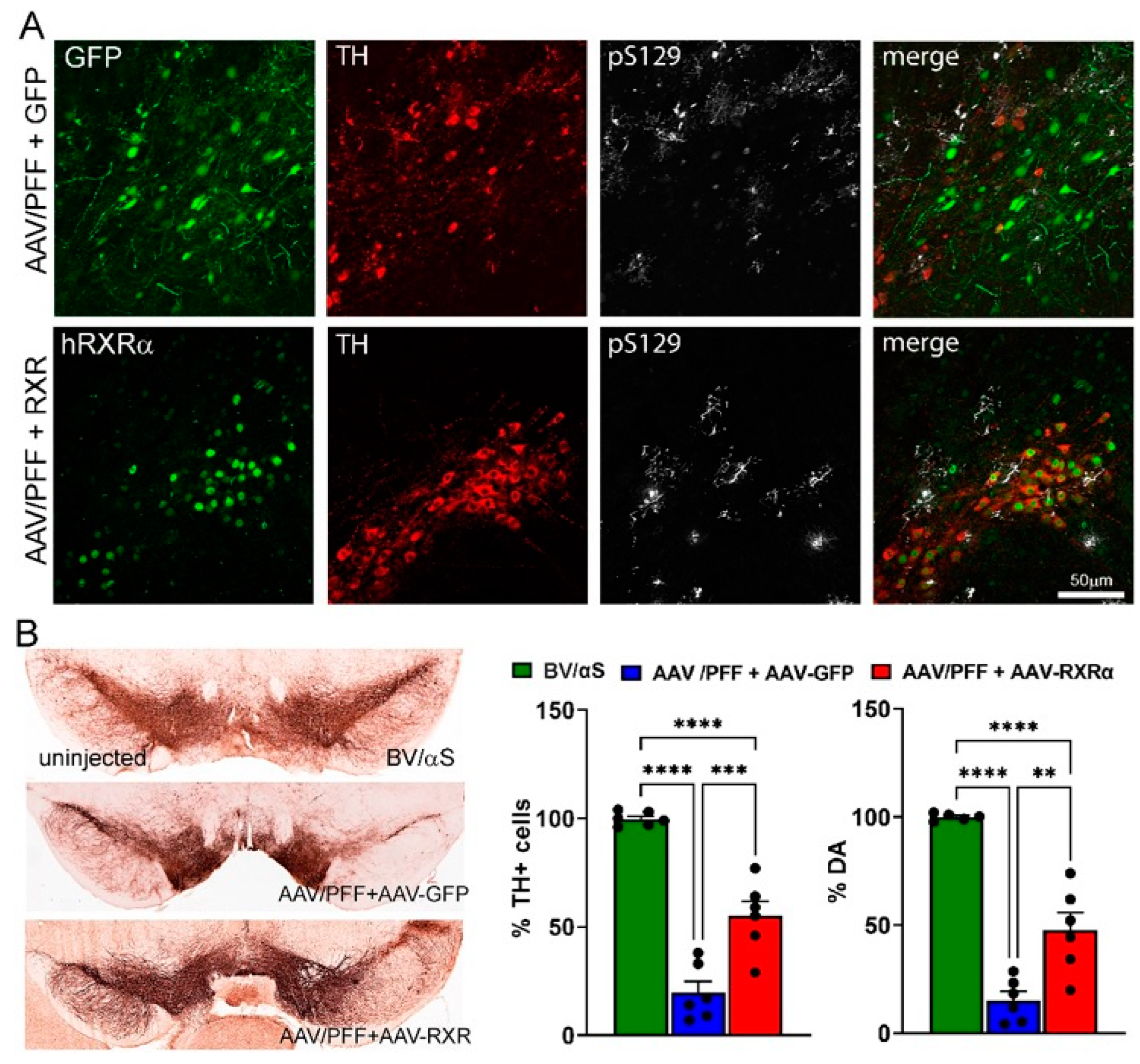
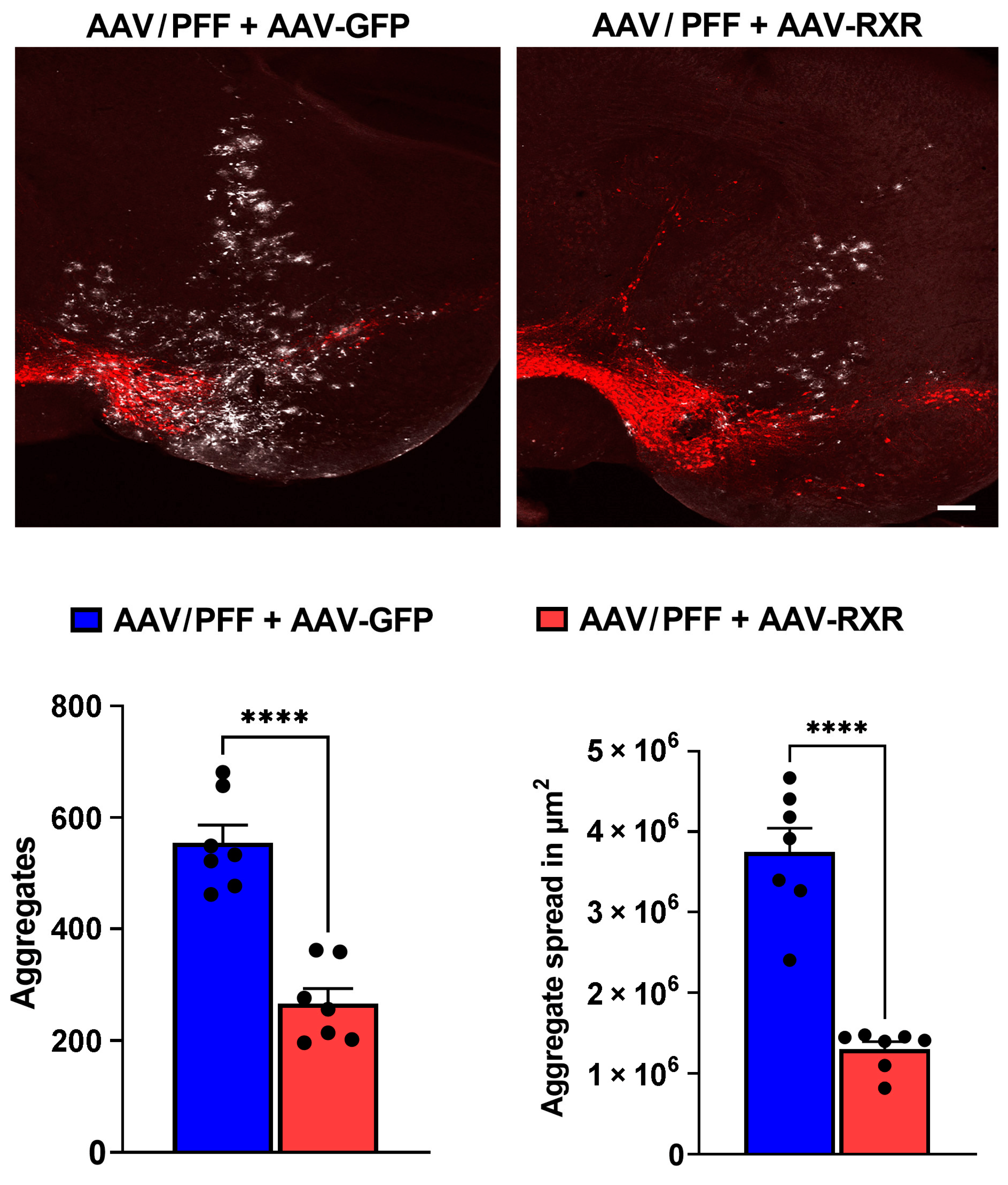
3.4. RXR Overexpression Significantly Ameliorates PD-Associated Pathology in Mouse PD Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bostrom, F.; Jonsson, L.; Minthon, L.; Londos, E. Patients with Lewy body dementia use more resources than those with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2007, 22, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Lamotte, G.; Singer, W. Synucleinopathies. Handb. Clin. Neurol. 2023, 196, 175–202. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Park. Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]
- Spathis, A.D.; Asvos, X.; Ziavra, D.; Karampelas, T.; Topouzis, S.; Cournia, Z.; Qing, X.; Alexakos, P.; Smits, L.M.; Dalla, C.; et al. Nurr1:RXRalpha heterodimer activation as monotherapy for Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 3999–4004. [Google Scholar] [CrossRef] [PubMed]
- Friling, S.; Bergsland, M.; Kjellander, S. Activation of Retinoid X Receptor increases dopamine cell survival in models for Parkinson’s disease. BMC Neurosci. 2009, 10, 146. [Google Scholar] [CrossRef]
- Al-Nusaif, M.; Yang, Y.; Li, S.; Cheng, C.; Le, W. The role of NURR1 in metabolic abnormalities of Parkinson’s disease. Mol. Neurodegener. 2022, 17, 46. [Google Scholar] [CrossRef]
- Sharma, S.; Shen, T.; Chitranshi, N.; Gupta, V.; Basavarajappa, D.; Sarkar, S.; Mirzaei, M.; You, Y.; Krezel, W.; Graham, S.L.; et al. Correction to: Retinoid X Receptor: Cellular and Biochemical Roles of Nuclear Receptor with a Focus on Neuropathological Involvement. Mol. Neurobiol. 2022, 59, 2051. [Google Scholar] [CrossRef] [PubMed]
- van Neerven, S.; Kampmann, E.; Mey, J. RAR/RXR and PPAR/RXR signaling in neurological and psychiatric diseases. Prog. Neurobiol. 2008, 85, 433–451. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARgamma and PGC-1alpha as therapeutic targets in Parkinson’s. Neurochem Res 2015, 40, 308–316. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARgamma as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef]
- Rahimi, A.; Faizi, M.; Talebi, F.; Noorbakhsh, F.; Kahrizi, F.; Naderi, N. Interaction between the protective effects of cannabidiol and palmitoylethanolamide in experimental model of multiple sclerosis in C57BL/6 mice. Neuroscience 2015, 290, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Ferret-Sena, V.; Capela, C.; Sena, A. Metabolic Dysfunction and Peroxisome Proliferator-Activated Receptors (PPAR) in Multiple Sclerosis. Int. J. Mol. Sci. 2018, 19, 1639. [Google Scholar] [CrossRef]
- Le, W.D.; Xu, P.; Jankovic, J.; Jiang, H.; Appel, S.H.; Smith, R.G.; Vassilatis, D.K. Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 2003, 33, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Perez-Segura, I.; Santiago-Balmaseda, A.; Rodriguez-Hernandez, L.D.; Morales-Martinez, A.; Martinez-Becerril, H.A.; Martinez-Gomez, P.A.; Delgado-Minjares, K.M.; Salinas-Lara, C.; Martinez-Davila, I.A.; Guerra-Crespo, M.; et al. PPARs and Their Neuroprotective Effects in Parkinson’s Disease: A Novel Therapeutic Approach in alpha-Synucleinopathy? Int. J. Mol. Sci. 2023, 24, 3264. [Google Scholar] [CrossRef]
- Al-Nusaif, M.; Lin, Y.; Li, T.; Cheng, C.; Le, W. Advances in NURR1-Regulated Neuroinflammation Associated with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 16184. [Google Scholar] [CrossRef]
- Loppi, S.; Kolosowska, N.; Karkkainen, O.; Korhonen, P.; Huuskonen, M.; Grubman, A.; Dhungana, H.; Wojciechowski, S.; Pomeshchik, Y.; Giordano, M.; et al. HX600, a synthetic agonist for RXR-Nurr1 heterodimer complex, prevents ischemia-induced neuronal damage. Brain Behav. Immun. 2018, 73, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bi, W.; Zhao, W.; Varghese, M.; Koch, R.J.; Walker, R.H.; Chandraratna, R.A.; Sanders, M.E.; Janesick, A.; Blumberg, B.; et al. Selective brain penetrable Nurr1 transactivator for treating Parkinson’s disease. Oncotarget 2016, 7, 7469–7479. [Google Scholar] [CrossRef]
- Machado, M.M.F.; Bassani, T.B.; Coppola-Segovia, V.; Moura, E.L.R.; Zanata, S.M.; Andreatini, R.; Vital, M. PPAR-gamma agonist pioglitazone reduces microglial proliferation and NF-kappaB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson’s disease. Pharmacol. Rep. 2019, 71, 556–564. [Google Scholar] [CrossRef]
- Argyrofthalmidou, M.; Spathis, A.D.; Maniati, M.; Poula, A.; Katsianou, M.A.; Sotiriou, E.; Manousaki, M.; Perier, C.; Papapanagiotou, I.; Papadopoulou-Daifoti, Z.; et al. Nurr1 repression mediates cardinal features of Parkinson’s disease in alpha-synuclein transgenic mice. Hum. Mol. Genet. 2021, 30, 1469–1483. [Google Scholar] [CrossRef]
- Kim, W.; Tripathi, M.; Kim, C.; Vardhineni, S.; Cha, Y.; Kandi, S.K.; Feitosa, M.; Kholiya, R.; Sah, E.; Thakur, A.; et al. An optimized Nurr1 agonist provides disease-modifying effects in Parkinson’s disease models. Nat. Commun. 2023, 14, 4283. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Salganik, M.; Sergeyev, V.G.; Shinde, V.; Meyers, C.A.; Gorbatyuk, M.S.; Lin, J.H.; Zolotukhin, S.; Gorbatyuk, O.S. The loss of glucose-regulated protein 78 (GRP78) during normal aging or from siRNA knockdown augments human alpha-synuclein (alpha-syn) toxicity to rat nigral neurons. Neurobiol. Aging 2015, 36, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.I.; Marcus, J.M.; Lee, J.H.; Garcia, P.L.; Singh, V.; Shacka, J.J.; Zhang, J.; Gropen, T.I.; Falany, C.N.; Andrabi, S.A. Restoration of CTSD (cathepsin D) and lysosomal function in stroke is neuroprotective. Autophagy 2021, 17, 1330–1348. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef]
- Magno, L.A.V.; Collodetti, M.; Tenza-Ferrer, H.; Romano-Silva, M.A. Cylinder Test to Assess Sensory-motor Function in a Mouse Model of Parkinson’s Disease. Bio Protoc. 2019, 9, e3337. [Google Scholar] [CrossRef]
- Maidana, D.E.; Tsoka, P.; Tian, B.; Dib, B.; Matsumoto, H.; Kataoka, K.; Lin, H.; Miller, J.W.; Vavvas, D.G. A Novel ImageJ Macro for Automated Cell Death Quantitation in the Retina. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6701–6708. [Google Scholar] [CrossRef]
- Kirik, D.; Rosenblad, C.; Burger, C.; Lundberg, C.; Johansen, T.E.; Muzyczka, N.; Mandel, R.J.; Bjorklund, A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci. 2002, 22, 2780–2791. [Google Scholar] [CrossRef]
- Thakur, P.; Breger, L.S.; Lundblad, M.; Wan, O.W.; Mattsson, B.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q.; Bjorklund, A. Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc. Natl. Acad. Sci. USA 2017, 114, E8284–E8293. [Google Scholar] [CrossRef]
- Bjorklund, A.; Nilsson, F.; Mattsson, B.; Hoban, D.B.; Parmar, M. A Combined alpha-Synuclein/Fibril (SynFib) Model of Parkinson-Like Synucleinopathy Targeting the Nigrostriatal Dopamine System. J. Park. Dis. 2022, 12, 2307–2320. [Google Scholar] [CrossRef]
- Gully, J.C.; Sergeyev, V.G.; Bhootada, Y.; Mendez-Gomez, H.; Meyers, C.A.; Zolotukhin, S.; Gorbatyuk, M.S.; Gorbatyuk, O.S. Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett. 2016, 627, 36–41. [Google Scholar] [CrossRef]
- Bobela, W.; Aebischer, P.; Schneider, B.L. Alphalpha-Synuclein as a Mediator in the Interplay between Aging and Parkinson’s Disease. Biomolecules 2015, 5, 2675–2700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yan, Z.; Xin, H.; Shao, S.; Xue, S.; Cespuglio, R.; Wang, S. Relationship among alpha-synuclein, aging and inflammation in Parkinson’s disease (Review). Exp. Ther. Med. 2024, 27, 23. [Google Scholar] [CrossRef]
- Eo, H.; Kim, S.; Jung, U.J.; Kim, S.R. Alpha-Synuclein and Microglia in Parkinson’s Disease: From Pathogenesis to Therapeutic Prospects. J. Clin. Med. 2024, 13, 7243. [Google Scholar] [CrossRef] [PubMed]
- Harackiewicz, O.; Grembecka, B. The Role of Microglia and Astrocytes in the Pathomechanism of Neuroinflammation in Parkinson’s Disease-Focus on Alpha-Synuclein. J. Integr. Neurosci. 2024, 23, 203. [Google Scholar] [CrossRef]
- Burger, C.; Gorbatyuk, O.S.; Velardo, M.J.; Peden, C.S.; Williams, P.; Zolotukhin, S.; Reier, P.J.; Mandel, R.J.; Muzyczka, N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 2004, 10, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nguyen, F.N.; Manfredsson, F.P.; Kondrikova, G.; Sullivan, L.F.; Meyers, C.; Chen, W.; Mandel, R.J.; Muzyczka, N. alpha-Synuclein expression in rat substantia nigra suppresses phospholipase D2 toxicity and nigral neurodegeneration. Mol. Ther. 2010, 18, 1758–1768. [Google Scholar] [CrossRef]
- Wojtowicz, S.; Strosznajder, A.K.; Jezyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef]
- Koivisto, A.M.; Helisalmi, S.; Pihlajamaki, J.; Hiltunen, M.; Koivisto, K.; Moilanen, L.; Kuusisto, J.; Helkala, E.L.; Hanninen, T.; Kervinen, K.; et al. Association analysis of peroxisome proliferator-activated receptor gamma polymorphisms and late onset Alzheimer’s disease in the Finnish population. Dement. Geriatr. Cogn. Disord. 2006, 22, 449–453. [Google Scholar] [CrossRef]
- Hamilton, G.; Proitsi, P.; Jehu, L.; Morgan, A.; Williams, J.; O’Donovan, M.C.; Owen, M.J.; Powell, J.F.; Lovestone, S. Candidate gene association study of insulin signaling genes and Alzheimer’s disease: Evidence for SOS2, PCK1, and PPARgamma as susceptibility loci. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 508–516. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Park, S.; Yu, B.P.; Chung, H.Y. Amelioration of age-related inflammation and oxidative stress by PPARgamma activator: Suppression of NF-kappaB by 2,4-thiazolidinedione. Exp. Gerontol. 2006, 41, 590–599. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Landreth, G.E.; Willson, T.M.; Klockgether, T.; van Leuven, F.; Heneka, M.T. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 2003, 23, 9796–9804. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Camacho, I.E.; Serneels, L.; Spittaels, K.; Merchiers, P.; Dominguez, D.; De Strooper, B. Peroxisome-proliferator-activated receptor gamma induces a clearance mechanism for the amyloid-beta peptide. J. Neurosci. 2004, 24, 10908–10917. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Xiong, C.; Dong, W.; Qu, Y.; Ning, W.; Liu, W.; Han, F.; Ma, Y.; Wang, R.; Fei, Z.; et al. Activation of peroxisome proliferator-activated receptor beta/delta attenuates acute ischemic stroke on middle cerebral ischemia occlusion in rats. J. Stroke Cerebrovasc. Dis. 2014, 23, 1396–1402. [Google Scholar] [CrossRef]
- Kalra, P.; Khan, H.; Kaur, A.; Singh, T.G. Mechanistic Insight on Autophagy Modulated Molecular Pathways in Cerebral Ischemic Injury: From Preclinical to Clinical Perspective. Neurochem. Res. 2022, 47, 825–843. [Google Scholar] [CrossRef]
- Prashantha Kumar, B.R.; Kumar, A.P.; Jose, J.A.; Prabitha, P.; Yuvaraj, S.; Chipurupalli, S.; Jeyarani, V.; Manisha, C.; Banerjee, S.; Jeyabalan, J.B.; et al. Minutes of PPAR-gamma agonism and neuroprotection. Neurochem. Int. 2020, 140, 104814. [Google Scholar] [CrossRef]
- Tong, Q.; Wu, L.; Gao, Q.; Ou, Z.; Zhu, D.; Zhang, Y. PPARbeta/delta Agonist Provides Neuroprotection by Suppression of IRE1alpha-Caspase-12-Mediated Endoplasmic Reticulum Stress Pathway in the Rotenone Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 3822–3831. [Google Scholar] [CrossRef]
- Falcone, R.; Florio, T.M.; Di Giacomo, E.; Benedetti, E.; Cristiano, L.; Antonosante, A.; Fidoamore, A.; Massimi, M.; Alecci, M.; Ippoliti, R.; et al. PPARbeta/delta and gamma in a rat model of Parkinson’s disease: Possible involvement in PD symptoms. J. Cell Biochem. 2015, 116, 844–855. [Google Scholar] [CrossRef]
- Lee, Y.; Cho, J.H.; Lee, S.; Lee, W.; Chang, S.C.; Chung, H.Y.; Moon, H.R.; Lee, J. Neuroprotective effects of MHY908, a PPAR alpha/gamma dual agonist, in a MPTP-induced Parkinson’s disease model. Brain Res. 2019, 1704, 47–58. [Google Scholar] [CrossRef]
- Bonato, J.M.; Bassani, T.B.; Milani, H.; Vital, M.; de Oliveira, R.M.W. Pioglitazone reduces mortality, prevents depressive-like behavior, and impacts hippocampal neurogenesis in the 6-OHDA model of Parkinson’s disease in rats. Exp. Neurol. 2018, 300, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, J.K.; Santiago, R.M.; Persike, D.S.; da Silva Fernandes, M.J.; Tonin, F.S.; da Cunha, C.; Lucio Boschen, S.; Lima, M.M.; Vital, M.A. Neuroprotective effects of peroxisome proliferator-activated receptor alpha and gamma agonists in model of parkinsonism induced by intranigral 1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine. Behav. Brain Res. 2014, 274, 390–399. [Google Scholar] [CrossRef]
- Hassanzadeh, K.; Rahimmi, A.; Moloudi, M.R.; Maccarone, R.; Corbo, M.; Izadpanah, E.; Feligioni, M. Effect of lobeglitazone on motor function in rat model of Parkinson’s disease with diabetes co-morbidity. Brain Res. Bull. 2021, 173, 184–192. [Google Scholar] [CrossRef]
- Chu, Y.; Le, W.; Kompoliti, K.; Jankovic, J.; Mufson, E.J.; Kordower, J.H. Nurr1 in Parkinson’s disease and related disorders. J. Comp. Neurol. 2006, 494, 495–514. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tan, X.; Tian, L.; Jia, C.; Cheng, C.; Chen, X.; Wei, M.; Wang, Y.; Hu, Y.; Jia, Q.; et al. The role of Nurr1-miR-30e-5p-NLRP3 axis in inflammation-mediated neurodegeneration: Insights from mouse models and patients’ studies in Parkinson’s disease. J. Neuroinflammation 2023, 20, 274. [Google Scholar] [CrossRef]
- Li, W.; Liu, X.; Tu, Y.; Ding, D.; Yi, Q.; Sun, X.; Wang, Y.; Wang, K.; Zhu, M.; Mao, J. Dysfunctional Nurr1 promotes high glucose-induced Muller cell activation by up-regulating the NF-kappaB/NLRP3 inflammasome axis. Neuropeptides 2020, 82, 102057. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Briand, O.; Helleboid-Chapman, A.; Ploton, M.; Hennuyer, N.; Carpentier, R.; Pattou, F.; Vandewalle, B.; Moerman, E.; Gmyr, V.; Kerr-Conte, J.; et al. The nuclear orphan receptor Nur77 is a lipotoxicity sensor regulating glucose-induced insulin secretion in pancreatic beta-cells. Mol. Endocrinol. 2012, 26, 399–413. [Google Scholar] [CrossRef]
- Tripathi, A.; Alnakhala, H.; Brontesi, L.; Selkoe, D.; Dettmer, U. RXR nuclear receptor signaling modulates lipid metabolism and triggers lysosomal clearance of alpha-synuclein in neuronal models of synucleinopathy. Cell Mol. Life Sci. 2024, 81, 362. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhylkibayev, A.; Starr, C.R.; Hossain, M.I.; Kumar, S.; Andrabi, S.A.; Grant, M.B.; Atigadda, V.R.; Gorbatyuk, M.S.; Gorbatyuk, O.S. Retinoid X Receptor as a Therapeutic Target to Treat Neurological Disorders Associated with α-Synucleinopathy. Cells 2025, 14, 685. https://doi.org/10.3390/cells14100685
Zhylkibayev A, Starr CR, Hossain MI, Kumar S, Andrabi SA, Grant MB, Atigadda VR, Gorbatyuk MS, Gorbatyuk OS. Retinoid X Receptor as a Therapeutic Target to Treat Neurological Disorders Associated with α-Synucleinopathy. Cells. 2025; 14(10):685. https://doi.org/10.3390/cells14100685
Chicago/Turabian StyleZhylkibayev, Assylbek, Christopher R. Starr, M. Iqbal Hossain, Sandeep Kumar, Shaida A. Andrabi, Maria B. Grant, Venkatram R. Atigadda, Marina S. Gorbatyuk, and Oleg S. Gorbatyuk. 2025. "Retinoid X Receptor as a Therapeutic Target to Treat Neurological Disorders Associated with α-Synucleinopathy" Cells 14, no. 10: 685. https://doi.org/10.3390/cells14100685
APA StyleZhylkibayev, A., Starr, C. R., Hossain, M. I., Kumar, S., Andrabi, S. A., Grant, M. B., Atigadda, V. R., Gorbatyuk, M. S., & Gorbatyuk, O. S. (2025). Retinoid X Receptor as a Therapeutic Target to Treat Neurological Disorders Associated with α-Synucleinopathy. Cells, 14(10), 685. https://doi.org/10.3390/cells14100685