

β2-Adrenoceptor Activation Favor Acquisition of Tumorigenic Properties in Non-Tumorigenic MCF-10A Breast Epithelial Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cells, Culture Conditions and Treatments
2.3. Cell Adhesion Assays
2.4. Cell Proliferation Assay
2.5. Random Motility Assay
2.6. Wound Healing Assay
2.7. F-Actin Staining, Cell Morphometric Analysis, and Cell Protrusions Quantification
2.8. mRrNA Extraction and RT-PCR
2.9. Resistance to Cell Death under Low Attachment Conditions
2.10. Statistical Analysis
3. Results
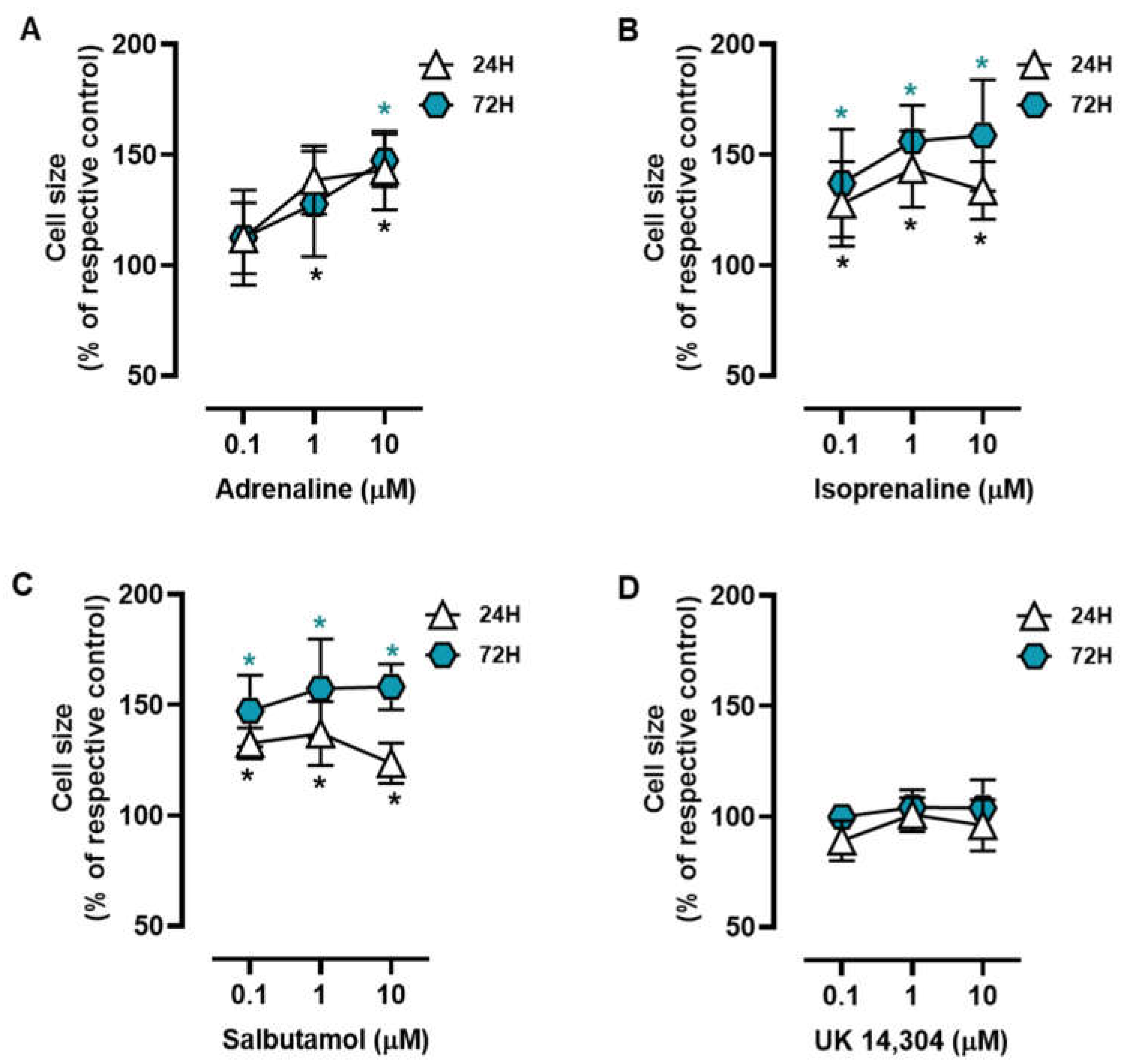
3.1. β2-Adrenoceptor Activation Alters in MCF-10A Cell Morphology
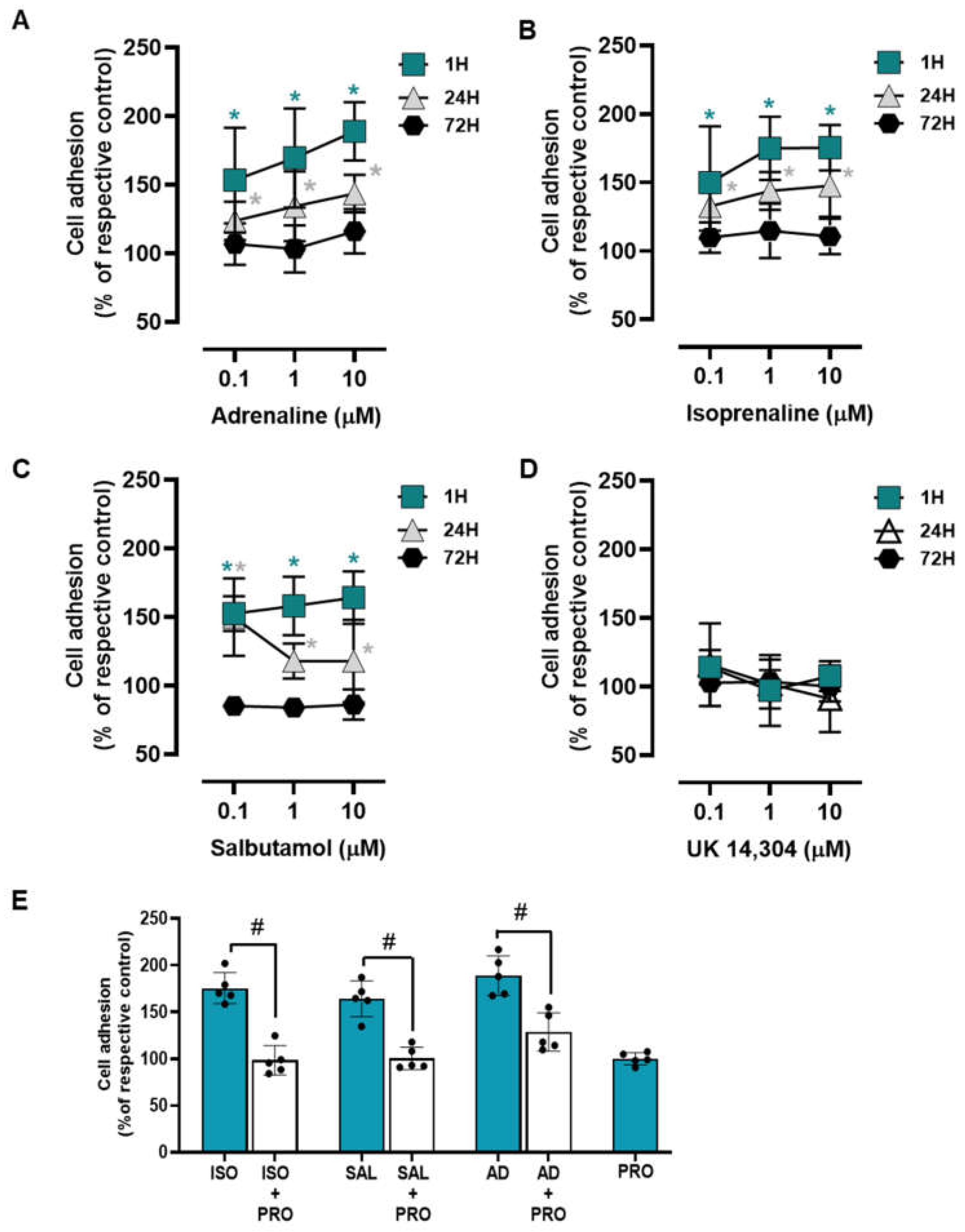
3.2. β2-Adrenoceptor Activation Increases MCF-10A Cell-to-Matrix Adhesion
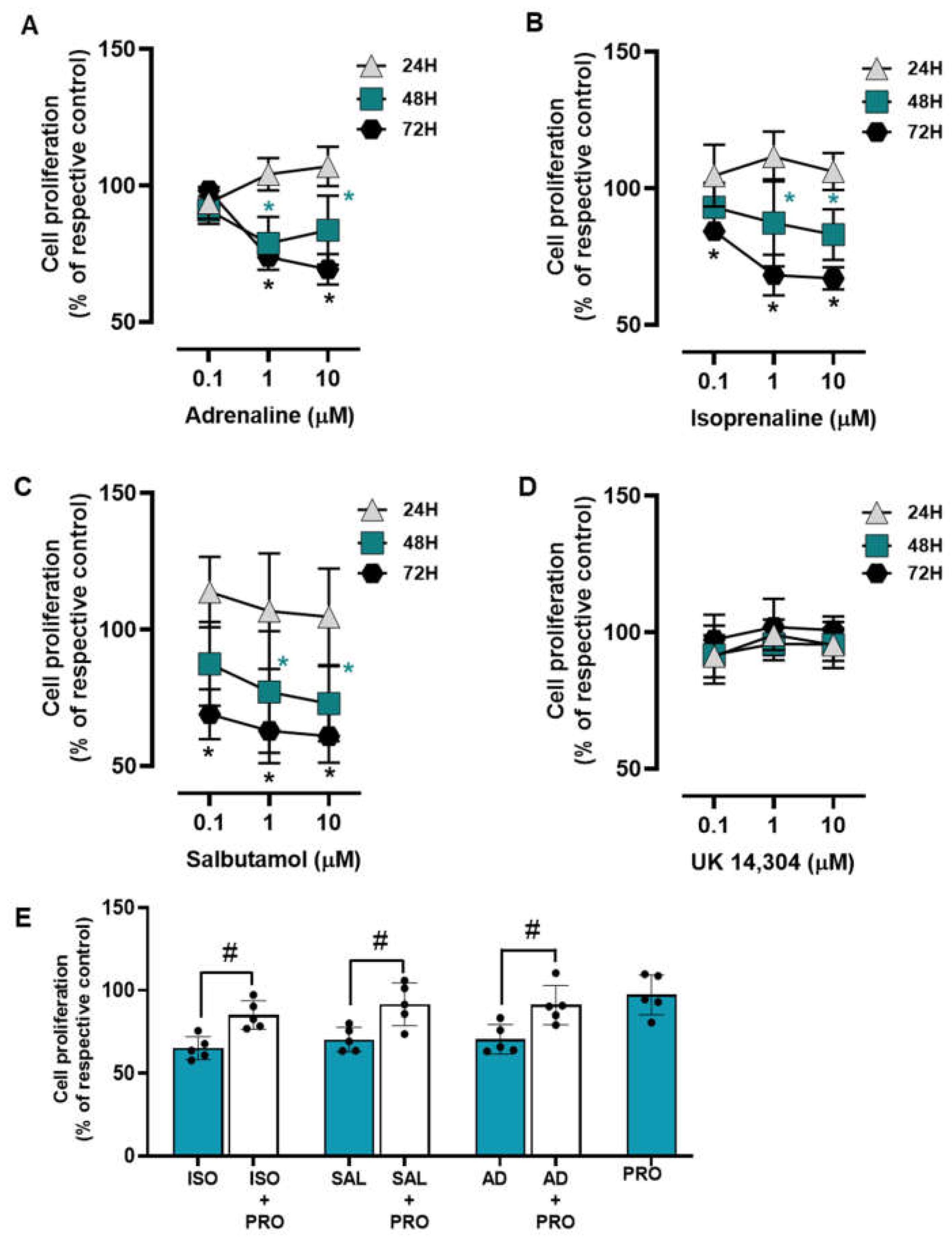
3.3. β2-Adrenoceptor Activation Decreases MCF-10A Cell Proliferation
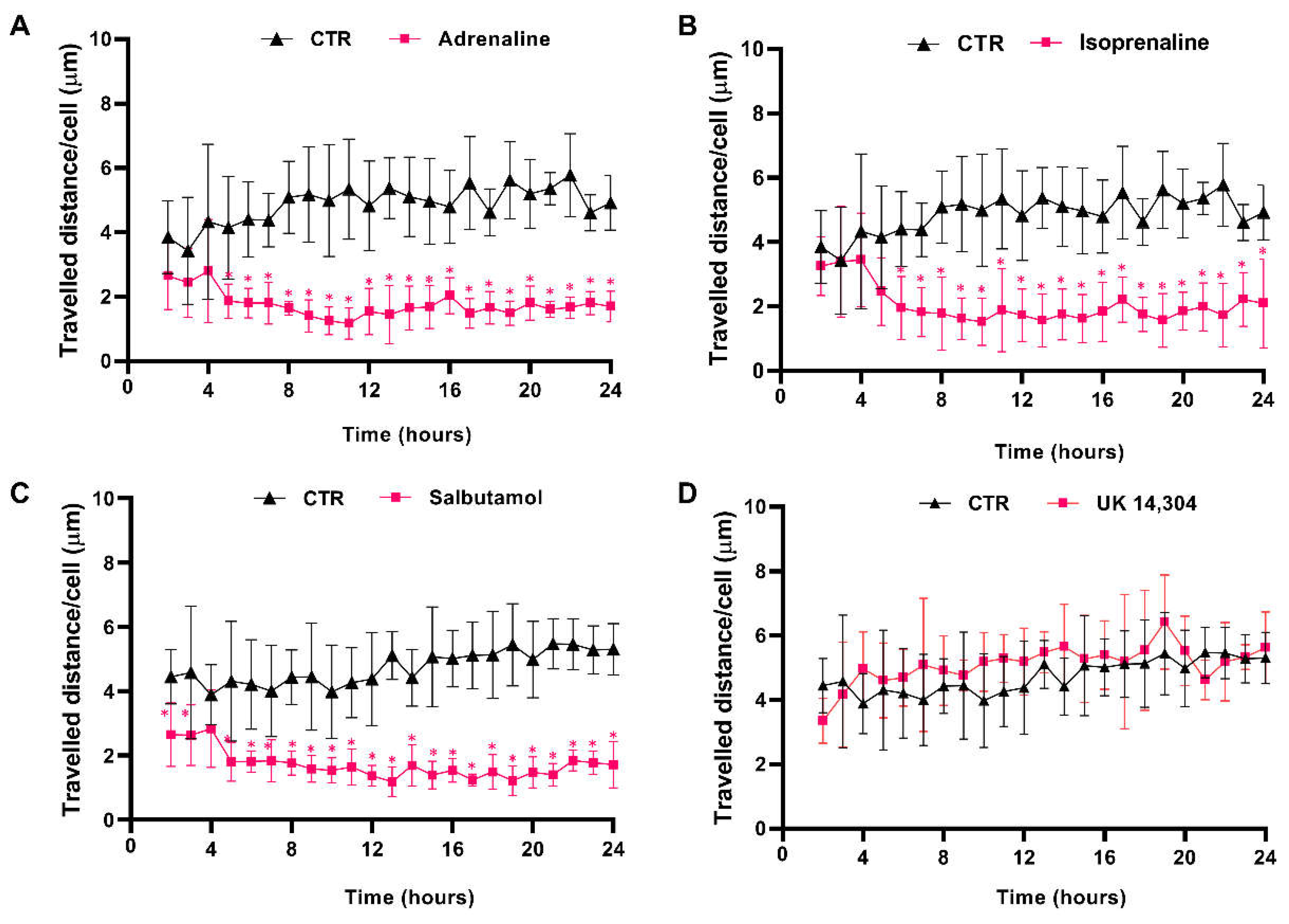
3.4. β2-Adrenoceptor Activation Decreases MCF-10A Cell Motility
3.5. β2-Adrenoceptor Activation Decreases MCF-10A Cell Migration
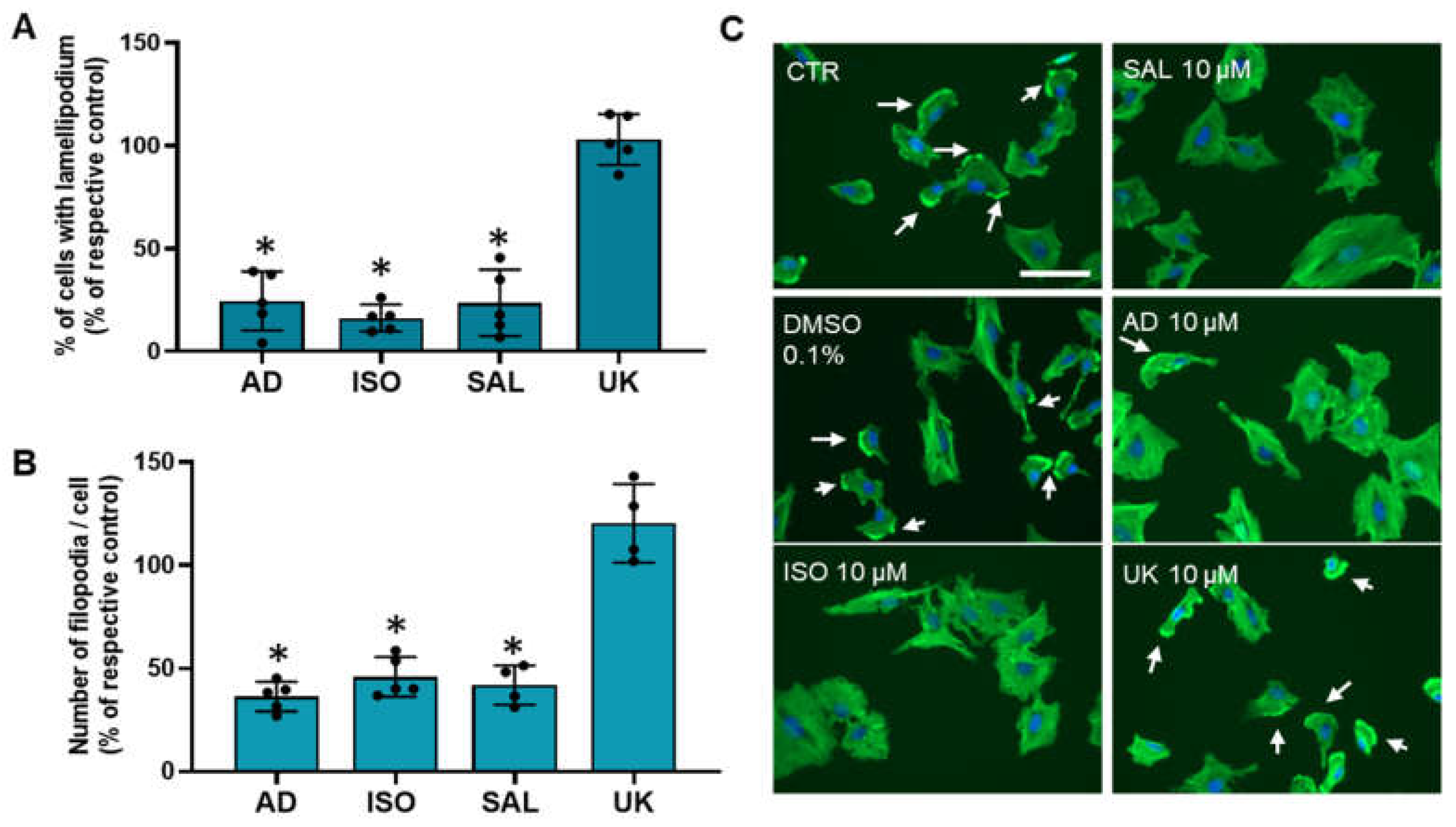
3.6. β2-Adrenoceptor Activation Decreases MCF-10A Cell Protrusions
3.7. β-Adrenoceptor Activation Increases MET Markers in MCF-10A Non-Tumorigenic Cells
3.8. β2-Adrenoceptor Activation Confers Protection against Cell Death in MCF-10A Cells under Low Attachment Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.; Quintas, C.; Goncalves, J.; Fresco, P. Contribution of adrenergic mechanisms for the stress-induced breast cancer carcinogenesis. J. Cell Physiol. 2022, 237, 2107–2127. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, D.; Duan, H.; Qian, L.; Wang, L.; Niu, L.; Zhang, H.; Yong, Z.; Gong, Z.; Song, L.; et al. The beta2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells. Breast Cancer Res. Treat. 2011, 125, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Z.; Wang, T.; Yang, Z.; Chen, H.; Hu, Y.; Hu, C.; Guo, L.; Deng, Q.; Liu, Y.; et al. beta2-AR signaling controls trastuzumab resistance-dependent pathway. Oncogene 2016, 35, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Parkin, R.; Neale, S. The effect of isoprenaline on induction of tumours by methyl nitrosourea in the salivary and mammary glands of female wistar rats. Br. J. Cancer 1976, 34, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Altosaar, K.; Balaji, P.; Bond, R.; Bylund, D.; Cotecchia, S.; Devost, D.; Doze, V.; Eikenburg, D.; Gora, S.; Goupil, E.; et al. Adrenoceptors (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. CITE 2019, 2019. [Google Scholar] [CrossRef]
- Chiba, T.; Maeda, T.; Fujita, Y.; Takeda, R.; Kikuchi, A.; Kudo, K. Stress-Induced Suppression of Milk Protein Is Involved in a Noradrenergic Mechanism in the Mammary Gland. Endocrinology 2019, 160, 2074–2084. [Google Scholar] [CrossRef]
- Song, S.L.; Crowley, W.R.; Grosvenor, C.E. Evidence for involvement of an adrenal catecholamine in the beta-adrenergic inhibition of oxytocin release in lactating rats. Brain Res. 1988, 457, 303–309. [Google Scholar] [CrossRef]
- Silberstein, G.B.; Strickland, P.; Trumpbour, V.; Coleman, S.; Daniel, C.W. In vivo, cAMP stimulates growth and morphogenesis of mouse mammary ducts. Proc. Natl. Acad. Sci. USA 1984, 81, 4950–4954. [Google Scholar] [CrossRef]
- Gargiulo, L.; May, M.; Rivero, E.M.; Copsel, S.; Lamb, C.; Lydon, J.; Davio, C.; Lanari, C.; Luthy, I.A.; Bruzzone, A. A Novel Effect of beta-Adrenergic Receptor on Mammary Branching Morphogenesis and its Possible Implications in Breast Cancer. J. Mammary Gland Biol. Neoplasia 2017, 22, 43–57. [Google Scholar] [CrossRef]
- Eriksson, M.; Lindh, B.; Uvnas-Moberg, K.; Hokfelt, T. Distribution and origin of peptide-containing nerve fibres in the rat and human mammary gland. Neuroscience 1996, 70, 227–245. [Google Scholar] [CrossRef]
- Donoso, E.A.; Sapag-Hagar, M.; Lara, H.E. Neurochemical evidence for the presence of sympathetic nerve terminals in the rat mammary gland: Changes during the lactogenic cycle. Mol. Cell. Neurosci. 1992, 3, 23–28. [Google Scholar] [CrossRef]
- Clapp, C.; Martinez-Escalera, G.; Morales, M.T.; Shyr, S.W.; Grosvenor, C.E.; Mena, F. Release of catecholamines follows suckling or electrical stimulation of mammary nerve in lactating rats. Endocrinology 1985, 117, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bisset, G.W.; Clark, B.J.; Lewis, G.P. The mechanism of the inhibitory action of adrenaline on the mammary gland. Br. J. Pharmacol. Chemother. 1967, 31, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Scott-Solomon, E.; Boehm, E.; Kuruvilla, R. The sympathetic nervous system in development and disease. Nat. Rev. Neurosci. 2021, 22, 685–702. [Google Scholar] [CrossRef] [PubMed]
- Duijts, S.F.; Zeegers, M.P.; Borne, B.V. The association between stressful life events and breast cancer risk: A meta-analysis. Int. J. Cancer 2003, 107, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Antonova, L.; Aronson, K.; Mueller, C.R. Stress and breast cancer: From epidemiology to molecular biology. Breast Cancer Res. 2011, 13, 208. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.J.; Fernandez Poole, S.; White, M.; Lyn, R.; Flores, D.A.; Haile, H.G.; Williams, D.R. The Role of Stress in Breast Cancer Incidence: Risk Factors, Interventions, and Directions for the Future. Int. J. Environ. Res. Public Health 2021, 18, 1871. [Google Scholar] [CrossRef] [PubMed]
- Adamekova, E.; Markova, M.; Kubatka, P.; Bojkova, B.; Ahlers, I.; Ahlersova, E. NMU-induced mammary carcinogenesis in female rats is influenced by repeated psychoemotional stress. Neoplasma 2003, 50, 428–432. [Google Scholar]
- Boyd, A.L.; Salleh, A.; Humber, B.; Yee, J.; Tomes, L.; Kerr, L.R. Neonatal experiences differentially influence mammary gland morphology, estrogen receptor {alpha} protein levels, and carcinogenesis in BALB/c mice. Cancer Prev. Res. 2010, 3, 1398–1408. [Google Scholar] [CrossRef]
- Riley, V. Mouse mammary tumors: Alteration of incidence as apparent function of stress. Science 1975, 189, 465–467. [Google Scholar] [CrossRef]
- Bahri, N.; Fathi Najafi, T.; Homaei Shandiz, F.; Tohidinik, H.R.; Khajavi, A. The relation between stressful life events and breast cancer: A systematic review and meta-analysis of cohort studies. Breast Cancer Res. Treat. 2019, 176, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol Reduces Cancer Risk: A Population-Based Cohort Study. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.; Raytis, J.L.; Smith, D.D.; Duenas, M.; Neman, J.; Jandial, R.; Lew, M.W. Inhibition of beta2-adrenergic receptor reduces triple-negative breast cancer brain metastases: The potential benefit of perioperative beta-blockade. Oncol. Rep. 2016, 35, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Luo, Y.; Tian, P.; Peng, F.; Lu, J.; Yang, Y.; Su, Q.; Liu, B.; Yu, J.; Luo, X.; et al. Stress-induced epinephrine enhances lactate dehydrogenase A and promotes breast cancer stem-like cells. J. Clin. Investig. 2019, 129, 1030–1046. [Google Scholar] [CrossRef]
- Du, P.; Zeng, H.; Xiao, Y.; Zhao, Y.; Zheng, B.; Deng, Y.; Liu, J.; Huang, B.; Zhang, X.; Yang, K.; et al. Chronic stress promotes EMT-mediated metastasis through activation of STAT3 signaling pathway by miR-337-3p in breast cancer. Cell Death Dis. 2020, 11, 761. [Google Scholar] [CrossRef]
- Gruet, M.; Cotton, D.; Coveney, C.; Boocock, D.J.; Wagner, S.; Komorowski, L.; Rees, R.C.; Pockley, A.G.; Garner, A.C.; Wallis, J.D.; et al. beta2-Adrenergic Signalling Promotes Cell Migration by Upregulating Expression of the Metastasis-Associated Molecule LYPD3. Biology 2020, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Gill, N.K.; Nyberg, K.D.; Nguyen, A.V.; Hohlbauch, S.V.; Geisse, N.A.; Nowell, C.J.; Sloan, E.K.; Rowat, A.C. Cancer cells become less deformable and more invasive with activation of beta-adrenergic signaling. J. Cell Sci. 2016, 129, 4563–4575. [Google Scholar] [CrossRef] [PubMed]
- Pon, C.K.; Lane, J.R.; Sloan, E.K.; Halls, M.L. The beta2-adrenoceptor activates a positive cAMP-calcium feedforward loop to drive breast cancer cell invasion. FASEB J. 2016, 30, 1144–1154. [Google Scholar] [CrossRef]
- Tibensky, M.; Cernackova, A.; Horvathova, L.; Macejova, D.; Tillinger, A.; Mravec, B. Chronic propranolol treatment moderately attenuated development of N-methyl-N-nitrosourea-induced mammary carcinoma in female rats. Anticancer Drugs 2021, 32, 1011–1018. [Google Scholar] [CrossRef]
- Gargiulo, L.; Copsel, S.; Rivero, E.M.; Gales, C.; Senard, J.M.; Luthy, I.A.; Davio, C.; Bruzzone, A. Differential beta(2)-adrenergic receptor expression defines the phenotype of non-tumorigenic and malignant human breast cell lines. Oncotarget 2014, 5, 10058–10069. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Sakakitani, S.; Podyma-Inoue, K.A.; Takayama, R.; Takahashi, K.; Ishigami-Yuasa, M.; Kagechika, H.; Harada, H.; Watabe, T. Activation of beta2-adrenergic receptor signals suppresses mesenchymal phenotypes of oral squamous cell carcinoma cells. Cancer Sci. 2021, 112, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Liang, F.; Wang, R.; Du, Q.; Zhu, S. An Epithelial-Mesenchymal Transition Hallmark Gene-Based Risk Score System in Head and Neck Squamous-Cell Carcinoma. Int. J. Gen. Med. 2021, 14, 4219–4227. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Hinkal, G.W.; Thomas, C.; Fauvet, F.; Courtois-Cox, S.; Wierinckx, A.; Devouassoux-Shisheboran, M.; Treilleux, I.; Tissier, A.; Gras, B.; et al. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet. 2012, 8, e1002723. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Frisch, S.M.; Vuori, K.; Ruoslahti, E.; Chan-Hui, P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996, 134, 793–799. [Google Scholar] [CrossRef]
- Parsons, M.J.; Patel, P.; Brat, D.J.; Colbert, L.; Vertino, P.M. Silencing of TMS1/ASC promotes resistance to anoikis in breast epithelial cells. Cancer Res. 2009, 69, 1706–1711. [Google Scholar] [CrossRef]
- Menard, R.E.; Jovanovski, A.P.; Mattingly, R.R. Active p21-activated kinase 1 rescues MCF10A breast epithelial cells from undergoing anoikis. Neoplasia 2005, 7, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Mailleux, A.A.; Overholtzer, M.; Brugge, J.S. Lumen formation during mammary epithelial morphogenesis: Insights from in vitro and in vivo models. Cell Cycle 2008, 7, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Haenssen, K.K.; Caldwell, S.A.; Shahriari, K.S.; Jackson, S.R.; Whelan, K.A.; Klein-Szanto, A.J.; Reginato, M.J. ErbB2 requires integrin alpha5 for anoikis resistance via Src regulation of receptor activity in human mammary epithelial cells. J. Cell Sci. 2010, 123, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Oudenaarden, C.R.L.; van de Ven, R.A.H.; Derksen, P.W.B. Re-inforcing the cell death army in the fight against breast cancer. J. Cell Sci. 2018, 131, jcs212563. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Khalili, A.A.; Ahmad, M.R. A Review of Cell Adhesion Studies for Biomedical and Biological Applications. Int. J. Mol. Sci. 2015, 16, 18149–18184. [Google Scholar] [CrossRef]
- Shi, L.; Liu, B.; Shen, D.D.; Yan, P.; Zhang, Y.; Tian, Y.; Hou, L.; Jiang, G.; Zhu, Y.; Liang, Y.; et al. A tumor-suppressive circular RNA mediates uncanonical integrin degradation by the proteasome in liver cancer. Sci. Adv. 2021, 7, eabe5043. [Google Scholar] [CrossRef]
- Held, P.G.; Clayton, J.; Banks, P. Kinetic Proliferation Assay Using Label-Free Cell Counting. Available online: https://www.agilent.com/cs/library/technicaloverviews/public/high-contrast-brightfield-5994-3444EN-agilent.pdf (accessed on 18 June 2022).
- Soares, A.S.; Costa, V.M.; Diniz, C.; Fresco, P. The combination of Cl-IB-MECA with paclitaxel: A new anti-metastatic therapeutic strategy for melanoma. Cancer Chemother. Pharmacol. 2014, 74, 847–860. [Google Scholar] [CrossRef]
- Amaro, F.; Silva, D.; Reguengo, H.; Oliveira, J.C.; Quintas, C.; Vale, N.; Goncalves, J.; Fresco, P. beta-Adrenoceptor Activation in Breast MCF-10A Cells Induces a Pattern of Catecholamine Production Similar to that of Tumorigenic MCF-7 Cells. Int. J. Mol. Sci. 2020, 21, 7968. [Google Scholar] [CrossRef]
- Ke, X.; Li, L.; Dong, H.L.; Chen, Z.N. Acquisition of anoikis resistance through CD147 upregulation: A new mechanism underlying metastasis of hepatocellular carcinoma cells. Oncol. Lett. 2012, 3, 1249–1254. [Google Scholar] [CrossRef]
- Vazquez, S.M.; Mladovan, A.G.; Perez, C.; Bruzzone, A.; Baldi, A.; Luthy, I.A. Human breast cell lines exhibit functional alpha2-adrenoceptors. Cancer Chemother. Pharmacol. 2006, 58, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Beckham, Y.; Vasquez, R.J.; Stricker, J.; Sayegh, K.; Campillo, C.; Gardel, M.L. Arp2/3 inhibition induces amoeboid-like protrusions in MCF10A epithelial cells by reduced cytoskeletal-membrane coupling and focal adhesion assembly. PLoS ONE 2014, 9, e100943. [Google Scholar] [CrossRef] [PubMed]
- Goulimari, P.; Kitzing, T.M.; Knieling, H.; Brandt, D.T.; Offermanns, S.; Grosse, R. Galpha12/13 is essential for directed cell migration and localized Rho-Dia1 function. J. Biol. Chem. 2005, 280, 42242–42251. [Google Scholar] [CrossRef] [PubMed]
- Bryce, N.S.; Clark, E.S.; Leysath, J.L.; Currie, J.D.; Webb, D.J.; Weaver, A.M. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr. Biol. 2005, 15, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Mogilner, A.; Rubinstein, B. The physics of filopodial protrusion. Biophys. J. 2005, 89, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Leggett, S.E.; Hruska, A.M.; Guo, M.; Wong, I.Y. The epithelial-mesenchymal transition and the cytoskeleton in bioengineered systems. Cell Commun. Signal. 2021, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, S.; Vourda, A.; Syggelos, S.; Gyftopoulos, K. Cell Plasticity and Prostate Cancer: The Role of Epithelial-Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance. Cancers 2021, 13, 2795. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Sung, J.Y.; Kim, S.H.; Yun, U.J.; Kim, H.; Jang, E.J.; Yoo, H.E.; Hong, E.K.; Goh, S.H.; Moon, A.; et al. Fibronectin regulates anoikis resistance via cell aggregate formation. Cancer Lett. 2021, 508, 59–72. [Google Scholar] [CrossRef]
- Rayavarapu, R.R.; Heiden, B.; Pagani, N.; Shaw, M.M.; Shuff, S.; Zhang, S.; Schafer, Z.T. The role of multicellular aggregation in the survival of ErbB2-positive breast cancer cells during extracellular matrix detachment. J. Biol. Chem. 2015, 290, 8722–8733. [Google Scholar] [CrossRef]
- McCorry, L.K. Physiology of the autonomic nervous system. Am. J. Pharm. Educ. 2007, 71, 78. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adelaide, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006, 25, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Han, B.; Yu, Y.; Yao, W.; Bose, S.; Karlan, B.Y.; Giuliano, A.E.; Cui, X. Evaluation of MCF10A as a Reliable Model for Normal Human Mammary Epithelial Cells. PLoS ONE 2015, 10, e0131285. [Google Scholar] [CrossRef] [PubMed]
- Vedoya, G.M.; Galarza, T.E.; Mohamad, N.A.; Cricco, G.P.; Martin, G.A. Non-tumorigenic epithelial breast cells and ionizing radiation cooperate in the enhancement of mesenchymal traits in tumorigenic breast cancer cells. Life Sci. 2022, 307, 120853. [Google Scholar] [CrossRef]
- Puleo, J.; Polyak, K. The MCF10 Model of Breast Tumor Progression. Cancer Res. 2021, 81, 4183–4185. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Kainuma, K.; Kobayashi, T.; D’Alessandro-Gabazza, C.N.; Toda, M.; Yasuma, T.; Nishihama, K.; Fujimoto, H.; Kuwabara, Y.; Hosoki, K.; Nagao, M.; et al. beta(2) adrenergic agonist suppresses eosinophil-induced epithelial-to-mesenchymal transition of bronchial epithelial cells. Respir. Res. 2017, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Cao, Q.; Mehra, R.; Laxman, B.; Yu, J.; Tomlins, S.A.; Creighton, C.J.; Dhanasekaran, S.M.; Shen, R.; Chen, G.; et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007, 12, 419–431. [Google Scholar] [CrossRef]
- Dinicola, S.; Fabrizi, G.; Masiello, M.G.; Proietti, S.; Palombo, A.; Minini, M.; Harrath, A.H.; Alwasel, S.H.; Ricci, G.; Catizone, A.; et al. Inositol induces mesenchymal-epithelial reversion in breast cancer cells through cytoskeleton rearrangement. Exp. Cell Res. 2016, 345, 37–50. [Google Scholar] [CrossRef]
- Peitzman, E.R.; Zaidman, N.A.; Maniak, P.J.; O’Grady, S.M. Agonist binding to beta-adrenergic receptors on human airway epithelial cells inhibits migration and wound repair. Am. J. Physiol. Cell Physiol. 2015, 309, C847–C855. [Google Scholar] [CrossRef]
- Steenhuis, P.; Huntley, R.E.; Gurenko, Z.; Yin, L.; Dale, B.A.; Fazel, N.; Isseroff, R.R. Adrenergic signaling in human oral keratinocytes and wound repair. J. Dent. Res. 2011, 90, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Pullar, C.E.; Grahn, J.C.; Liu, W.; Isseroff, R.R. Beta2-adrenergic receptor activation delays wound healing. FASEB J. 2006, 20, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, L.; Rivero, E.M.; di Siervi, N.; Buzzi, E.D.; Buffone, M.G.; Davio, C.A.; Luthy, I.A.; Bruzzone, A. Agonist Effects of Propranolol on Non-Tumor Human Breast Cells. Cells 2020, 9, 1036. [Google Scholar] [CrossRef] [PubMed]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Asokan, S.B.; Berginski, M.E.; Haynes, E.M.; Sharpless, N.E.; Griffith, J.D.; Gomez, S.M.; Bear, J.E. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 2012, 148, 973–987. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Guerrero, A.M.; Espinosa-Bermejo, N.; Sanchez-Lopez, I.; Macartney, T.; Pascual-Caro, C.; Orantos-Aguilera, Y.; Rodriguez-Ruiz, L.; Perez-Oliva, A.B.; Mulero, V.; Pozo-Guisado, E.; et al. RAC1-Dependent ORAI1 Translocation to the Leading Edge Supports Lamellipodia Formation and Directional Persistence. Sci. Rep. 2020, 10, 6580. [Google Scholar] [CrossRef] [PubMed]
- Janik, M.E.; Szlezak, D.; Surman, M.; Golas, A.; Litynska, A.; Przybylo, M. Diversified beta-2-adrenergic Receptor Expression and Action in Melanoma Cells. Anticancer Res. 2017, 37, 3025–3033. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pullar, C.E.; Zhao, M.; Song, B.; Pu, J.; Reid, B.; Ghoghawala, S.; McCaig, C.; Isseroff, R.R. Beta-adrenergic receptor agonists delay while antagonists accelerate epithelial wound healing: Evidence of an endogenous adrenergic network within the corneal epithelium. J. Cell Physiol. 2007, 211, 261–272. [Google Scholar] [CrossRef]
- Bravo-Calderon, D.M.; Assao, A.; Garcia, N.G.; Coutinho-Camillo, C.M.; Roffe, M.; Germano, J.N.; Oliveira, D.T. Beta adrenergic receptor activation inhibits oral cancer migration and invasiveness. Arch. Oral. Biol. 2020, 118, 104865. [Google Scholar] [CrossRef]
- Rivero, E.M.; Pinero, C.P.; Gargiulo, L.; Entschladen, F.; Zanker, K.; Bruzzone, A.; Luthy, I.A. The beta 2-Adrenergic Agonist Salbutamol Inhibits Migration, Invasion and Metastasis of the Human Breast Cancer MDA-MB- 231 Cell Line. Curr. Cancer Drug Targets 2017, 17, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Gardel, M.L.; Schneider, I.C.; Aratyn-Schaus, Y.; Waterman, C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010, 26, 315–333. [Google Scholar] [CrossRef]
- Kondratyeva, L.; Chernov, I.; Kopantzev, E.; Didych, D.; Kuzmich, A.; Alekseenko, I.; Kostrov, S.; Sverdlov, E. Pancreatic Lineage Specifier PDX1 Increases Adhesion and Decreases Motility of Cancer Cells. Cancers 2021, 13, 4390. [Google Scholar] [CrossRef]
- Palecek, S.P.; Loftus, J.C.; Ginsberg, M.H.; Lauffenburger, D.A.; Horwitz, A.F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 1997, 385, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, A.; Sauliere, A.; Finana, F.; Senard, J.M.; Luthy, I.; Gales, C. Dosage-dependent regulation of cell proliferation and adhesion through dual beta2-adrenergic receptor/cAMP signals. FASEB J. 2014, 28, 1342–1354. [Google Scholar] [CrossRef]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef]
- Li, E.; Wei, B.; Wang, X.; Kang, R. METTL3 enhances cell adhesion through stabilizing integrin beta1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am. J. Cancer Res. 2020, 10, 1012–1025. [Google Scholar]
- Oltean, S.; Sorg, B.S.; Albrecht, T.; Bonano, V.I.; Brazas, R.M.; Dewhirst, M.W.; Garcia-Blanco, M.A. Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc. Natl. Acad. Sci. USA 2006, 103, 14116–14121. [Google Scholar] [CrossRef]
- Melhem-Bertrandt, A.; Chavez-Macgregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645–2652. [Google Scholar] [CrossRef]
- Mulcrone, P.L.; Campbell, J.P.; Clement-Demange, L.; Anbinder, A.L.; Merkel, A.R.; Brekken, R.A.; Sterling, J.A.; Elefteriou, F. Skeletal Colonization by Breast Cancer Cells Is Stimulated by an Osteoblast and beta2AR-Dependent Neo-Angiogenic Switch. J. Bone Miner. Res. 2017, 32, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Madel, M.B.; Elefteriou, F. Mechanisms Supporting the Use of Beta-Blockers for the Management of Breast Cancer Bone Metastasis. Cancers 2021, 13, 2887. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, R.C.; Krajewska, M.; Krnacik, S.; Jaeger, R.; Weiher, H.; Krajewski, S.; Reed, J.C.; Rosen, J.M. Apoptosis in the terminal endbud of the murine mammary gland: A mechanism of ductal morphogenesis. Development 1996, 122, 4013–4022. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.C.; Avivar-Valderas, A.; Sosa, M.S.; Girnius, N.; Farias, E.F.; Davis, R.J.; Aguirre-Ghiso, J.A. p38alpha Signaling Induces Anoikis and Lumen Formation During Mammary Morphogenesis. Sci. Signal. 2011, 4, ra34. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, S.; Sadaoui, N.C.; Hu, W.; Dasari, S.K.; Mangala, L.S.; Sun, Y.; Zhao, S.; Wang, L.; Liu, Y.; et al. Sustained Adrenergic Activation of YAP1 Induces Anoikis Resistance in Cervical Cancer Cells. iScience 2020, 23, 101289. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Armaiz-Pena, G.N.; Halder, J.; Nick, A.M.; Stone, R.L.; Hu, W.; Carroll, A.R.; Spannuth, W.A.; Deavers, M.T.; Allen, J.K.; et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Investig. 2010, 120, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, X.M.; Wang, Y.H.; Feng, M.X.; Liu, X.J.; Zhang, Y.L.; Huang, S.; Wu, Z.; Xue, F.; Qin, W.X.; et al. Monoamine oxidase A suppresses hepatocellular carcinoma metastasis by inhibiting the adrenergic system and its transactivation of EGFR signaling. J. Hepatol. 2014, 60, 1225–1234. [Google Scholar] [CrossRef]
- Dal Monte, M.; Fornaciari, I.; Nicchia, G.P.; Svelto, M.; Casini, G.; Bagnoli, P. beta3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn. Schmiedebergs Arch. Pharmacol. 2014, 387, 533–543. [Google Scholar] [CrossRef]
- Dal Monte, M.; Calvani, M.; Cammalleri, M.; Favre, C.; Filippi, L.; Bagnoli, P. beta-Adrenoceptors as drug targets in melanoma: Novel preclinical evidence for a role of beta3 -adrenoceptors. Br. J. Pharmacol. 2019, 176, 2496–2508. [Google Scholar] [CrossRef]
- Bergin, E.; Levine, J.S.; Koh, J.S.; Lieberthal, W. Mouse proximal tubular cell-cell adhesion inhibits apoptosis by a cadherin-dependent mechanism. Am. J. Physiol. Renal. Physiol. 2000, 278, F758–F768. [Google Scholar] [CrossRef]
- Aoshiba, K.; Rennard, S.I.; Spurzem, J.R. Cell-matrix and cell-cell interactions modulate apoptosis of bronchial epithelial cells. Am. J. Physiol. 1997, 272, L28–L37. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, X.; Ou, Y.; Zou, L.; Zhang, D.; Yang, Q.; Qin, Y.; Du, X.; Li, W.; Yuan, Z.; et al. Anoikis resistance--protagonists of breast cancer cells survive and metastasize after ECM detachment. Cell Commun. Signal. 2023, 21, 190. [Google Scholar] [CrossRef]
- Toll, L.; Jimenez, L.; Waleh, N.; Jozwiak, K.; Woo, A.Y.; Xiao, R.P.; Bernier, M.; Wainer, I.W. Beta2-adrenergic receptor agonists inhibit the proliferation of 1321N1 astrocytoma cells. J. Pharmacol. Exp. Ther. 2011, 336, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Perez Pinero, C.; Bruzzone, A.; Sarappa, M.G.; Castillo, L.F.; Luthy, I.A. Involvement of alpha2- and beta2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br. J. Pharmacol. 2012, 166, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.V.; Kim, S.J.; Donovan, E.L.; Chen, M.; Gross, A.C.; Webster Marketon, J.I.; Barsky, S.H.; Glaser, R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: Implications for stress-related enhancement of tumor progression. Brain Behav. Immun. 2009, 23, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A.; Park, J.M.; Lee, K.S.; Xu, C.; Stella, N.; Hague, C. Label-Free Dynamic Mass Redistribution Reveals Low-Density, Prosurvival alpha(1B)-Adrenergic Receptors in Human SW480 Colon Carcinoma Cells. J. Pharmacol. Exp. Ther. 2017, 361, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Cros, G. Mechanisms of desensitization of beta-adrenergic receptors. Bull. Eur. Physiopathol. Respir. 1985, 21, 35s–43s. [Google Scholar]
- Kwon, Y.; Mehta, S.; Clark, M.; Walters, G.; Zhong, Y.; Lee, H.N.; Sunahara, R.K.; Zhang, J. Non-canonical beta-adrenergic activation of ERK at endosomes. Nature 2022, 611, 173–179. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. beta2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell 2018, 33, 75–90.e77. [Google Scholar] [CrossRef]
- Jang, H.J.; Boo, H.J.; Lee, H.J.; Min, H.Y.; Lee, H.Y. Chronic Stress Facilitates Lung Tumorigenesis by Promoting Exocytosis of IGF2 in Lung Epithelial Cells. Cancer Res. 2016, 76, 6607–6619. [Google Scholar] [CrossRef]
- Saul, A.N.; Oberyszyn, T.M.; Daugherty, C.; Kusewitt, D.; Jones, S.; Jewell, S.; Malarkey, W.B.; Lehman, A.; Lemeshow, S.; Dhabhar, F.S. Chronic stress and susceptibility to skin cancer. J. Natl. Cancer Inst. 2005, 97, 1760–1767. [Google Scholar] [CrossRef]
- Huan, H.B.; Wen, X.D.; Chen, X.J.; Wu, L.; Wu, L.L.; Zhang, L.; Yang, D.P.; Zhang, X.; Bie, P.; Qian, C.; et al. Sympathetic nervous system promotes hepatocarcinogenesis by modulating inflammation through activation of alpha1-adrenergic receptors of Kupffer cells. Brain Behav. Immun. 2017, 59, 118–134. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, D.; Quintas, C.; Gonçalves, J.; Fresco, P. β2-Adrenoceptor Activation Favor Acquisition of Tumorigenic Properties in Non-Tumorigenic MCF-10A Breast Epithelial Cells. Cells 2024, 13, 262. https://doi.org/10.3390/cells13030262
Silva D, Quintas C, Gonçalves J, Fresco P. β2-Adrenoceptor Activation Favor Acquisition of Tumorigenic Properties in Non-Tumorigenic MCF-10A Breast Epithelial Cells. Cells. 2024; 13(3):262. https://doi.org/10.3390/cells13030262
Chicago/Turabian StyleSilva, Dany, Clara Quintas, Jorge Gonçalves, and Paula Fresco. 2024. "β2-Adrenoceptor Activation Favor Acquisition of Tumorigenic Properties in Non-Tumorigenic MCF-10A Breast Epithelial Cells" Cells 13, no. 3: 262. https://doi.org/10.3390/cells13030262
APA StyleSilva, D., Quintas, C., Gonçalves, J., & Fresco, P. (2024). β2-Adrenoceptor Activation Favor Acquisition of Tumorigenic Properties in Non-Tumorigenic MCF-10A Breast Epithelial Cells. Cells, 13(3), 262. https://doi.org/10.3390/cells13030262