
An Efficient Method for Isolating and Purifying Nuclei from Mice Brain for Single-Molecule Imaging Using High-Speed Atomic Force Microscopy

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
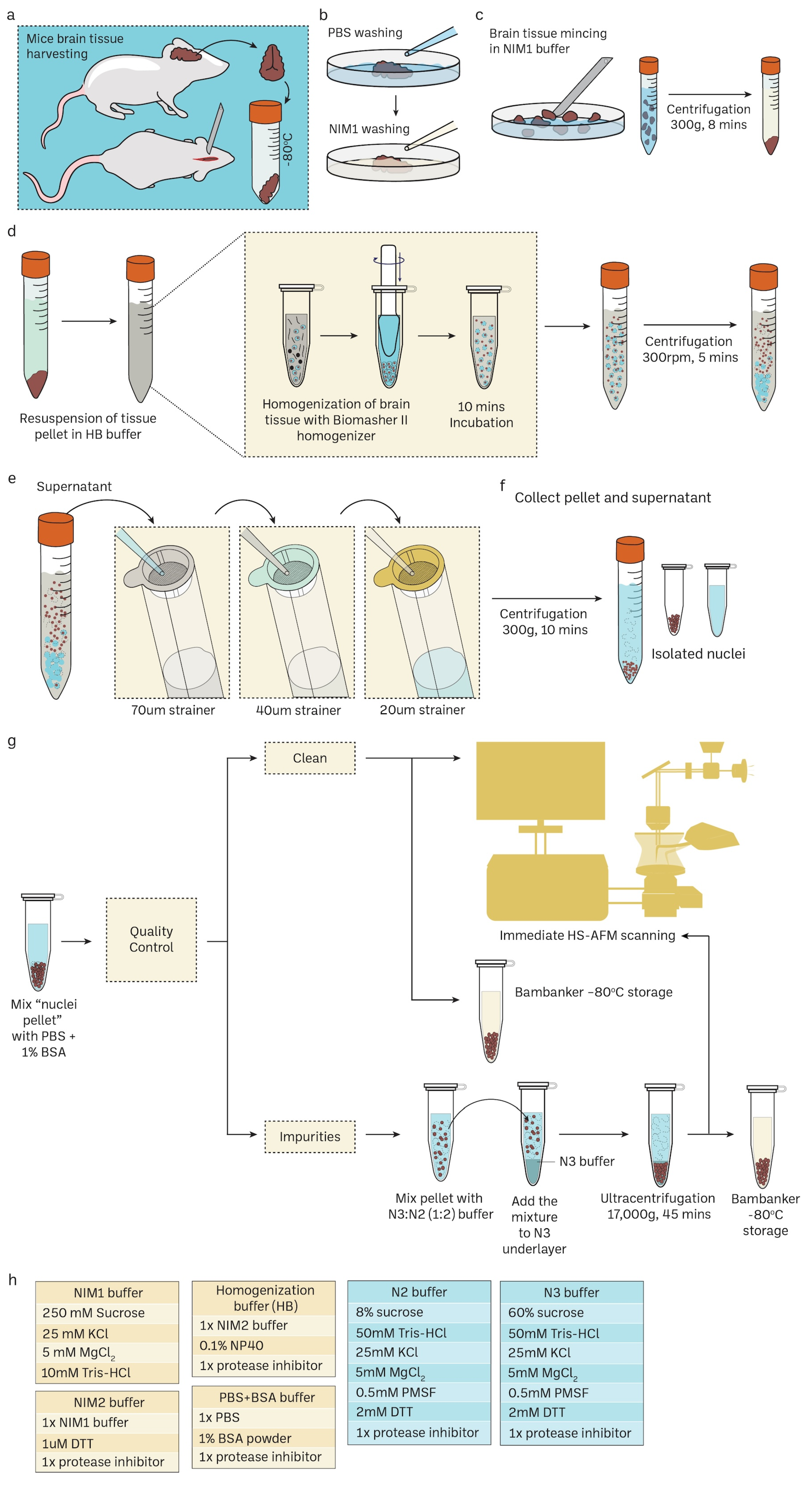
2.1. Nuclei Isolation
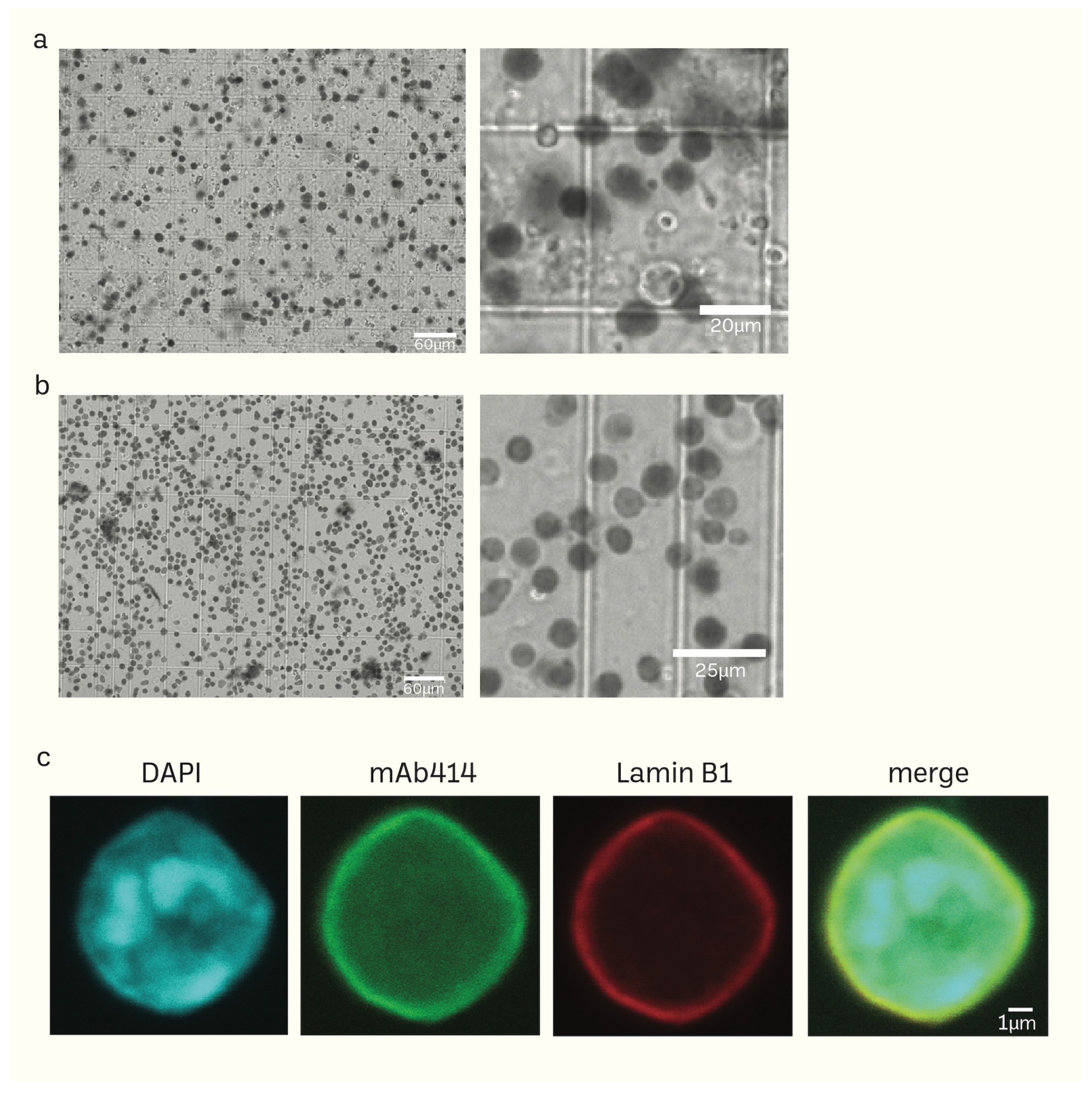
2.2. Immunostaining and Confocal Microscopic Analysis
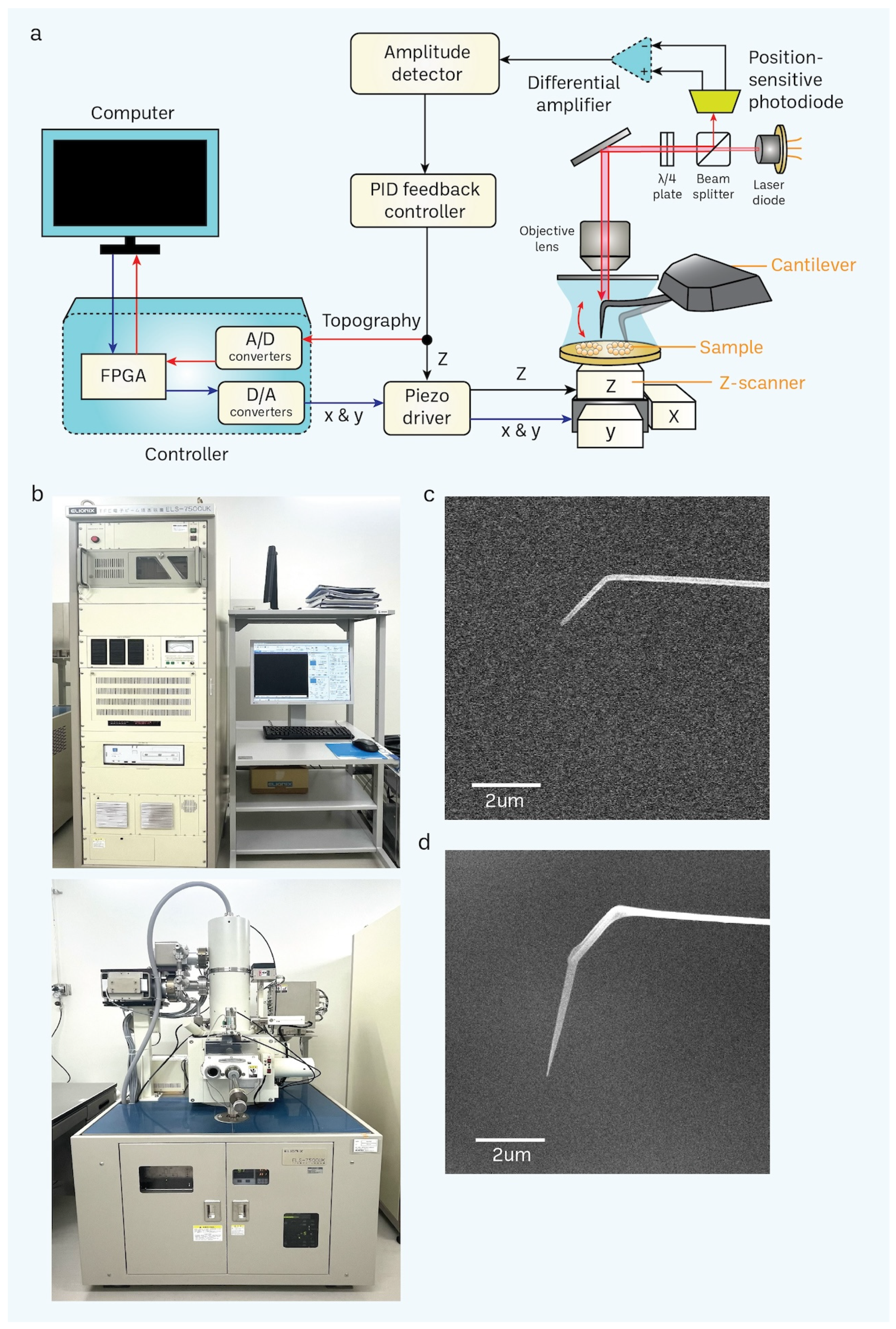
2.3. Electron-Beam Deposition (EBD) for the Nanofabrication of Cantilever Tips
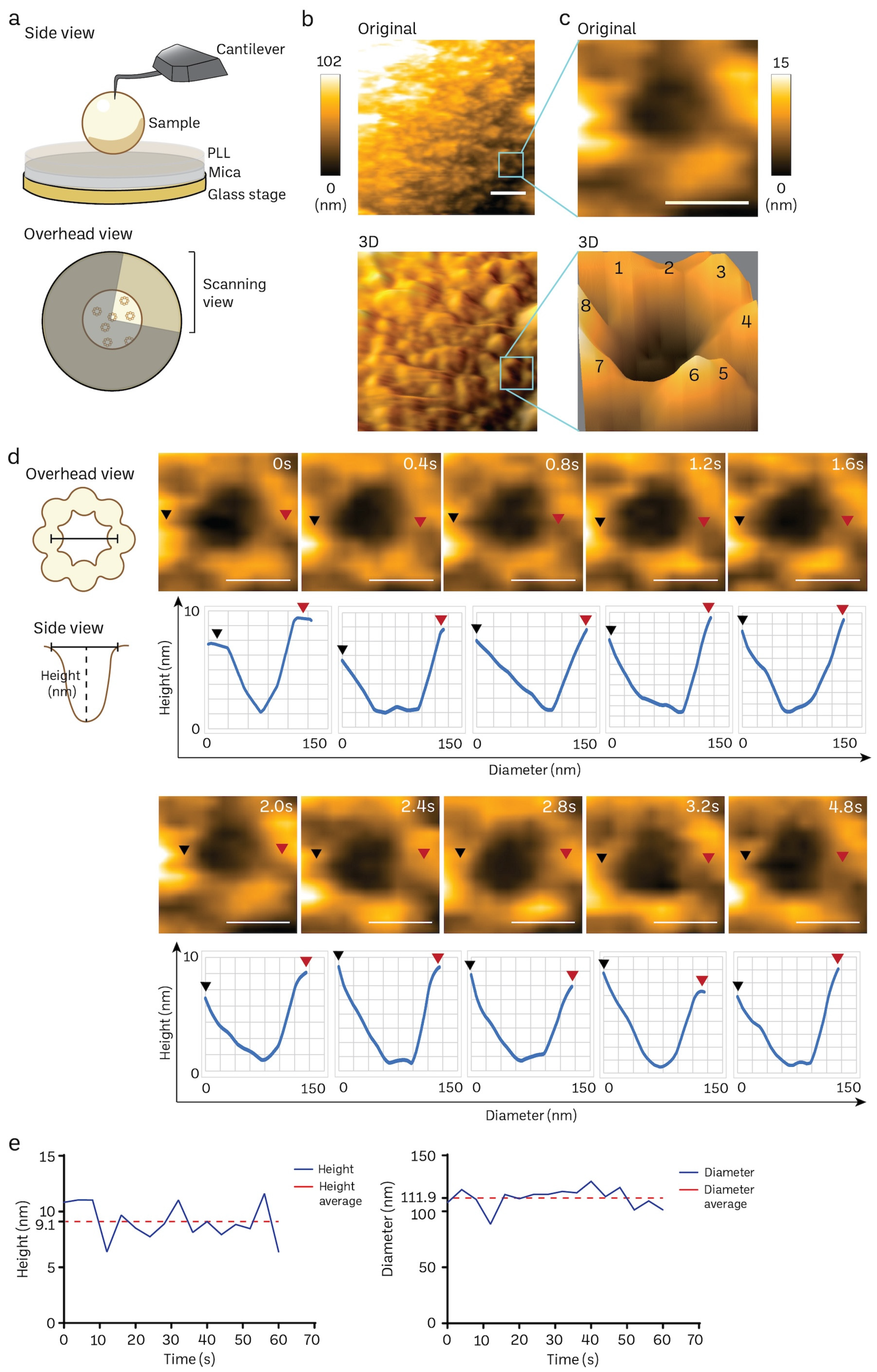
2.4. HS-AFM Imaging
2.5. HS-AFM Image Processing
2.6. Analysis of FG-Nups
3. Results
3.1. Preparation of Mice Brain Nuclei
3.2. Nanofabrication of Cantilevers Using Electron-Beam Deposition (EBD)
3.3. Acquisition of Nanoscopic Nuclear Pore Complex Topology of Normal Mice Brain Nuclei Using HS-AFM
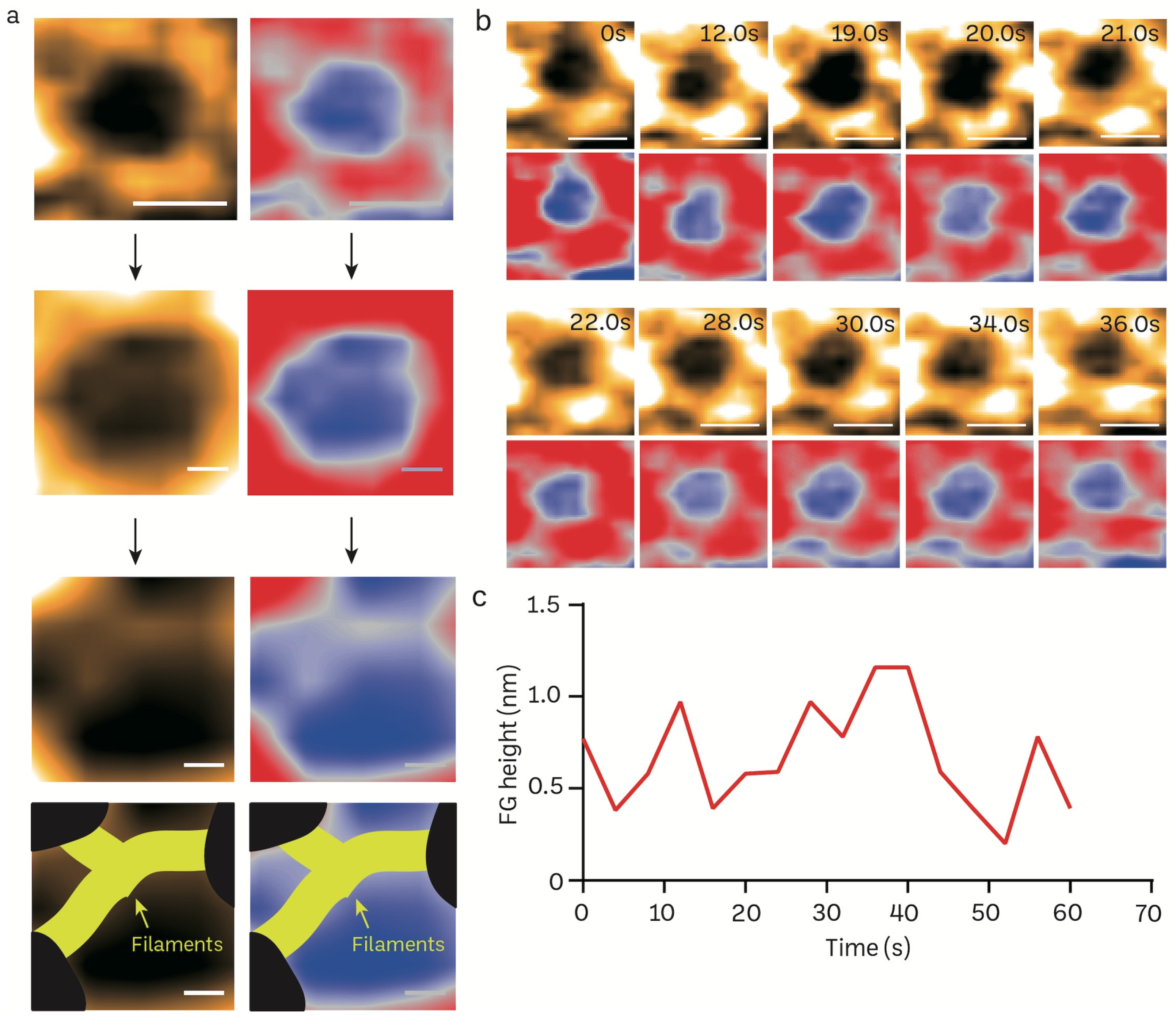
3.4. Transient Rapid Conformational Dynamics of FG-NUP Complexes Inside the Native Mice Brain Nuclear Pores
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hara, Y. Physical forces modulate interphase nuclear size. Curr. Opin. Cell Biol. 2023, 85, 102253. [Google Scholar] [CrossRef]
- Imamoto, N.; Funakoshi, T. Nuclear pore dynamics during the cell cycle. Curr. Opin. Cell Biol. 2012, 24, 453–459. [Google Scholar] [CrossRef]
- Maeshima, K.; Tamura, S.; Hansen, J.C.; Itoh, Y. Fluid-like chromatin: Toward understanding the real chromatin organization present in the cell. Curr. Opin. Cell Biol. 2020, 64, 77–89. [Google Scholar] [CrossRef]
- Hoelz, A.; Glavy, J.S.; Beck, M. Toward the atomic structure of the nuclear pore complex: When top down meets bottom up. Nat. Struct. Mol. Biol. 2016, 23, 624–630. [Google Scholar] [CrossRef]
- Dultz, E.; Wojtynek, M.; Medalia, O.; Onischenko, E. The Nuclear Pore Complex: Birth, Life, and Death of a Cellular Behemoth. Cells 2022, 11, 1456. [Google Scholar] [CrossRef] [PubMed]
- Cronshaw, J.M.; Krutchinsky, A.N.; Zhang, W.; Chait, B.T.; Matunis, M.J. Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 2002, 158, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Rout, M.P.; Aitchison, J.D.; Suprapto, A.; Hjertaas, K.; Zhao, Y.; Chait, B.T. The yeast nuclear pore complex: Composition, architecture, and transport mechanism. J. Cell Biol. 2000, 148, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Paci, G.; Caria, J.; Lemke, E.A. Cargo transport through the nuclear pore complex at a glance. J. Cell Sci. 2021, 134, jcs247874. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Svirelis, J.; Medin, J.; Järlebark, J.; Hailes, R.; Dahlin, A. Pore performance: Artificial nanoscale constructs that mimic the biomolecular transport of the nuclear pore complex. Nanoscale Adv. 2022, 4, 4925–4937. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, C.E.; Allegretti, M.; Rantos, V.; Goetz, S.K.; Obarska-Kosinska, A.; Zagoriy, I.; Halavatyi, A.; Hummer, G.; Mahamid, J.; Kosinski, J.; et al. Nuclear pores dilate and constrict in cellulo. Science 2021, 374, eabd9776. [Google Scholar] [CrossRef]
- Allegretti, M.; Zimmerli, C.E.; Rantos, V.; Wilfling, F.; Ronchi, P.; Fung, H.K.H.; Lee, C.W.; Hagen, W.; Turoňová, B.; Karius, K.; et al. In-cell architecture of the nuclear pore and snapshots of its turnover. Nature 2020, 586, 796–800. [Google Scholar] [CrossRef]
- Goswami, R.; Gupta, A.; Bednova, O.; Coulombe, G.; Patel, D.; Rotello, V.M.; Leyton, J.V. Nuclear localization signal-tagged systems: Relevant nuclear import principles in the context of current therapeutic design. Chem. Soc. Rev. 2024, 53, 204–226. [Google Scholar] [CrossRef]
- Manda, N.K.; Golla, U.; Sesham, K.; Desai, P.; Joshi, S.; Patel, S.; Nalla, S.; Kondam, S.; Singh, L.; Dewansh, D.; et al. Tuning between Nuclear Organization and Functionality in Health and Disease. Cells 2023, 12, 706. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, L.; Chen, L.; Gong, B.; Jia, D.; Sun, Q. Nuclear transport proteins: Structure, function, and disease relevance. Signal Transduct. Target. Ther. 2023, 8, 425. [Google Scholar] [CrossRef] [PubMed]
- Kutay, U.; Jühlen, R.; Antonin, W. Mitotic disassembly and reassembly of nuclear pore complexes. Trends Cell Biol. 2021, 31, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Capelson, M. You are who your friends are-nuclear pore proteins as components of chromatin-binding complexes. FEBS Lett. 2023, 597, 2769–2781. [Google Scholar] [CrossRef] [PubMed]
- Fahrenkrog, B.; Gasser, S.M. Structure and function of the nuclear envelope and nuclear pores. FEBS Lett. 2023, 597, 2703–2704. [Google Scholar] [CrossRef]
- Veldsink, A.C.; Gallardo, P.; Lusk, C.P.; Veenhoff, L.M. Changing the guard-nuclear pore complex quality control. FEBS Lett. 2023, 597, 2739–2749. [Google Scholar] [CrossRef]
- Rush, C.; Jiang, Z.; Tingey, M.; Feng, F.; Yang, W. Unveiling the complexity: Assessing models describing the structure and function of the nuclear pore complex. Front. Cell Dev. Biol. 2023, 11, 1245939. [Google Scholar] [CrossRef]
- Mohamed, M.S.; Kobayashi, A.; Taoka, A.; Watanabe-Nakayama, T.; Kikuchi, Y.; Hazawa, M.; Minamoto, T.; Fukumori, Y.; Kodera, N.; Uchihashi, T.; et al. High-Speed Atomic Force Microscopy Reveals Loss of Nuclear Pore Resilience as a Dying Code in Colorectal Cancer Cells. ACS Nano 2017, 11, 5567–5578. [Google Scholar] [CrossRef]
- Shevelyov, Y.Y. Interactions of Chromatin with the Nuclear Lamina and Nuclear Pore Complexes. Int. J. Mol. Sci. 2023, 24, 15771. [Google Scholar] [CrossRef]
- Hoogenboom, B.W.; Hough, L.E.; Lemke, E.A.; Lim, R.Y.H.; Onck, P.R.; Zilman, A. Physics of the Nuclear Pore Complex: Theory, Modeling and Experiment. Phys. Rep. 2021, 921, 1–53. [Google Scholar] [CrossRef]
- Minasbekyan, L.A.; Badalyan, H.G. Physical model of the nuclear membrane permeability mechanism. Biophys. Rev. 2023, 15, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Penzo, A.; Palancade, B. Puzzling out nuclear pore complex assembly. FEBS Lett. 2023, 597, 2705–2727. [Google Scholar] [CrossRef]
- Davis, L.K.; Ford, I.J.; Hoogenboom, B.W. Crowding-induced phase separation of nuclear transport receptors in FG nucleoporin assemblies. eLife 2022, 11, e72627. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, S.; Mobbs, G.W.; Bley, C.J.; Nie, S.; Patke, A.; Hoelz, A. Structure and Function of the Nuclear Pore Complex. Cold Spring Harb. Perspect. Biol. 2022, 14, a041264. [Google Scholar] [CrossRef] [PubMed]
- Wing, C.E.; Fung, H.Y.J.; Chook, Y.M. Karyopherin-mediated nucleocytoplasmic transport. Nat. Rev. Mol. Cell Biol. 2022, 23, 307–328. [Google Scholar] [CrossRef]
- Wang, C.; Wojtynek, M.; Medalia, O. Structural investigation of eukaryotic cells: From the periphery to the interior by cryo-electron tomography. Adv. Biol. Regul. 2023, 87, 100923. [Google Scholar] [CrossRef]
- Tai, L.; Yin, G.; Sun, F.; Zhu, Y. Cryo-electron Microscopy Reveals the Structure of the Nuclear Pore Complex. J. Mol. Biol. 2023, 435, 168051. [Google Scholar] [CrossRef]
- Wong, R.W. New Activities of the Nuclear Pore Complexes. Cells 2021, 10, 2123. [Google Scholar] [CrossRef]
- Coyne, A.N.; Rothstein, J.D. Nuclear pore complexes—A doorway to neural injury in neurodegeneration. Nat. Rev. Neurol. 2022, 18, 348–362. [Google Scholar] [CrossRef]
- Sajidah, E.S.; Lim, K.; Wong, R.W. How SARS-CoV-2 and Other Viruses Build an Invasion Route to Hijack the Host Nucleocytoplasmic Trafficking System. Cells 2021, 10, 1424. [Google Scholar] [CrossRef]
- Kato, K.; Ikliptikawati, D.K.; Kobayashi, A.; Kondo, H.; Lim, K.; Hazawa, M.; Wong, R.W. Overexpression of SARS-CoV-2 protein ORF6 dislocates RAE1 and NUP98 from the nuclear pore complex. Biochem. Biophys. Res. Commun. 2021, 536, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Makiyama, K.; Hazawa, M.; Kobayashi, A.; Lim, K.; Voon, D.C.; Wong, R.W. NSP9 of SARS-CoV-2 attenuates nuclear transport by hampering nucleoporin 62 dynamics and functions in host cells. Biochem. Biophys. Res. Commun. 2022, 586, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Döhner, K.; Serrero, M.C.; Sodeik, B. The role of nuclear pores and importins for herpes simplex virus infection. Curr. Opin. Virol. 2023, 62, 101361. [Google Scholar] [CrossRef] [PubMed]
- Esparza, M.; Bhat, P.; Fontoura, B.M. Viral-host interactions during splicing and nuclear export of influenza virus mRNAs. Curr. Opin. Virol. 2022, 55, 101254. [Google Scholar] [CrossRef]
- Lucic, B.; de Castro, I.J.; Lusic, M. Viruses in the Nucleus. Cold Spring Harb. Perspect. Biol. 2021, 13, a039446. [Google Scholar] [CrossRef]
- Ikliptikawati, D.K.; Hirai, N.; Makiyama, K.; Sabit, H.; Kinoshita, M.; Matsumoto, K.; Lim, K.; Meguro-Horike, M.; Horike, S.I.; Hazawa, M.; et al. Nuclear transport surveillance of p53 by nuclear pores in glioblastoma. Cell Rep. 2023, 42, 112882. [Google Scholar] [CrossRef]
- Hazawa, M.; Ikliptikawati, D.K.; Iwashima, Y.; Lin, D.C.; Jiang, Y.; Qiu, Y.; Makiyama, K.; Matsumoto, K.; Kobayashi, A.; Nishide, G.; et al. Super-enhancer trapping by the nuclear pore via intrinsically disordered regions of proteins in squamous cell carcinoma cells. Cell Chem. Biol. 2023, 31, 1–13. [Google Scholar] [CrossRef]
- Zhu, X.; Qi, C.; Wang, R.; Lee, J.H.; Shao, J.; Bei, L.; Xiong, F.; Nguyen, P.T.; Li, G.; Krakowiak, J.; et al. Acute depletion of human core nucleoporin reveals direct roles in transcription control but dispensability for 3D genome organization. Cell Rep. 2022, 41, 111576. [Google Scholar] [CrossRef]
- Chua, E.Y.D.; Mendez, J.H.; Rapp, M.; Ilca, S.L.; Tan, Y.Z.; Maruthi, K.; Kuang, H.; Zimanyi, C.M.; Cheng, A.; Eng, E.T.; et al. Better, Faster, Cheaper: Recent Advances in Cryo-Electron Microscopy. Annu. Rev. Biochem. 2022, 91, 1–32. [Google Scholar] [CrossRef]
- Burke, D.F.; Bryant, P.; Barrio-Hernandez, I.; Memon, D.; Pozzati, G.; Shenoy, A.; Zhu, W.; Dunham, A.S.; Albanese, P.; Keller, A.; et al. Towards a structurally resolved human protein interaction network. Nat. Struct. Mol. Biol. 2023, 30, 216–225. [Google Scholar] [CrossRef]
- Bryant, P. Deep learning for protein complex structure prediction. Curr. Opin. Struct. Biol. 2023, 79, 102529. [Google Scholar] [CrossRef] [PubMed]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic force microscope. Phys. Rev. Lett. 1986, 56, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, B.W. Stretching the resolution limit of atomic force microscopy. Nat. Struct. Mol. Biol. 2021, 28, 629–630. [Google Scholar] [CrossRef] [PubMed]
- Liashkovich, I.; Rosso, G.; Shahin, V. Atomic Force Microscopy for Structural and Biophysical Investigations on Nuclear Pore Complexes. Methods Mol. Biol. 2022, 2502, 299–310. [Google Scholar] [CrossRef]
- Vial, A.; Costa, L.; Dosset, P.; Rosso, P.; Boutières, G.; Faklaris, O.; Haschke, H.; Milhiet, P.E.; Doucet, C.M. Structure and mechanics of the human nuclear pore complex basket using correlative AFM-fluorescence superresolution microscopy. Nanoscale 2023, 15, 5756–5770. [Google Scholar] [CrossRef]
- Shahin, V. Cellular transport: Gatekeepers of the nucleus. Nat. Nanotechnol. 2016, 11, 658–659. [Google Scholar] [CrossRef]
- Sakiyama, Y.; Panatala, R.; Lim, R.Y.H. Structural dynamics of the nuclear pore complex. Semin. Cell Dev. Biol. 2017, 68, 27–33. [Google Scholar] [CrossRef]
- Heath, G.R.; Kots, E.; Robertson, J.L.; Lansky, S.; Khelashvili, G.; Weinstein, H.; Scheuring, S. Localization atomic force microscopy. Nature 2021, 594, 385–390. [Google Scholar] [CrossRef]
- Heath, G.R.; Lin, Y.C.; Matin, T.R.; Scheuring, S. Structural dynamics of channels and transporters by high-speed atomic force microscopy. Methods Enzymol. 2021, 652, 127–159. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Ruan, Y.; Scheuring, S. High-speed atomic force microscopy to study pore-forming proteins. Methods Enzymol. 2021, 649, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Jukic, N.; Perrino, A.P.; Redondo-Morata, L.; Scheuring, S. Structure and dynamics of ESCRT-III membrane remodeling proteins by high-speed atomic force microscopy. J. Biol. Chem. 2023, 299, 104575. [Google Scholar] [CrossRef] [PubMed]
- Amyot, R.; Kodera, N.; Flechsig, H. BioAFMviewer software for simulation atomic force microscopy of molecular structures and conformational dynamics. J. Struct. Biol. X 2023, 7, 100086. [Google Scholar] [CrossRef]
- Casuso, I.; Redondo-Morata, L.; Rico, F. Biological physics by high-speed atomic force microscopy. Philos. Trans. A Math. Phys. Eng. Sci. 2020, 378, 20190604. [Google Scholar] [CrossRef] [PubMed]
- Ganser, C.; Uchihashi, T. Measuring mechanical properties with high-speed atomic force microscopy. Microscopy 2023. corrected proof. [Google Scholar] [CrossRef]
- Lyubchenko, Y.L.; Shlyakhtenko, L.S. Imaging of DNA and Protein-DNA Complexes with Atomic Force Microscopy. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 63–96. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Noi, K.; Inaba, K. Visualization of structural dynamics of protein disulfide isomerase enzymes in catalysis of oxidative folding and reductive unfolding. Curr. Opin. Struct. Biol. 2021, 66, 49–57. [Google Scholar] [CrossRef]
- Suzuki, Y.; Endo, M.; Sugiyama, H. Studying RNAP-promoter interactions using atomic force microscopy. Methods 2015, 86, 4–9. [Google Scholar] [CrossRef]
- Uchihashi, T.; Ganser, C. Recent advances in bioimaging with high-speed atomic force microscopy. Biophys. Rev. 2020, 12, 363–369. [Google Scholar] [CrossRef]
- Uchihashi, T.; Scheuring, S. Applications of high-speed atomic force microscopy to real-time visualization of dynamic biomolecular processes. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 229–240. [Google Scholar] [CrossRef]
- Umakoshi, T. Near-field optical microscopy toward its applications for biological studies. Biophys. Physicobiol. 2023, 20, e200011. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Ono, K. Single-molecule observation of self-propagating amyloid fibrils. Microscopy 2022, 71, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Sahoo, B.R.; Ramamoorthy, A.; Ono, K. High-Speed Atomic Force Microscopy Reveals the Structural Dynamics of the Amyloid-β and Amylin Aggregation Pathways. Int. J. Mol. Sci. 2020, 21, 4287. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Youcef-Toumi, K. Review: Advanced Atomic Force Microscopy Modes for Biomedical Research. Biosensors 2022, 12, 1116. [Google Scholar] [CrossRef] [PubMed]
- Puppulin, L.; Kanayama, D.; Terasaka, N.; Sakai, K.; Kodera, N.; Umeda, K.; Sumino, A.; Marchesi, A.; Weilin, W.; Tanaka, H.; et al. Macrocyclic Peptide-Conjugated Tip for Fast and Selective Molecular Recognition Imaging by High-Speed Atomic Force Microscopy. ACS Appl. Mater. Interfaces 2021, 13, 54817–54829. [Google Scholar] [CrossRef]
- Ando, T.; Kodera, N.; Takai, E.; Maruyama, D.; Saito, K.; Toda, A. A high-speed atomic force microscope for studying biological macromolecules. Proc. Natl. Acad. Sci. USA 2001, 98, 12468–12472. [Google Scholar] [CrossRef] [PubMed]
- Umeda, K.; McArthur, S.J.; Kodera, N. Spatiotemporal resolution in high-speed atomic force microscopy for studying biological macromolecules in action. Microscopy 2023, 72, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Fukuda, S.; Ngo, K.X.; Flechsig, H. High-Speed Atomic Force Microscopy for Filming Protein Molecules in Dynamic Action. Annu. Rev. Biophys. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Akey, C.W.; Singh, D.; Ouch, C.; Echeverria, I.; Nudelman, I.; Varberg, J.M.; Yu, Z.; Fang, F.; Shi, Y.; Wang, J.; et al. Comprehensive structure and functional adaptations of the yeast nuclear pore complex. Cell 2022, 185, 361–378. [Google Scholar] [CrossRef]
- Schuller, A.P.; Wojtynek, M.; Mankus, D.; Tatli, M.; Kronenberg-Tenga, R.; Regmi, S.G.; Dip, P.V.; Lytton-Jean, A.K.R.; Brignole, E.J.; Dasso, M.; et al. The cellular environment shapes the nuclear pore complex architecture. Nature 2021, 598, 667–671. [Google Scholar] [CrossRef]
- Mohamed, M.S.; Hazawa, M.; Kobayashi, A.; Guillaud, L.; Watanabe-Nakayama, T.; Nakayama, M.; Wang, H.; Kodera, N.; Oshima, M.; Ando, T.; et al. Spatiotemporally tracking of nano-biofilaments inside the nuclear pore complex core. Biomaterials 2020, 256, 120198. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.; Potter, V.R. Nuclei from rat liver: Isolation method that combines purity with high yield. Science 1966, 154, 1662–1665. [Google Scholar] [CrossRef] [PubMed]
- Diguilio, A.L.; Glavy, J.S. Depletion of nucleoporins from HeLa nuclear pore complexes to facilitate the production of ghost pores for in vitro reconstitution. Cytotechnology 2013, 65, 469–479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nadelmann, E.R.; Gorham, J.M.; Reichart, D.; Delaughter, D.M.; Wakimoto, H.; Lindberg, E.L.; Litviňukova, M.; Maatz, H.; Curran, J.J.; Ischiu Gutierrez, D.; et al. Isolation of Nuclei from Mammalian Cells and Tissues for Single-Nucleus Molecular Profiling. Curr. Protoc. 2021, 1, e132. [Google Scholar] [CrossRef]
- Shibata, M.; Watanabe, H.; Uchihashi, T.; Ando, T.; Yasuda, R. High-speed atomic force microscopy imaging of live mammalian cells. Biophys. Physicobiol. 2017, 14, 127–135. [Google Scholar] [CrossRef]
- Shibata, M.; Uchihashi, T.; Ando, T.; Yasuda, R. Long-tip high-speed atomic force microscopy for nanometer-scale imaging in live cells. Sci. Rep. 2015, 5, 8724. [Google Scholar] [CrossRef] [PubMed]
- Sajidah, E.S.; Lim, K.; Yamano, T.; Nishide, G.; Qiu, Y.; Yoshida, T.; Wang, H.; Kobayashi, A.; Hazawa, M.; Dewi, F.R.P.; et al. Spatiotemporal tracking of small extracellular vesicle nanotopology in response to physicochemical stresses revealed by HS-AFM. J. Extracell. Vesicles 2022, 11, e12275. [Google Scholar] [CrossRef]
- Lim, K.; Nishide, G.; Sajidah, E.S.; Yamano, T.; Qiu, Y.; Yoshida, T.; Kobayashi, A.; Hazawa, M.; Ando, T.; Hanayama, R.; et al. Nanoscopic Assessment of Anti-SARS-CoV-2 Spike Neutralizing Antibody Using High-Speed AFM. Nano Lett. 2023, 23, 619–628. [Google Scholar] [CrossRef]
- Lim, K.; Nishide, G.; Yoshida, T.; Watanabe-Nakayama, T.; Kobayashi, A.; Hazawa, M.; Hanayama, R.; Ando, T.; Wong, R.W. Millisecond dynamic of SARS-CoV-2 spike and its interaction with ACE2 receptor and small extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12170. [Google Scholar] [CrossRef]
- Senichkin, V.V.; Prokhorova, E.A.; Zhivotovsky, B.; Kopeina, G.S. Simple and Efficient Protocol for Subcellular Fractionation of Normal and Apoptotic Cells. Cells 2021, 10, 852. [Google Scholar] [CrossRef]
- Herrmann, C.; Avgousti, D.C.; Weitzman, M.D. Differential Salt Fractionation of Nuclei to Analyze Chromatin-associated Proteins from Cultured Mammalian Cells. Bio Protoc. 2017, 7, e2175. [Google Scholar] [CrossRef] [PubMed]
- Penedo, M.; Miyazawa, K.; Okano, N.; Furusho, H.; Ichikawa, T.; Alam, M.S.; Miyata, K.; Nakamura, C.; Fukuma, T. Visualizing intracellular nanostructures of living cells by nanoendoscopy-AFM. Sci. Adv. 2021, 7, eabj4990. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.; Sajidah, E.S.; Kondo, S.; Narimatsu, S.; Sandira, M.I.; Higashiguchi, Y.; Nishide, G.; Taoka, A.; Hazawa, M.; Inaba, Y.; et al. An Efficient Method for Isolating and Purifying Nuclei from Mice Brain for Single-Molecule Imaging Using High-Speed Atomic Force Microscopy. Cells 2024, 13, 279. https://doi.org/10.3390/cells13030279
Qiu Y, Sajidah ES, Kondo S, Narimatsu S, Sandira MI, Higashiguchi Y, Nishide G, Taoka A, Hazawa M, Inaba Y, et al. An Efficient Method for Isolating and Purifying Nuclei from Mice Brain for Single-Molecule Imaging Using High-Speed Atomic Force Microscopy. Cells. 2024; 13(3):279. https://doi.org/10.3390/cells13030279
Chicago/Turabian StyleQiu, Yujia, Elma Sakinatus Sajidah, Sota Kondo, Shinnosuke Narimatsu, Muhammad Isman Sandira, Yoshiki Higashiguchi, Goro Nishide, Azuma Taoka, Masaharu Hazawa, Yuka Inaba, and et al. 2024. "An Efficient Method for Isolating and Purifying Nuclei from Mice Brain for Single-Molecule Imaging Using High-Speed Atomic Force Microscopy" Cells 13, no. 3: 279. https://doi.org/10.3390/cells13030279
APA StyleQiu, Y., Sajidah, E. S., Kondo, S., Narimatsu, S., Sandira, M. I., Higashiguchi, Y., Nishide, G., Taoka, A., Hazawa, M., Inaba, Y., Inoue, H., Matsushima, A., Okada, Y., Nakada, M., Ando, T., Lim, K., & Wong, R. W. (2024). An Efficient Method for Isolating and Purifying Nuclei from Mice Brain for Single-Molecule Imaging Using High-Speed Atomic Force Microscopy. Cells, 13(3), 279. https://doi.org/10.3390/cells13030279