
Comprehensive Co-Inhibitory Receptor (Co-IR) Expression on T Cells and Soluble Proteins in Rheumatoid Arthritis


Abstract
1. Introduction
2. Patients and Methods
2.1. Descriptive Attributes of Cohorts and Study Populations
2.2. Cell Isolation
2.3. Antibodies for Immune-Phenotype
2.4. Flow Cytometric Analysis
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Statistical Analysis
3. Results
3.1. Co-IR Expression on T Cell Characterization with Taiwanese RA
3.2. Correlation between RA Disease Activity and Co-IRs Expression on T Cells
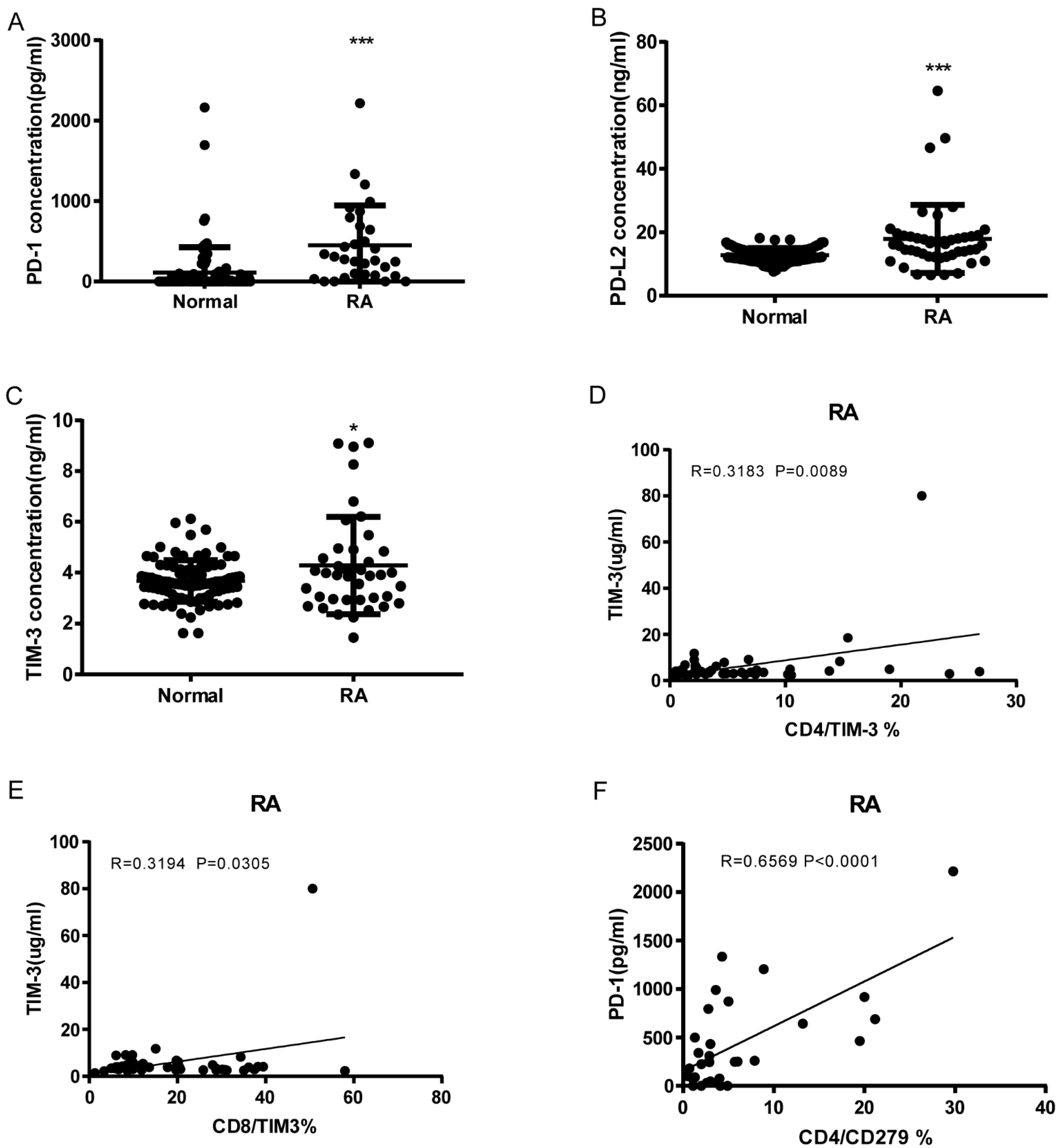
3.3. Soluble PD-1, PDL-2, and TIM3 Levels in RA and Correlation with Cell Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology 2012, 51 (Suppl. S5), v3–v11. [Google Scholar] [CrossRef]
- Mellado, M.; Martinez-Munoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodriguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.-K.; Cho, M.-L.; Park, M.-K.; Oh, H.-J.; Park, J.-S.; Her, Y.-M.; Lee, S.-Y.; Youn, J.; Ju, J.H.; Park, K.S.; et al. Interleukin-21 promotes osteoclastogenesis in humans with rheumatoid arthritis and in mice with collagen-induced arthritis. Arthritis Rheum. 2012, 64, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Dubey, C. Accessory Molecule and Costimulation Requirements for CD4 T Cell Response. Crit. Rev. Immunol. 2017, 37, 261–290. [Google Scholar] [CrossRef]
- Burke, K.P.; Patterson, D.G.; Liang, D.; Sharpe, A.H. Immune checkpoint receptors in autoimmunity. Curr. Opin. Immunol. 2023, 80, 102283. [Google Scholar] [CrossRef]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Lu, J.; Wu, J.; Mao, L.; Xu, H.; Wang, S. Revisiting PD-1/PD-L pathway in T and B cell response: Beyond immunosuppression. Cytokine Growth Factor Rev. 2022, 67, 58–65. [Google Scholar] [CrossRef]
- Chen, R.-Y.; Zhu, Y.; Shen, Y.-Y.; Xu, Q.-Y.; Tang, H.-Y.; Cui, N.-X.; Jiang, L.; Dai, X.-M.; Chen, W.-Q.; Lin, Q.; et al. The role of PD-1 signaling in health and immune-related diseases. Front. Immunol. 2023, 14, 1163633. [Google Scholar] [CrossRef]
- Grebinoski, S.; Vignali, D.A. Inhibitory receptor agonists: The future of autoimmune disease therapeutics? Curr. Opin. Immunol. 2020, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Gao, S.; Li, S.; Xing, Z.; Qian, H.; Hu, Y.; Wang, W.; Hua, C. TIGIT as a Promising Therapeutic Target in Autoimmune Diseases. Front. Immunol. 2022, 13, 911919. [Google Scholar] [CrossRef] [PubMed]
- Barata, J.T.; Durum, S.K.; Seddon, B. Flip the coin: IL-7 and IL-7R in health and disease. Nat. Immunol. 2019, 20, 1584–1593. [Google Scholar] [CrossRef]
- Sun, L.; Gang, X.; Li, Z.; Zhao, X.; Zhou, T.; Zhang, S.; Wang, G. Advances in Understanding the Roles of CD244 (SLAMF4) in Immune Regulation and Associated Diseases. Front. Immunol. 2021, 12, 648182. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Raptopoulou, A.P.; Bertsias, G.; Makrygiannakis, D.; Verginis, P.; Kritikos, I.; Tzardi, M.; Klareskog, L.; Catrina, A.I.; Sidiropoulos, P.; Boumpas, D.T. The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum. 2010, 62, 1870–1880. [Google Scholar] [CrossRef]
- Luo, Q.; Ye, J.; Zeng, L.; Luo, Z.; Deng, Z.; Li, X.; Guo, Y.; Huang, Z.; Li, J. Elevated expression of PD-1 on T cells correlates with disease activity in rheumatoid arthritis. Mol. Med. Rep. 2018, 17, 3297–3305. [Google Scholar] [CrossRef]
- Koohini, Z.; Hossein-Nataj, H.; Mobini, M.; Hosseinian-Amiri, A.; Rafiei, A.; Asgarian-Omran, H. Analysis of PD-1 and Tim-3 expression on CD4+ T cells of patients with rheumatoid arthritis; negative association with DAS28. Clin. Rheumatol. 2018, 37, 2063–2071. [Google Scholar] [CrossRef]
- Tuttle, J.; Drescher, E.; Simón-Campos, J.A.; Emery, P.; Greenwald, M.; Kivitz, A.; Rha, H.; Yachi, P.; Kiley, C.; Nirula, A. A Phase 2 Trial of Peresolimab for Adults with Rheumatoid Arthritis. N. Engl. J. Med. 2023, 388, 1853–1862. [Google Scholar] [CrossRef]
- AldAldridge, J.; Andersson, K.; Gjertsson, I.; Hultgård Ekwall, A.K.; Hallström, M.; van Vollenhoven, R.; Lundell, A.C.; Rudin, A. Blood PD-1+TFh and CTLA-4+CD4+ T cells predict remission after CTLA-4Ig treatment in early rheumatoid arthritis. Rheumatology 2022, 61, 1233–1242. [Google Scholar] [CrossRef]
- Cao, G.; Chi, S.; Wang, X.; Sun, J.; Zhang, Y. CD4+CXCR5+PD-1+ T Follicular Helper Cells Play a Pivotal Role in the Development of Rheumatoid Arthritis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 3032–3040. [Google Scholar] [CrossRef]
- An, L.M.; Li, J.; Ji, L.L.; Li, G.T.; Zhang, Z.L. Detection of peripheral follicular helper T cells in rheumatoid arthritis. Beijing Da Xue Xue Bao Yi Xue Ban = J. Peking Univ. Health Sci. 2016, 48, 951–957. [Google Scholar]
- Wang, J.; Shan, Y.; Jiang, Z.; Feng, J.; Li, C.; Ma, L.; Jiang, Y. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Exp. Immunol. 2013, 174, 212–220. [Google Scholar] [CrossRef]
- Rao, D.A. T Cells That Help B Cells in Chronically Inflamed Tissues. Front. Immunol. 2018, 9, 1924. [Google Scholar] [CrossRef]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, K.; Mizoguchi, F.; et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017, 542, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, Z.; Zeng, X.; Xia, C.; Xu, L.; Xu, Q.; Song, Y.; Liu, C. Circulating CD4+ FoxP3− CXCR5− CXCR3+ PD-1(hi) cells are elevated in active rheumatoid arthritis and reflect the severity of the disease. Int. J. Rheum. Dis. 2021, 24, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Fortea-Gordo, P.; Nuño, L.; Villalba, A.; Peiteado, D.; Monjo, I.; Sánchez-Mateos, P.; Puig-Kröger, A.; Balsa, A.; Miranda-Carús, M.E. Two populations of circulating PD-1hiCD4 T cells with distinct B cell helping capacity are elevated in early rheumatoid arthritis. Rheumatology 2019, 58, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, C.; Xu, F.; Qi, L.; Wang, J.; Yang, P. Imbalance of circulating Tfr/Tfh ratio in patients with rheumatoid arthritis. Clin. Exp. Med. 2019, 19, 55–64. [Google Scholar] [CrossRef]
- Cao, G.; Wang, P.; Cui, Z.; Yue, X.; Chi, S.; Ma, A.; Zhang, Y. An imbalance between blood CD4+CXCR5+Foxp3+ Tfr cells and CD4+CXCR5+ Tfh cells may contribute to the immunopathogenesis of rheumatoid arthritis. Mol. Immunol. 2020, 125, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Wang, Y.; Hu, F.; Li, B.; Guo, Q.; Zheng, X.; Liu, Y.; Gao, C.; Li, X.; Wang, C. Altered Distribution of Circulating T Follicular Helper-Like Cell Subsets in Rheumatoid Arthritis Patients. Front. Med. 2021, 8, 690100. [Google Scholar] [CrossRef] [PubMed]
- Higashioka, K.; Yoshimura, M.; Sakuragi, T.; Ayano, M.; Kimoto, Y.; Mitoma, H.; Ono, N.; Arinobu, Y.; Kikukawa, M.; Yamada, H.; et al. Human PD-1(hi)CD8+ T Cells Are a Cellular Source of IL-21 in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 654623. [Google Scholar] [CrossRef]
- Wasén, C.; Turkkila, M.; Bossios, A.; Erlandsson, M.; Andersson, K.M.; Ekerljung, L.; Malmhäll, C.; Brisslert, M.; Töyrä Silfverswärd, S.; Lundbäck, B.; et al. Smoking activates cytotoxic CD8+ T cells and causes survivin release in rheumatoid arthritis. J. Autoimmun. 2017, 78, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Bartosińska, J.; Zakrzewska, E.; Król, A.; Raczkiewicz, D.; Purkot, J.; Majdan, M.; Krasowska, D.; Chodorowska, G.; Giannopoulos, K. Differential expression of programmed death 1 (PD-1) on CD4+ and CD8+ T cells in rheumatoid arthritis and psoriatic arthritis. Pol. Arch. Intern. Med. 2017, 127, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, Y.; Sui, Y.; Sun, J.; Li, D.; Li, G.; Liu, J.; Li, T.; Shu, Q. High Interleukin-37 (IL-37) Expression and Increased Mucin-Domain Containing-3 (TIM-3) on Peripheral T Cells in Patients with Rheumatoid Arthritis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 5660–5667. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shu, Q.; Gao, L.; Hou, N.; Zhao, D.; Liu, X.; Zhang, X.; Xu, L.; Yue, X.; Zhu, F.; et al. Increased Tim-3 expression on peripheral lymphocytes from patients with rheumatoid arthritis negatively correlates with disease activity. Clin. Immunol. 2010, 137, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Peng, D.; He, Y.; Zhang, H.; Sun, H.; Shan, S.; Song, Y.; Zhang, S.; Xiao, H.; Song, H.; et al. Expression of TIM-3 on CD4+ and CD8+ T cells in the peripheral blood and synovial fluid of rheumatoid arthritis. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2014, 122, 899–904. [Google Scholar] [CrossRef]
- Skejoe, C.; Hansen, A.S.; Stengaard-Pedersen, K.; Junker, P.; Hoerslev-Pedersen, K.; Hetland, M.L.; Oestergaard, M.; Greisen, S.; Hvid, M.; Deleuran, M.; et al. T-cell immunoglobulin and mucin domain 3 is upregulated in rheumatoid arthritis, but insufficient in controlling inflammation. Am. J. Clin. Exp. Immunol. 2022, 11, 34–44. [Google Scholar]
- Nakazawa, M.; Suzuki, K.; Takeshita, M.; Inamo, J.; Kamata, H.; Ishii, M.; Oyamada, Y.; Oshima, H.; Takeuchi, T. Distinct Expression of Coinhibitory Molecules on Alveolar T Cells in Patients With Rheumatoid Arthritis-Associated and Idiopathic Inflammatory Myopathy-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2020, 73, 576–586. [Google Scholar] [CrossRef]
- Luo, Q.; Deng, Z.; Xu, C.; Zeng, L.; Ye, J.; Li, X.; Guo, Y.; Huang, Z.; Li, J. Elevated Expression of Immunoreceptor Tyrosine-Based Inhibitory Motif (TIGIT) on T Lymphocytes is Correlated with Disease Activity in Rheumatoid Arthritis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 1232–1241. [Google Scholar] [CrossRef]
- Zhao, W.; Dong, Y.; Wu, C.; Ma, Y.; Jin, Y.; Ji, Y. TIGIT overexpression diminishes the function of CD4 T cells and ameliorates the severity of rheumatoid arthritis in mouse models. Exp. Cell Res. 2016, 340, 132–138. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Y.; Mo, B.; Xue, Y.; Ye, C.; Jiang, Y.; Bi, X.; Liu, M.; Wu, Y.; Wang, J.; et al. Helios but not CD226, TIGIT and Foxp3 is a Potential Marker for CD4+ Treg Cells in Patients with Rheumatoid Arthritis. Cell. Physiol. Biochem. 2019, 52, 1178–1192. [Google Scholar] [CrossRef]
- Luo, Q.; Fu, P.; Guo, Y.; Fu, B.; Guo, Y.; Huang, Q.; Huang, Z.; Li, J. Increased TIGIT+PD-1+CXCR5−CD4+T cells are associated with disease activity in rheumatoid arthritis. Exp. Ther. Med. 2022, 24, 642. [Google Scholar] [CrossRef]
- Anaparti, V.; Tanner, S.; Zhang, C.; O’Neil, L.; Smolik, I.; Meng, X.; Marshall, A.J.; El-Gabalawy, H. Increased frequency of TIGIT+ CD4 T Cell subset in autoantibody-positive first-degree relatives of patients with rheumatoid arthritis. Front. Immunol. 2022, 13, 932627. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Seo, W.; Jerin, C.; Nishikawa, H. Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp. Mol. Med. 2021, 53, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Greisen, S.R.; Yan, Y.; Hansen, A.S.; Venø, M.T.; Nyengaard, J.R.; Moestrup, S.K.; Hvid, M.; Freeman, G.J.; Kjems, J.; Deleuran, B. Extracellular Vesicles Transfer the Receptor Programmed Death-1 in Rheumatoid Arthritis. Front. Immunol. 2017, 8, 851. [Google Scholar] [CrossRef]
- Schioppo, T.; Ubiali, T.; Ingegnoli, F.; Bollati, V.; Caporali, R. The role of extracellular vesicles in rheumatoid arthritis: A systematic review. Clin. Rheumatol. 2021, 40, 3481–3497. [Google Scholar] [CrossRef]
- Zeidler, J.D.; Hogan, K.A.; Agorrody, G.; Peclat, T.R.; Kashyap, S.; Kanamori, K.S.; Gomez, L.S.; Mazdeh, D.Z.; Warner, G.M.; Thompson, K.L.; et al. The CD38 glycohydrolase and the NAD sink: Implications for pathological conditions. Am. J. Physiol. Cell Physiol. 2022, 322, C521–C545. [Google Scholar] [CrossRef]
- Wang, H.; Fang, K.; Yan, W.; Chang, X. T-Cell Immune Imbalance in Rheumatoid Arthritis Is Associated with Alterations in NK Cells and NK-Like T Cells Expressing CD38. J. Innate Immun. 2022, 14, 148–166. [Google Scholar] [CrossRef]
- Chaturvedi, V.; Marsh, R.A.; Zoref-Lorenz, A.; Owsley, E.; Chaturvedi, V.; Nguyen, T.C.; Goldman, J.R.; Henry, M.M.; Greenberg, J.N.; Ladisch, S.; et al. T-cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood 2021, 137, 2337–2346. [Google Scholar] [CrossRef]
- Wang, R.; Singaraju, A.; Marks, K.E.; Shakib, L.; Dunlap, G.; Adejoorin, I.; Greisen, S.R.; Chen, L.; Tirpack, A.K.; Aude, C.; et al. Clonally expanded CD38(hi) cytotoxic CD8 T cells define the T cell infiltrate in checkpoint inhibitor-associated arthritis. Sci. Immunol. 2023, 8, eadd1591. [Google Scholar] [CrossRef]
- Huang, C.H.; Fan, J.H.; Jeng, W.J.; Chang, S.T.; Yang, C.K.; Teng, W.; Wu, T.H.; Hsieh, Y.C.; Chen, W.T.; Chen, Y.C.; et al. Innate-like bystander-activated CD38+ HLA-DR+ CD8+ T cells play a pathogenic role in patients with chronic hepatitis C. Hepatology 2022, 76, 803–818. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, L.; Nguyen, T.H.O.; Wan, Y.; Sant, S.; Quiñones-Parra, S.M.; Crawford, J.C.; Eltahla, A.A.; Rizzetto, S.; Bull, R.A.; et al. Clonally diverse CD38+HLA-DR+CD8+ T cells persist during fatal H7N9 disease. Nat. Commun. 2018, 9, 824. [Google Scholar] [CrossRef]
- Du, J.; Wei, L.; Li, G.; Hua, M.; Sun, Y.; Wang, D.; Han, K.; Yan, Y.; Song, C.; Song, R.; et al. Persistent High Percentage of HLA-DR+CD38(high) CD8+ T Cells Associated With Immune Disorder and Disease Severity of COVID-19. Front. Immunol. 2021, 12, 735125. [Google Scholar] [CrossRef] [PubMed]
- Bobcakova, A.; Barnova, M.; Vysehradsky, R.; Petriskova, J.; Kocan, I.; Diamant, Z.; Jesenak, M. Activated CD8+CD38+ Cells Are Associated With Worse Clinical Outcome in Hospitalized COVID-19 Patients. Front. Immunol. 2022, 13, 861666. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.; Chaturvedi, V.; Owsley, E.; Lane, A.; Heyenbruch, D.; Lutzko, C.M.; Leemhuis, T.; Grimley, M.S.; Nelson, A.S.; Davies, S.M.; et al. CD38(bright)CD8+ T Cells Associated with the Development of Acute GVHD Are Activated, Proliferating, and Cytotoxic Trafficking Cells. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2020, 26, 1–6. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Schou, M.D.; McMahon, J.H.; Deeks, S.G.; Fromentin, R.; Chomont, N.; Wykes, M.N.; Rasmussen, T.A.; Lewin, S.R. Soluble immune checkpoints as correlates for HIV persistence and T cell function in people with HIV on antiretroviral therapy. Front. Immunol. 2023, 14, 1123342. [Google Scholar] [CrossRef]
- Liu, C.; Jiang, J.; Gao, L.; Wang, X.; Hu, X.; Wu, M.; Wu, J.; Xu, T.; Shi, Q.; Zhang, X. Soluble PD-1 aggravates progression of collagen-induced arthritis through Th1 and Th17 pathways. Arthritis Res. Ther. 2015, 17, 340. [Google Scholar] [CrossRef]
- Matsumoto, H.; Fujita, Y.; Asano, T.; Matsuoka, N.; Temmoku, J.; Sato, S.; Yashiro-Furuya, M.; Watanabe, H.; Migita, K. T cell immunoglobulin and mucin domain-3 is associated with disease activity and progressive joint damage in rheumatoid arthritis patients. Medicine 2020, 99, e22892. [Google Scholar] [CrossRef] [PubMed]
- Greisen, S.R.; Rasmussen, T.K.; Stengaard-Pedersen, K.; Hetland, M.L.; Hørslev-Petersen, K.; Hvid, M.; Deleuran, B. Increased soluble programmed death-1 (sPD-1) is associated with disease activity and radiographic progression in early rheumatoid arthritis. Scand. J. Rheumatol. 2014, 43, 101–108. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, L.; Nie, L.; Zhang, S.; Liu, L.; Du, Y.; Xue, J. Soluble programmed death molecule 1 (sPD-1) as a predictor of interstitial lung disease in rheumatoid arthritis. BMC Immunol. 2021, 22, 69. [Google Scholar] [CrossRef]
- Wu, X.; Xu, L.; Cheng, Q.; Nie, L.; Zhang, S.; Du, Y.; Xue, J. Increased serum soluble programmed death ligand 1(sPD-L1) is associated with the presence of interstitial lung disease in rheumatoid arthritis: A monocentric cross-sectional study. Respir. Med. 2020, 166, 105948. [Google Scholar] [CrossRef]
- Wasén, C.; Erlandsson, M.C.; Bossios, A.; Ekerljung, L.; Malmhäll, C.; Töyrä Silfverswärd, S.; Pullerits, R.; Lundbäck, B.; Bokarewa, M.I. Smoking Is Associated With Low Levels of Soluble PD-L1 in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1677. [Google Scholar] [CrossRef]
- Bommarito, D.; Hall, C.; Taams, L.S.; Corrigall, V.M. Inflammatory cytokines compromise programmed cell death-1 (PD-1)-mediated T cell suppression in inflammatory arthritis through up-regulation of soluble PD-1. Clin. Exp. Immunol. 2017, 188, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Dang, Q.M.; Watanabe, R.; Shiomi, M.; Fukumoto, K.; Nobashi, T.W.; Okano, T.; Yamada, S.; Hashimoto, M. Rheumatic Immune-Related Adverse Events due to Immune Checkpoint Inhibitors-A 2023 Update. Int. J. Mol. Sci. 2023, 24, 5643. [Google Scholar] [CrossRef]
- Kim, S.T.; Chu, Y.; Misoi, M.; Suarez-Almazor, M.E.; Tayar, J.H.; Lu, H.; Buni, M.; Kramer, J.; Rodriguez, E.; Hussain, Z.; et al. Distinct molecular and immune hallmarks of inflammatory arthritis induced by immune checkpoint inhibitors for cancer therapy. Nat. Commun. 2022, 13, 1970. [Google Scholar] [CrossRef] [PubMed]
- Kostine, M.; Finckh, A.; Bingham, C.O.; Visser, K.; Leipe, J.; Schulze-Koops, H.; Choy, E.H.; Benesova, K.; Radstake, T.; Cope, A.P.; et al. EULAR points to consider for the diagnosis and management of rheumatic immune-related adverse events due to cancer immunotherapy with checkpoint inhibitors. Ann. Rheum. Dis. 2021, 80, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Cunningham-Bussel, A.; Wang, J.; Prisco, L.C.; Martin, L.W.; Vanni, K.M.M.; Zaccardelli, A.; Nasrallah, M.; Gedmintas, L.; MacFarlane, L.A.; Shadick, N.A.; et al. Predictors of Rheumatic Immune-Related Adverse Events and De Novo Inflammatory Arthritis After Immune Checkpoint Inhibitor Treatment for Cancer. Arthritis Rheumatol. 2022, 74, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.C.; Bingham, C.O., 3rd. Expert Perspective: Immune Checkpoint Inhibitors and Rheumatologic Complications. Arthritis Rheumatol. 2021, 73, 553–565. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers | Mean ± SD (Number) | p Value | |
|---|---|---|---|
| RA | Normal | ||
| CD4+CD279 | 4.6 (9.625) (N = 48) | 3.1 (6.9) (N = 27) | 0.0088 |
| CD4+TIM3+ | 4.75 (5.75) (N = 48) | 2.4 (2.225) (N = 26) | 0.0171 |
| CD4+CTLA4+ | 0.15 (0.3) (N = 46) | 0.1 (0.1) (N = 27) | 0.0522 |
| CD4+TIGIT+ | 25.35 (11.23) (N = 48) | 20.1 (5.73) (N = 26) | 0.0016 * |
| CD8+CD279+ | 5.7 (8.3) (N = 47) | 3.1 (6.2) (N = 27) | 0.0253 |
| CD8+TIM3+ | 12.9 (20.255) (N = 48) | 7.5 (8.8) (N = 27) | 0.0012 * |
| CD8+LAG3+ | 0.55 (1) (N = 46) | 0.1 (0.3) (N = 27) | 0.0015 * |
| CD8+CD127+ | 21.2 (22.63) (N = 48) | 40.2 (36.2) (N = 27) | 0.0012 * |
| CD8+TIGIT+ | 47.95 (23.65) (N = 48) | 25.4 (12.9) (N = 27) | <0.0001 * |
| CD8+CD160+ | 36.7 (18.1) (N = 47) | 28.6 (15.6) (N = 27) | 0.0513 |
| CD8+HLA-DR+ | 43.8 (24.8) (N = 31) | 15.4 (13.85) (N = 17) | <0.0001 * |
| CD8+CD38+ | 26.9 (15.8) (N = 31) | 23.9 (11.9) (N = 17) | 0.2493 |
| CD3+CD244+ | 8.65 (30.68) (N = 48) | 0.45 (8.45) (N = 26) | 0.0791 |
| CD3+CD279+ | 4.7 (9.33) (N = 46) | 2 (5.7) (N = 27) | 0.025 |
| CD3+TIGIT+ | 40 (31.8) (N = 47) | 22.5 (18.6) (N = 27) | 0.0001 * |
| Markers | Mean ± SD (Number) | p Value | |
|---|---|---|---|
| RA | Normal | ||
| CD4+CD279+TIM3+ | 0.4 (0.6) (N = 47) | 0.2 (0.4) (N = 27) | 0.0246 |
| CD8+CD279+TIM3+ | 0.2 (0.325) (N = 46) | 0.1 (0.1) (N = 26) | 0.0052 * |
| CD8+CD127+TIGIT+ | 2.3 (4.3) (N = 47) | 2 (3.5) (N = 27) | 0.4903 |
| CD8+CD160+CD244+ | 0.2 (0.4) (N = 47) | 0.1 (0.55) (N = 25) | 0.912 |
| CD8+HLA-DR+CD127+ | 0.6 (0.4) (N = 19) | 0.9 (2.3) (N = 19) | 0.3567 |
| CD8+HLA-DR+CD38+ | 7.5 (6.6) (N = 47) | 3.1 (3.3) (N = 27) | 0.0004 * |
| CD3+CD160+CD244+ | 0.2 (0.5) (N = 47) | 0.2 (0.2) (N = 27) | 0.1262 |
| CD3+CD279+TIGIT+ | 3.05 (4.95) (N = 46) | 1.7 (2.975) (N = 26) | 0.0657 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-M.; Jan Wu, Y.-J.; Huang, L.-Y.; Zheng, J.-W.; Chen, J.-Y. Comprehensive Co-Inhibitory Receptor (Co-IR) Expression on T Cells and Soluble Proteins in Rheumatoid Arthritis. Cells 2024, 13, 403. https://doi.org/10.3390/cells13050403
Wang C-M, Jan Wu Y-J, Huang L-Y, Zheng J-W, Chen J-Y. Comprehensive Co-Inhibitory Receptor (Co-IR) Expression on T Cells and Soluble Proteins in Rheumatoid Arthritis. Cells. 2024; 13(5):403. https://doi.org/10.3390/cells13050403
Chicago/Turabian StyleWang, Chin-Man, Yeong-Jian Jan Wu, Li-Yu Huang, Jian-Wen Zheng, and Ji-Yih Chen. 2024. "Comprehensive Co-Inhibitory Receptor (Co-IR) Expression on T Cells and Soluble Proteins in Rheumatoid Arthritis" Cells 13, no. 5: 403. https://doi.org/10.3390/cells13050403
APA StyleWang, C.-M., Jan Wu, Y.-J., Huang, L.-Y., Zheng, J.-W., & Chen, J.-Y. (2024). Comprehensive Co-Inhibitory Receptor (Co-IR) Expression on T Cells and Soluble Proteins in Rheumatoid Arthritis. Cells, 13(5), 403. https://doi.org/10.3390/cells13050403