
Filamin C Deficiency Impairs Sarcomere Stability and Activates Focal Adhesion Kinase through PDGFRA Signaling in Induced Pluripotent Stem Cell-Derived Cardiomyocytes

 ,
,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of iPSC Deficient of FLNC Using CRISRP-Cas 9 System
2.2. Culturing and Cardiac Differentiation of Induced Pluripotent Stem Cells
2.3. Quantitative Real-Time Polymerase Chain Reaction
2.4. Protein Extraction and Immunno Blotting
2.5. Immunofluorescence Staining
2.6. Image Analysis
2.7. RNA Sequencing
2.8. RNA-Seq Data Analysis
2.9. Statistics
3. Results
3.1. Modeling FLNC-Related Cardiomyopathy Using FLNC Knockout hiPSC-Derived Cardiomyocytes
3.2. FLNC Deficiency Causes Structural Remodeling of hiPSC-CMs
3.3. FLNC Deficiency Causes Calcium (Ca2+) Handling Abnormalities in hiPSC-Derived Cardiomyocytes
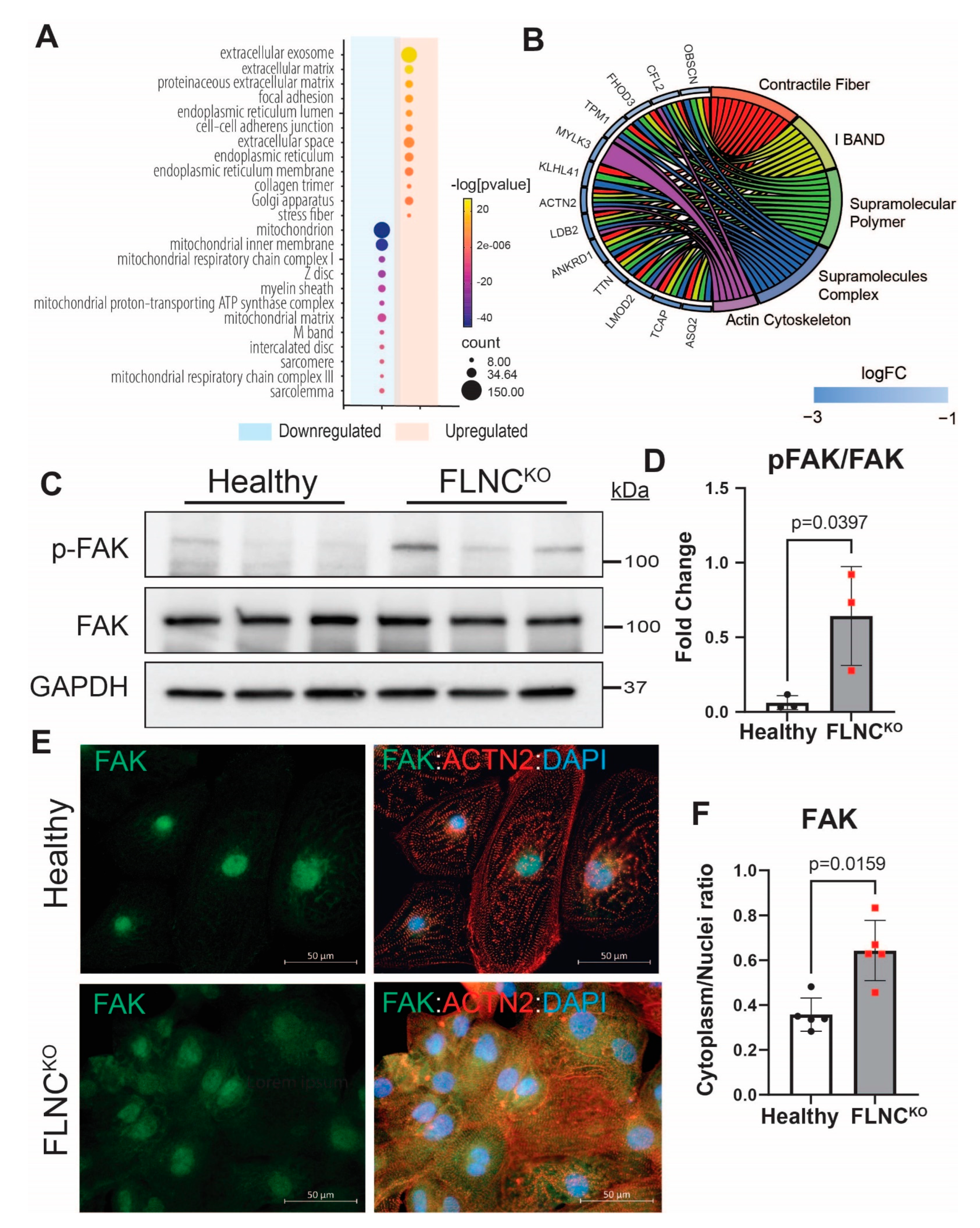
3.4. Focal Adhesion Kinase Signaling Pathway Is Upregulated in FLNC-Related Cardiomyopathy
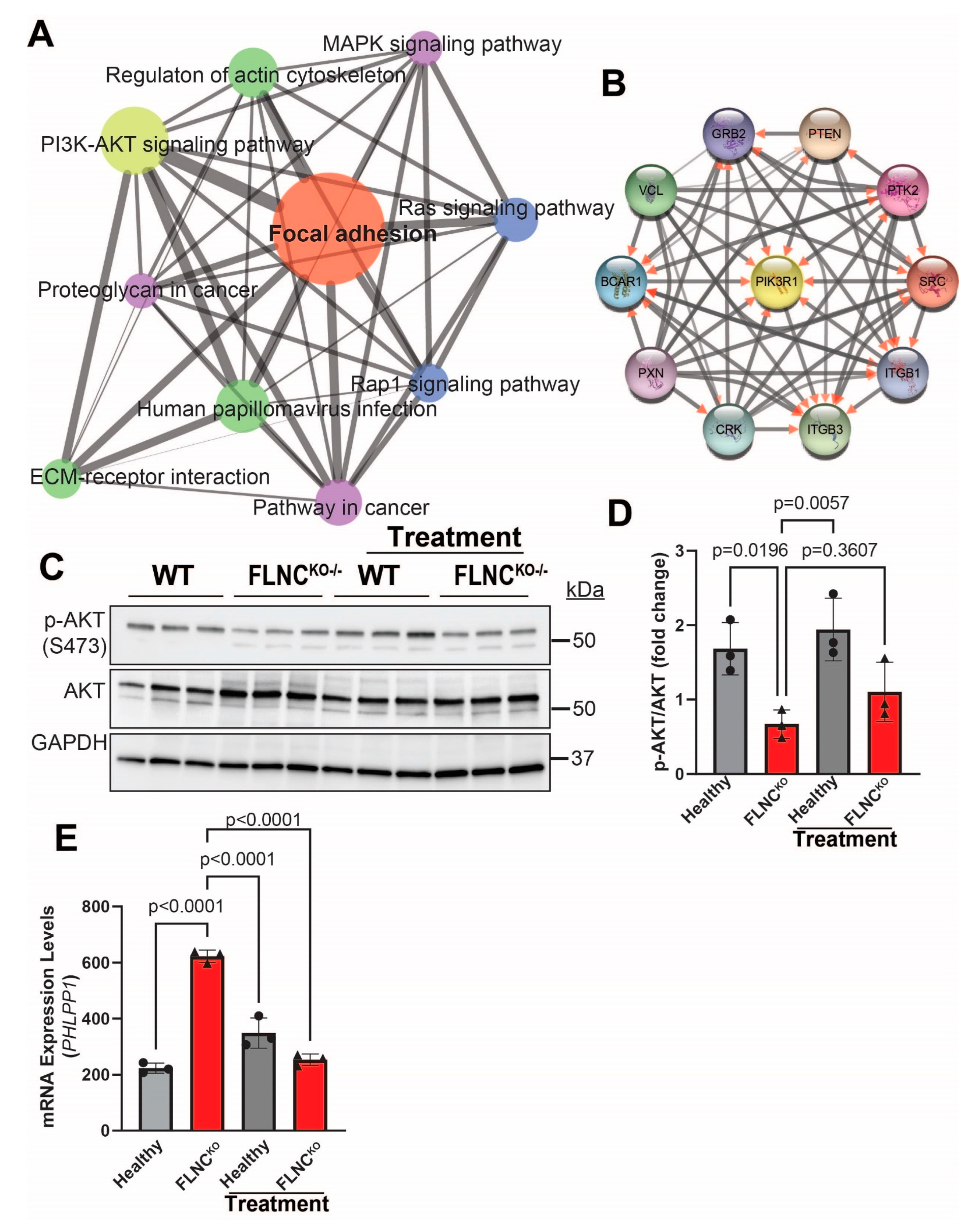
3.5. Inhibition of PDGFRA Signaling Attenuates FA Signaling Pathway Activation in FLNCKO Cardiomyocytes
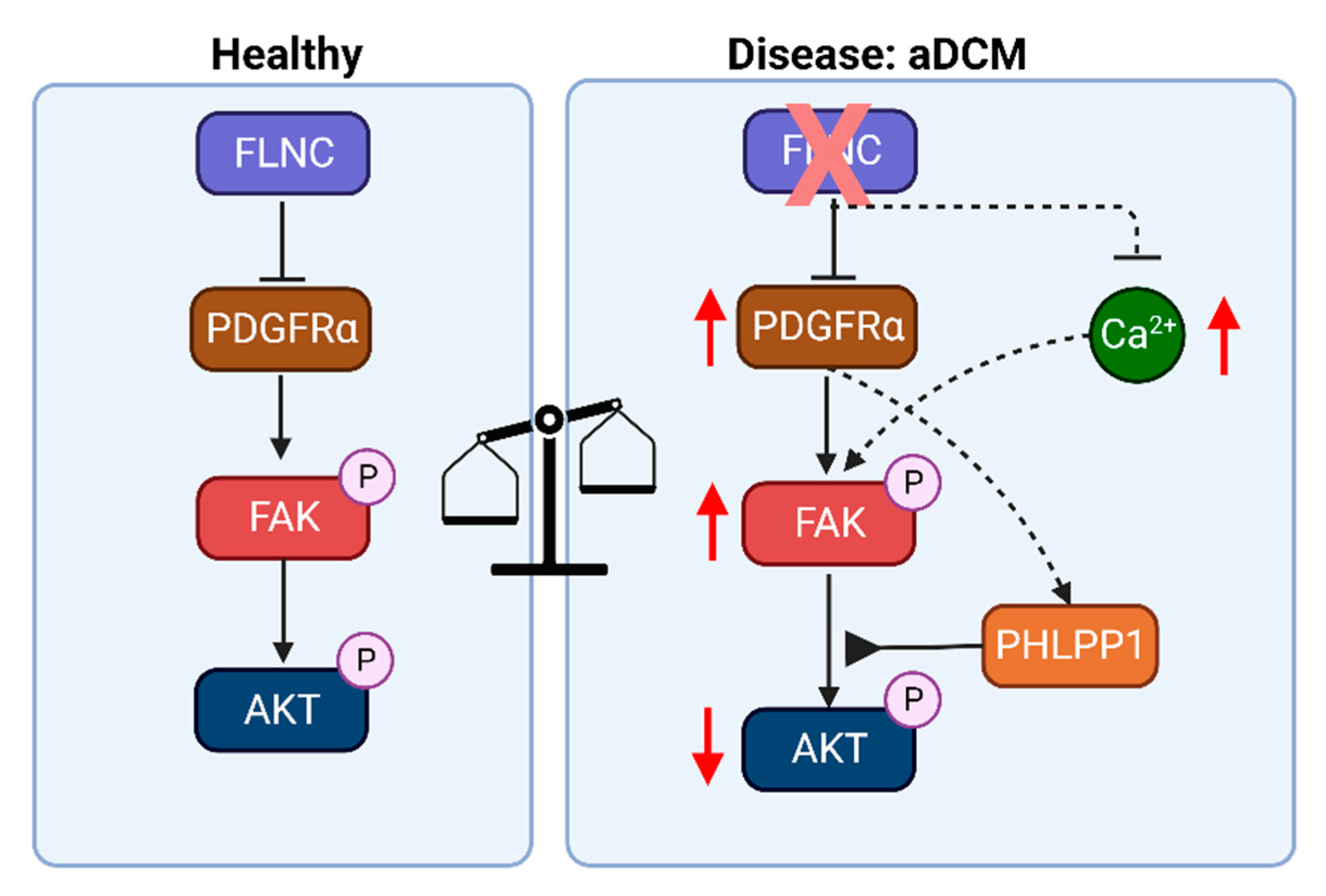
4. Discussion
5. Conclusions
6. Novelty and Significance
Author Contributions
Funding
Institutional Review Board
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell–Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Begay, R.L.; Tharp, C.A.; Martin, A.; Graw, S.L.; Sinagra, G.; Miani, D.; Sweet, M.E.; Slavov, D.B.; Stafford, N.; Zeller, M.J.; et al. FLNC Gene Splice Mutations Cause Dilated Cardiomyopathy. JACC Basic. Transl. Sci. 2016, 1, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Ferro, M.D.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Brun, F.; Gigli, M.; Graw, S.L.; Judge, D.P.; Merlo, M.; Murray, B.; Calkins, H.; Sinagra, G.; Taylor, M.R.; Mestroni, L.; et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2020, 57, 254–257. [Google Scholar] [CrossRef]
- Gigli, M.; Stolfo, D.; Graw, S.L.; Merlo, M.; Gregorio, C.; Nee Chen, S.; Dal Ferro, M.; Paldino, M.A.; De Angelis, G.; Brun, F.; et al. Phenotypic Expression, Natural History, and Risk Stratification of Cardiomyopathy Caused by Filamin C Truncating Variants. Circulation 2021, 144, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef]
- Valdes-Mas, R.; Gutierrez-Fernandez, A.; Gomez, J.; Coto, E.; Astudillo, A.; Puente, D.A.; Reguero, J.R.; Alvarez, V.; Moris, C.; Leon, D.; et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat. Commun. 2014, 5, 5326. [Google Scholar] [CrossRef]
- Agarwal, R.; Paulo, J.A.; Toepfer, C.N.; Ewoldt, J.K.; Sundaram, S.; Chopra, A.; Zhang, Q.; Gorham, J.; DePalma, S.R.; Chen, C.S.; et al. Filamin C Cardiomyopathy Variants Cause Protein and Lysosome Accumulation. Circ. Res. 2021, 129, 751–766. [Google Scholar] [CrossRef]
- Chen, S.N.; Lam, C.K.; Wan, Y.W.; Gao, S.; Malak, O.A.; Zhao, S.R.; Lombardi, R.; Ambardekar, A.V.; Bristow, M.R.; Cleveland, J.; et al. Activation of PDGFRA signaling contributes to filamin C-related arrhythmogenic cardiomyopathy. Sci. Adv. 2022, 8, eabk0052. [Google Scholar] [CrossRef]
- Zhou, A.X.; Hartwig, J.H.; Akyurek, L.M. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010, 20, 113–123. [Google Scholar] [CrossRef]
- Razinia, Z.; Makela, T.; Ylanne, J.; Calderwood, D.A. Filamins in mechanosensing and signaling. Annu. Rev. Biophys. 2012, 41, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Dalkilic, I.; Schienda, J.; Thompson, T.G.; Kunkel, L.M. Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Mol. Cell. Biol. 2006, 26, 6522–6534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Z.; Zhang, L.; Zhu, M.; Tan, C.; Zhou, X.; Evans, S.M.; Fang, X.; Feng, W.; Chen, J. Loss of Filamin C Is Catastrophic for Heart Function. Circulation 2020, 141, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Decker, M.L.; Simpson, D.G.; Behnke, M.; Cook, M.G.; Decker, R.S. Morphological analysis of contracting and quiescent adult rabbit cardiac myocytes in long-term culture. Anat. Rec. 1990, 227, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Samarel, A.M. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2291–H2301. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Meenderink, L.M.; Ryzhova, L.M.; Donato, D.M.; Gochberg, D.F.; Kaverina, I.; Hanks, S.K. P130Cas Src-binding and substrate domains have distinct roles in sustaining focal adhesion disassembly and promoting cell migration. PLoS ONE 2010, 5, e13412. [Google Scholar] [CrossRef]
- Wang, Y.; McNiven, M.A. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 2012, 196, 375–385. [Google Scholar] [CrossRef]
- Izaguirre, G.; Aguirre, L.; Hu, Y.P.; Lee, H.Y.; Schlaepfer, D.D.; Aneskievich, B.J.; Haimovich, B. The cytoskeletal/non-muscle isoform of alpha-actinin is phosphorylated on its actin-binding domain by the focal adhesion kinase. J. Biol. Chem. 2001, 276, 28676–28685. [Google Scholar] [CrossRef]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef]
- Grune, T.; Kehm, R.; Hohn, A.; Jung, T. “Cyt/Nuc”, a Customizable and Documenting ImageJ Macro for Evaluation of Protein Distributions Between Cytosol and Nucleus. Biotechnol. J. 2018, 13, e1700652. [Google Scholar] [CrossRef]
- Vezendi, K.; Krizsa, F.; Szabo, I.; Cserhati, I. The effect of platelet homogenate fractions on megakaryocytopoiesis in the mouse. Haematologia 1986, 19, 113–116. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Lombardi, R.; Dong, J.; Rodriguez, G.; Bell, A.; Leung, T.K.; Schwartz, R.J.; Willerson, J.T.; Brugada, R.; Marian, A.J. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2009, 104, 1076–1084. [Google Scholar] [CrossRef]
- Weiss, J.N.; Garfinkel, A.; Karagueuzian, H.S.; Chen, P.S.; Qu, Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010, 7, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.M.; Viola, M.C.; D’Amato, E.; Antolini, R.; Forti, S. Distinct patterns of calcium transients during early and delayed afterdepolarizations induced by isoproterenol in ventricular myocytes. Circulation 1995, 91, 2510–2515. [Google Scholar] [CrossRef] [PubMed]
- Furst, D.O.; Goldfarb, L.G.; Kley, R.A.; Vorgerd, M.; Olive, M.; van der Ven, P.F. Filamin C-related myopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 33–46. [Google Scholar] [CrossRef] [PubMed]
- van der Ven, P.F.; Obermann, W.M.; Lemke, B.; Gautel, M.; Weber, K.; Furst, D.O. Characterization of muscle filamin isoforms suggests a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil. Cytoskelet. 2000, 45, 149–162. [Google Scholar] [CrossRef]
- Zhou, X.; Fang, X.; Ithychanda, S.S.; Wu, T.; Gu, Y.; Chen, C.; Wang, L.; Bogomolovas, J.; Qin, J.; Chen, J. Interaction of Filamin C With Actin Is Essential for Cardiac Development and Function. Circ. Res. 2023, 133, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Iyengar, R.; Koshman, Y.E.; Kim, T.; Russell, B.; Martin, J.L.; Heroux, A.L.; Robia, S.L.; Samarel, A.M. Serine-910 phosphorylation of focal adhesion kinase is critical for sarcomere reorganization in cardiomyocyte hypertrophy. Cardiovasc. Res. 2011, 92, 409–419. [Google Scholar] [CrossRef]
- Hakim, Z.S.; DiMichele, L.A.; Doherty, J.T.; Homeister, J.W.; Beggs, H.E.; Reichardt, L.F.; Schwartz, R.J.; Brackhan, J.; Smithies, O.; Mack, C.P.; et al. Conditional deletion of focal adhesion kinase leads to defects in ventricular septation and outflow tract alignment. Mol. Cell. Biol. 2007, 27, 5352–5364. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Mitra, S.K.; Ilic, D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta 2004, 1692, 77–102. [Google Scholar] [CrossRef]
- Giannone, G.; Ronde, P.; Gaire, M.; Beaudouin, J.; Haiech, J.; Ellenberg, J.; Takeda, K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J. Biol. Chem. 2004, 279, 28715–28723. [Google Scholar] [CrossRef] [PubMed]
- Heerkens, E.H.; Quinn, L.; Withers, S.B.; Heagerty, A.M. beta Integrins mediate FAK Y397 autophosphorylation of resistance arteries during eutrophic inward remodeling in hypertension. J. Vasc. Res. 2014, 51, 305–314. [Google Scholar] [CrossRef]
- Mohanty, P.; Bhatnagar, S. Structure of focal adhesion kinase in healthy heart versus pathological cardiac hypertrophy: A modeling and simulation study. J. Mol. Graph. Model. 2018, 80, 15–24. [Google Scholar] [CrossRef]
- Tucci, A.R.; de Oliveira, F.O.R., Jr.; Lechuga, G.C.; de Oliveira, G.M.; Eleuterio, A.C.; Mesquita, L.B.; Farani, P.S.G.; Britto, C.; Moreira, O.C.; Pereira, M.C.S. Role of FAK signaling in chagasic cardiac hypertrophy. Braz. J. Infect. Dis. 2020, 24, 386–397. [Google Scholar] [CrossRef]
- Ghigo, A.; Laffargue, M.; Li, M.; Hirsch, E. PI3K and Calcium Signaling in Cardiovascular Disease. Circ. Res. 2017, 121, 282–292. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.; Treskov, I.; Lupu, T.S.; Weinheimer, C.; Kovacs, A.; Courtois, M.; Muslin, A.J. Akt1 is required for physiological cardiac growth. Circulation 2006, 113, 2097–2104. [Google Scholar] [CrossRef]
- Catalucci, D.; Latronico, M.V.G.; Ceci, M.; Rusconi, F.; Young, H.S.; Gallo, P.; Santonastasi, M.; Bellacosa, A.; Brown, J.H.; Condorelli, G. Akt increases sarcoplasmic reticulum Ca2+ cycling by direct phosphorylation of phospholamban at Thr17. J. Biol. Chem. 2009, 284, 28180–28187. [Google Scholar] [CrossRef]
- Catalucci, D.; Zhang, D.H.; DeSantiago, J.; Aimond, F.; Barbara, G.; Chemin, J.; Bonci, D.; Picht, E.; Rusconi, F.; Dalton, N.D.; et al. Akt regulates L-type Ca2+ channel activity by modulating Cavalpha1 protein stability. J. Cell Biol. 2009, 184, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Abdellatif, M.; Koul, S.; Crystal, G.J. Chronic treatment with insulin-like growth factor I enhances myocyte contraction by upregulation of Akt-SERCA2a signaling pathway. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H130–H135. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Mukhopadhyay, A.; Narasimhan, S.D.; Tesz, G.; Czech, M.P.; Tissenbaum, H.A. A PP2A regulatory subunit regulates C. elegans insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell 2009, 136, 939–951. [Google Scholar] [CrossRef]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef]
- Miyamoto, S.; Purcell, N.H.; Smith, J.M.; Gao, T.; Whittaker, R.; Huang, K.; Castillo, R.; Glembotski, C.C.; Sussman, M.A.; Newton, A.C.; et al. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ. Res. 2010, 107, 476–484. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; He, L.; Lam, C.K.; Taylor, M.R.G.; Mestroni, L.; Lombardi, R.; Chen, S.N. Filamin C Deficiency Impairs Sarcomere Stability and Activates Focal Adhesion Kinase through PDGFRA Signaling in Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cells 2024, 13, 278. https://doi.org/10.3390/cells13030278
Gao S, He L, Lam CK, Taylor MRG, Mestroni L, Lombardi R, Chen SN. Filamin C Deficiency Impairs Sarcomere Stability and Activates Focal Adhesion Kinase through PDGFRA Signaling in Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cells. 2024; 13(3):278. https://doi.org/10.3390/cells13030278
Chicago/Turabian StyleGao, Shanshan, Lingaonan He, Chi Keung Lam, Matthew R. G. Taylor, Luisa Mestroni, Raffaella Lombardi, and Suet Nee Chen. 2024. "Filamin C Deficiency Impairs Sarcomere Stability and Activates Focal Adhesion Kinase through PDGFRA Signaling in Induced Pluripotent Stem Cell-Derived Cardiomyocytes" Cells 13, no. 3: 278. https://doi.org/10.3390/cells13030278
APA StyleGao, S., He, L., Lam, C. K., Taylor, M. R. G., Mestroni, L., Lombardi, R., & Chen, S. N. (2024). Filamin C Deficiency Impairs Sarcomere Stability and Activates Focal Adhesion Kinase through PDGFRA Signaling in Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cells, 13(3), 278. https://doi.org/10.3390/cells13030278