
Preparation of Viable Human Neurites for Neurobiological and Neurodegeneration Studies

Abstract
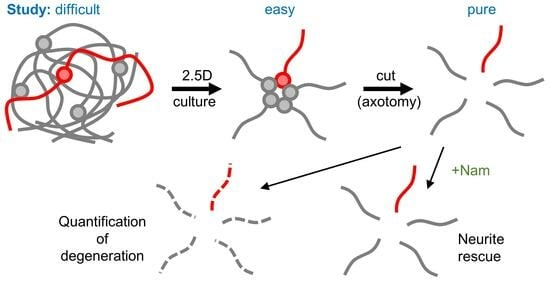
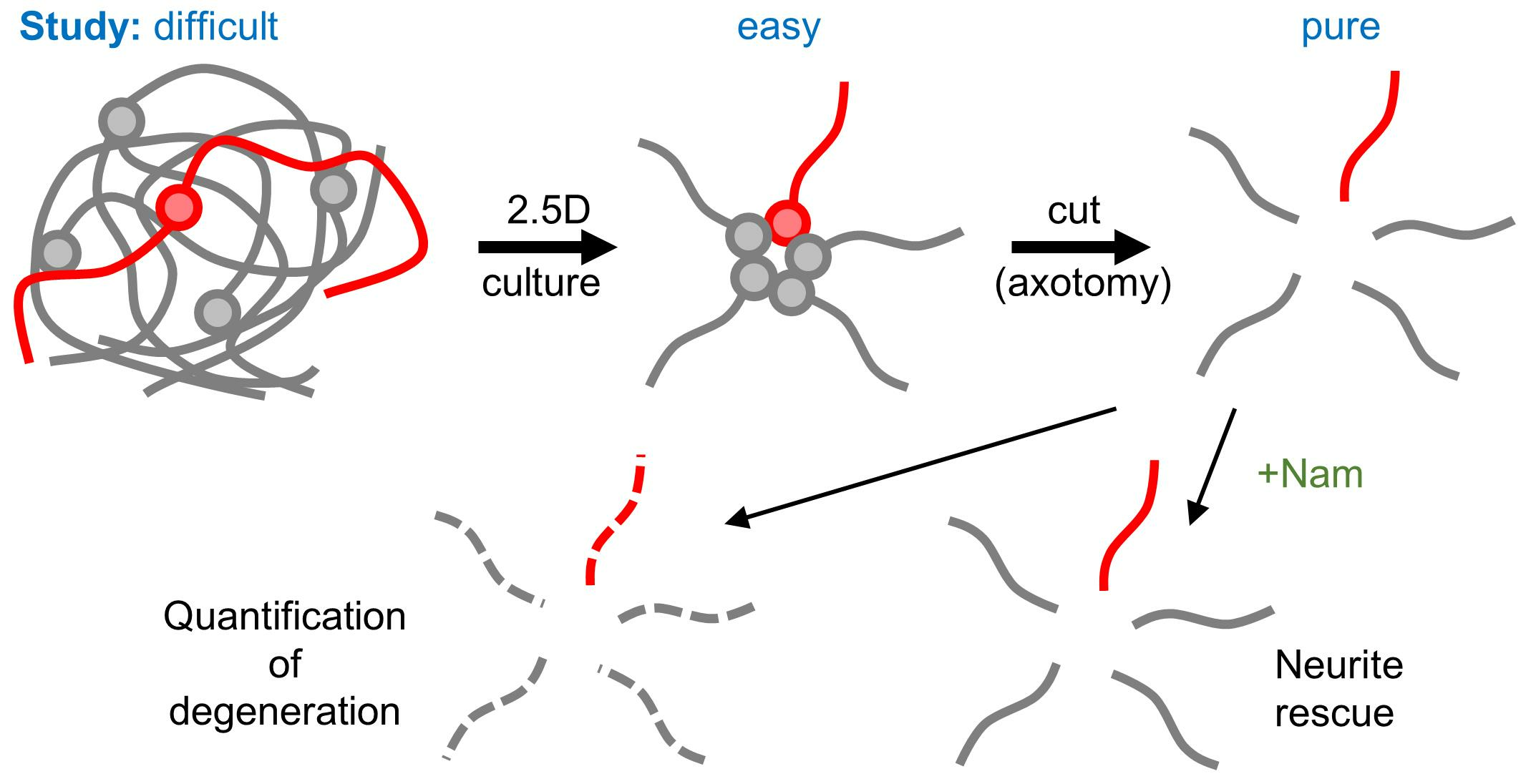{kind=link}
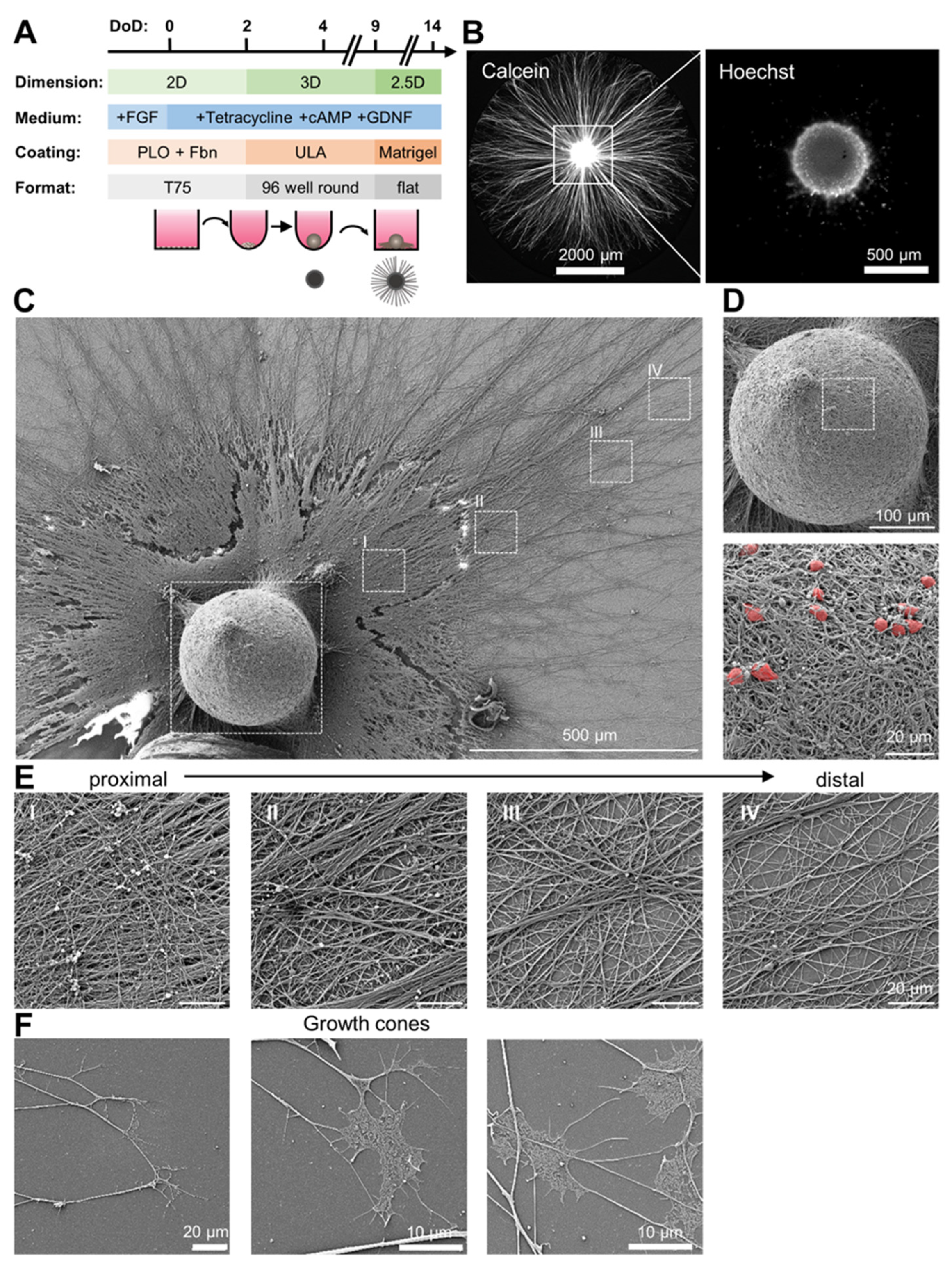{kind=link}
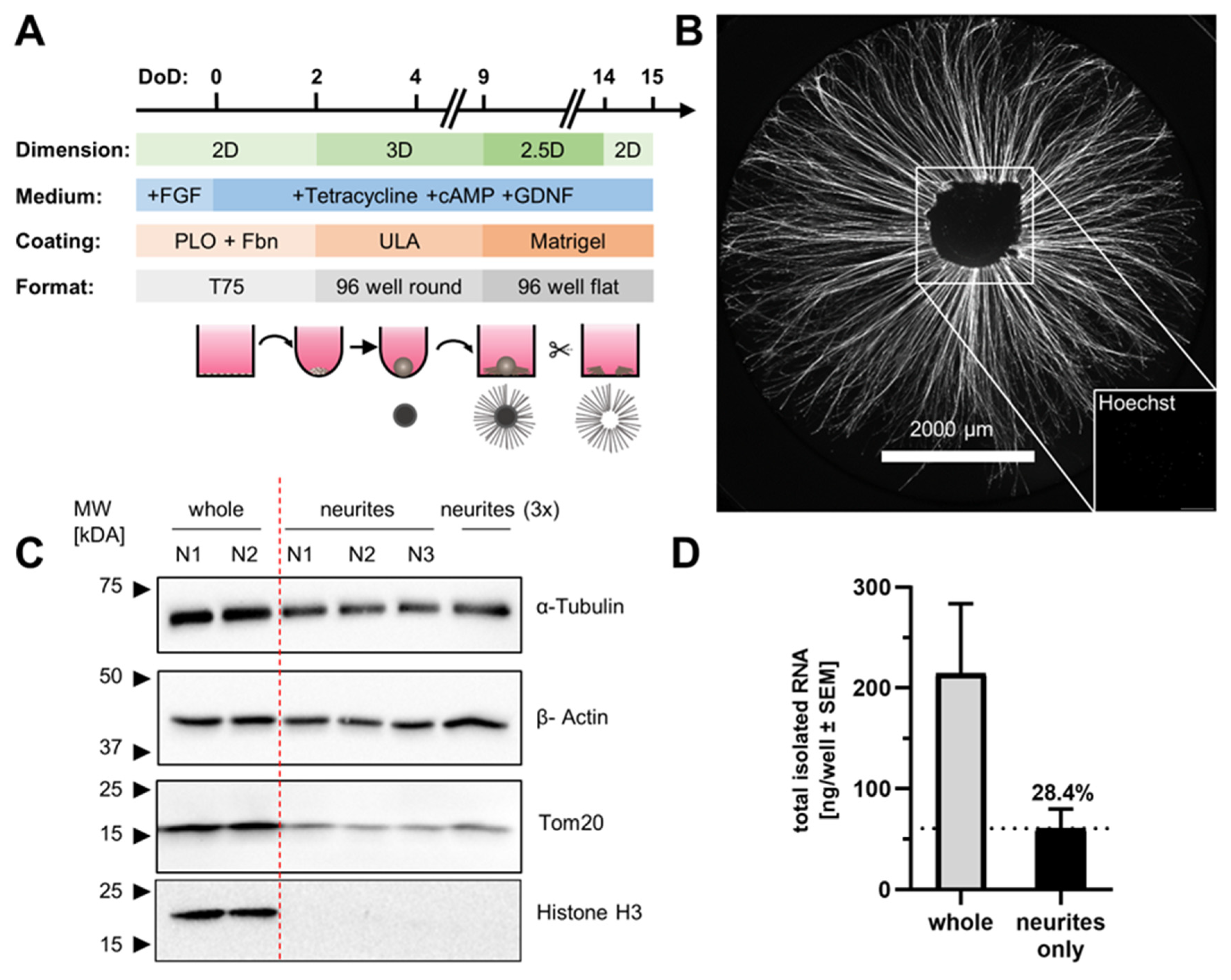{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Coating
2.3. Spheroid Generation and Plating
2.4. Neurite Isolation
2.5. Ribonucleic Acid (RNA) Isolation, Reverse Transcription and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.6. Western Blots
2.7. Immunocytochemistry
2.8. Scanning Electron Microscopy
2.9. Measurement and Quantification of Degeneration
2.10. Adenosine Triphosphate (ATP) Assay
2.11. Nicotinamide Adenine Dinucleotide (NAD(H)) Assay
2.12. Statistical Analysis
3. Results and Discussion
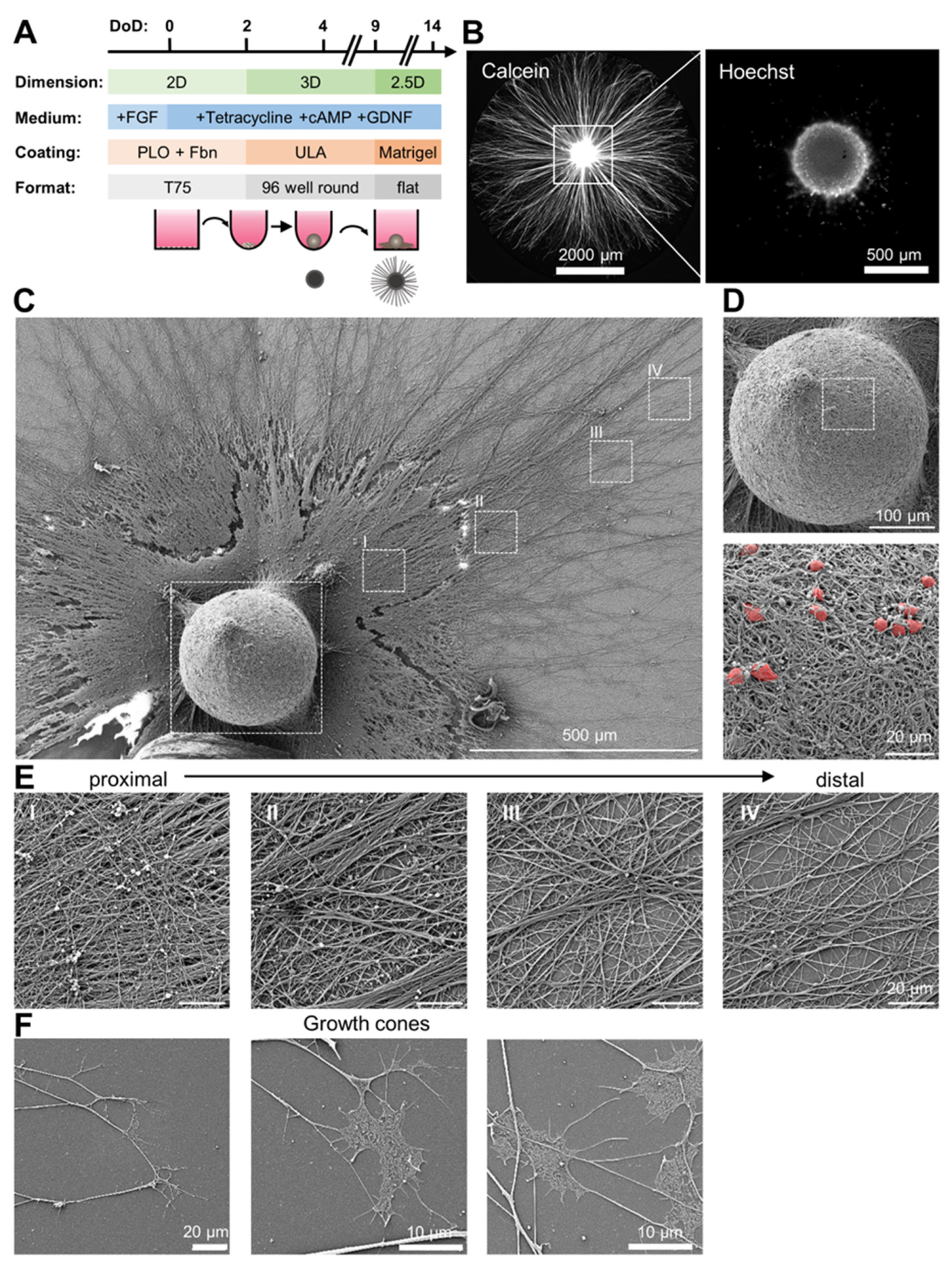
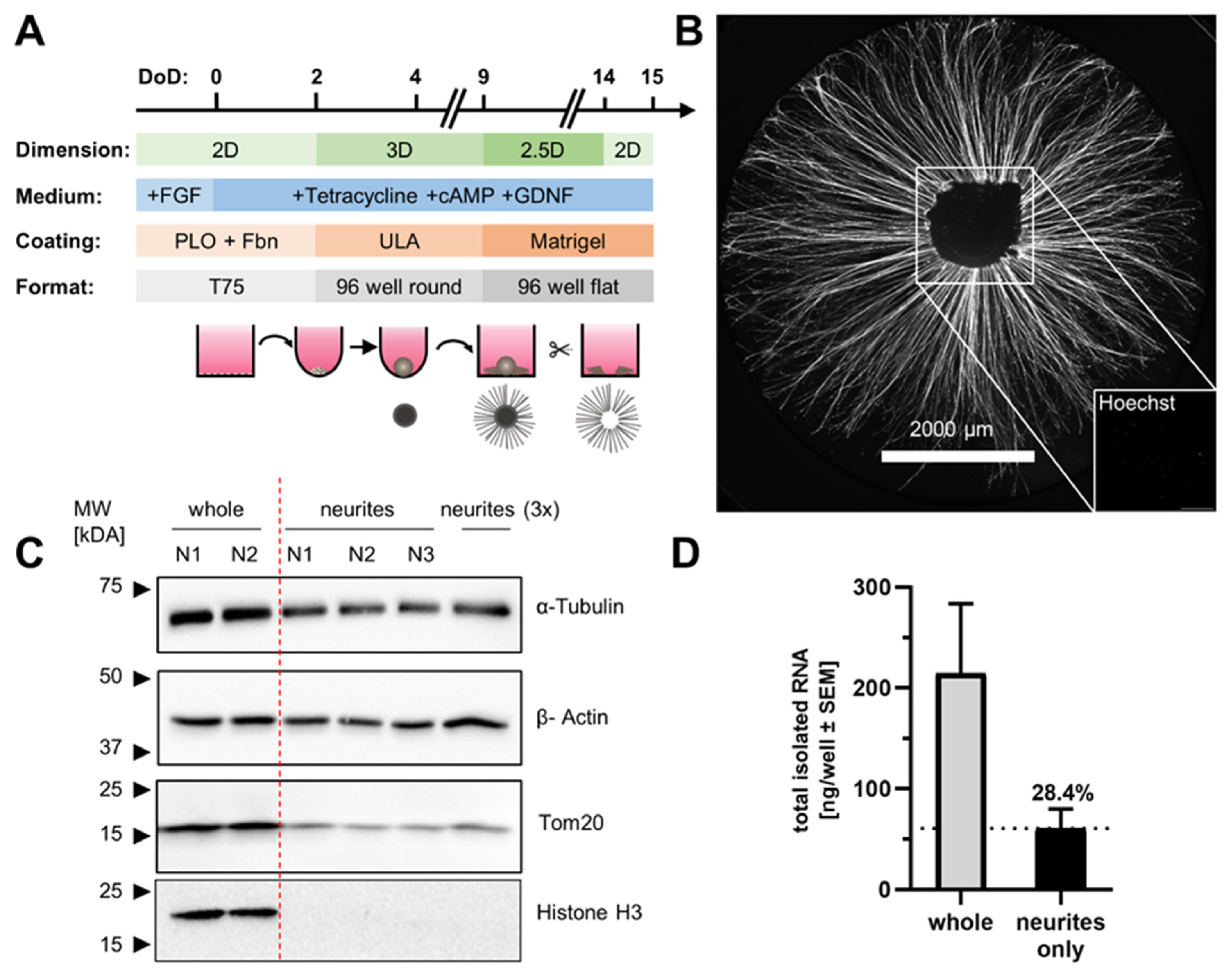
3.1. Preparation of Organized Neuronal Cultures with Distinct Neurite-Only Areas
3.2. Neurite-Only Cultures (NOC)
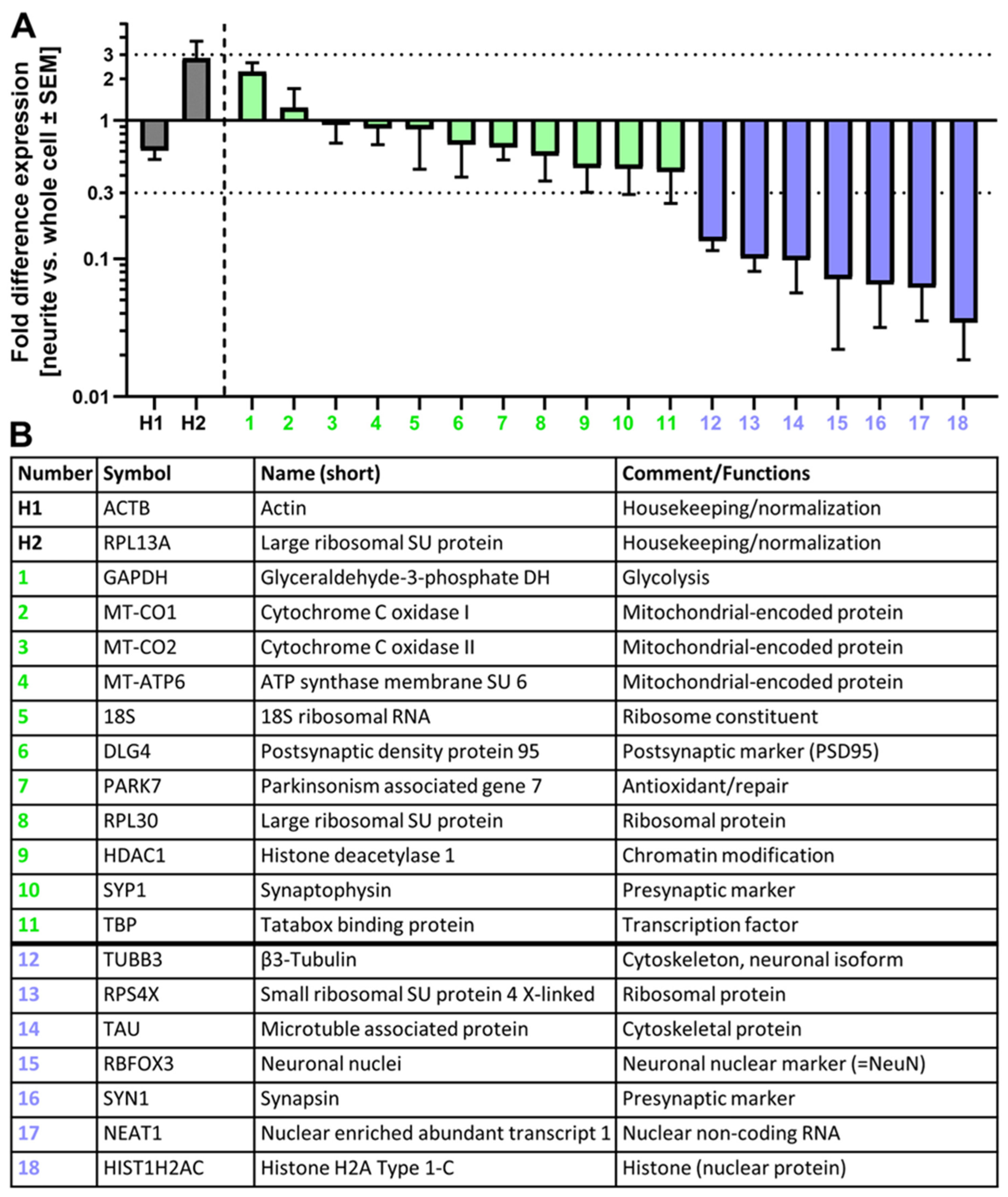
3.3. Quantitative Analysis of Neurite RNA
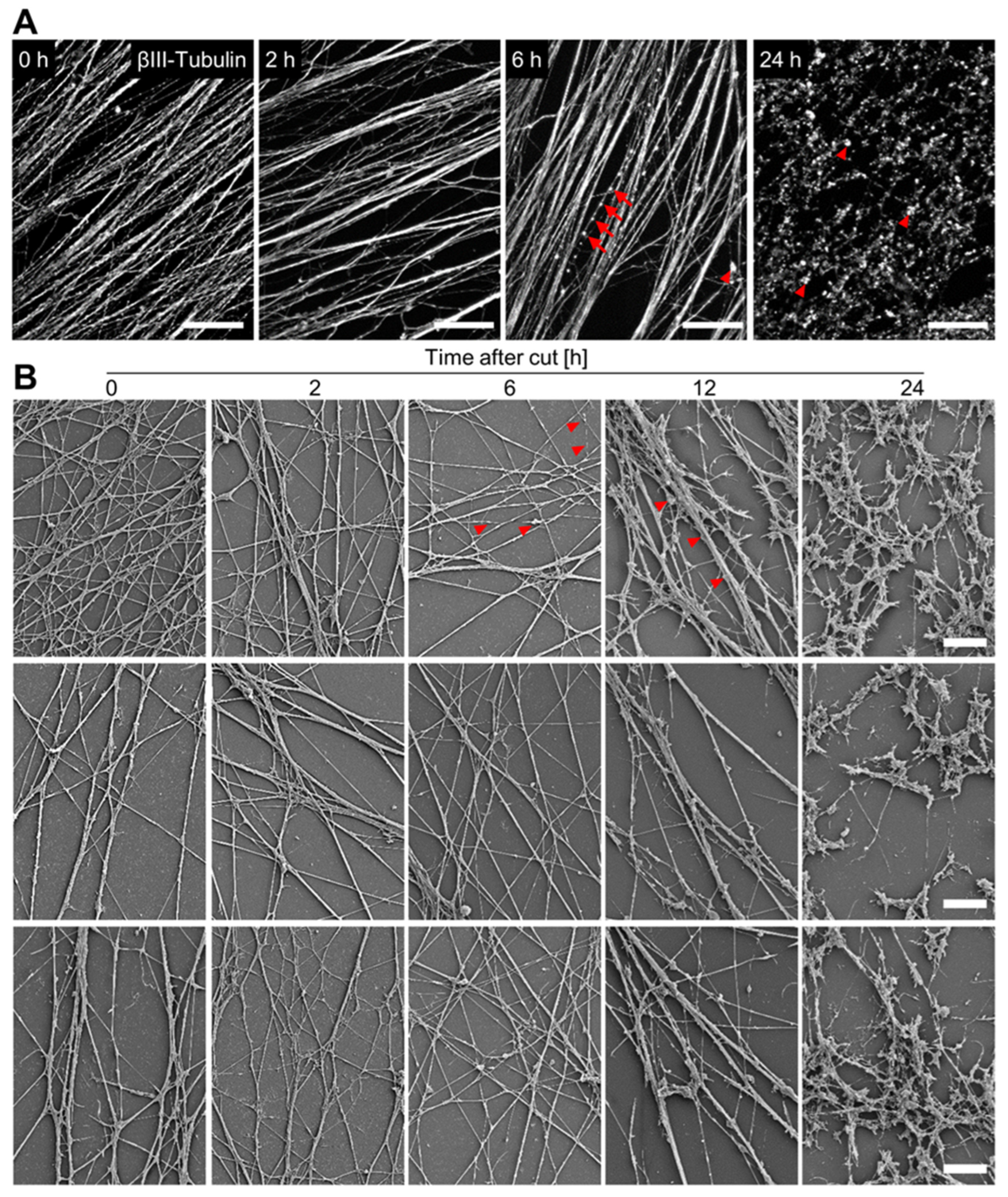
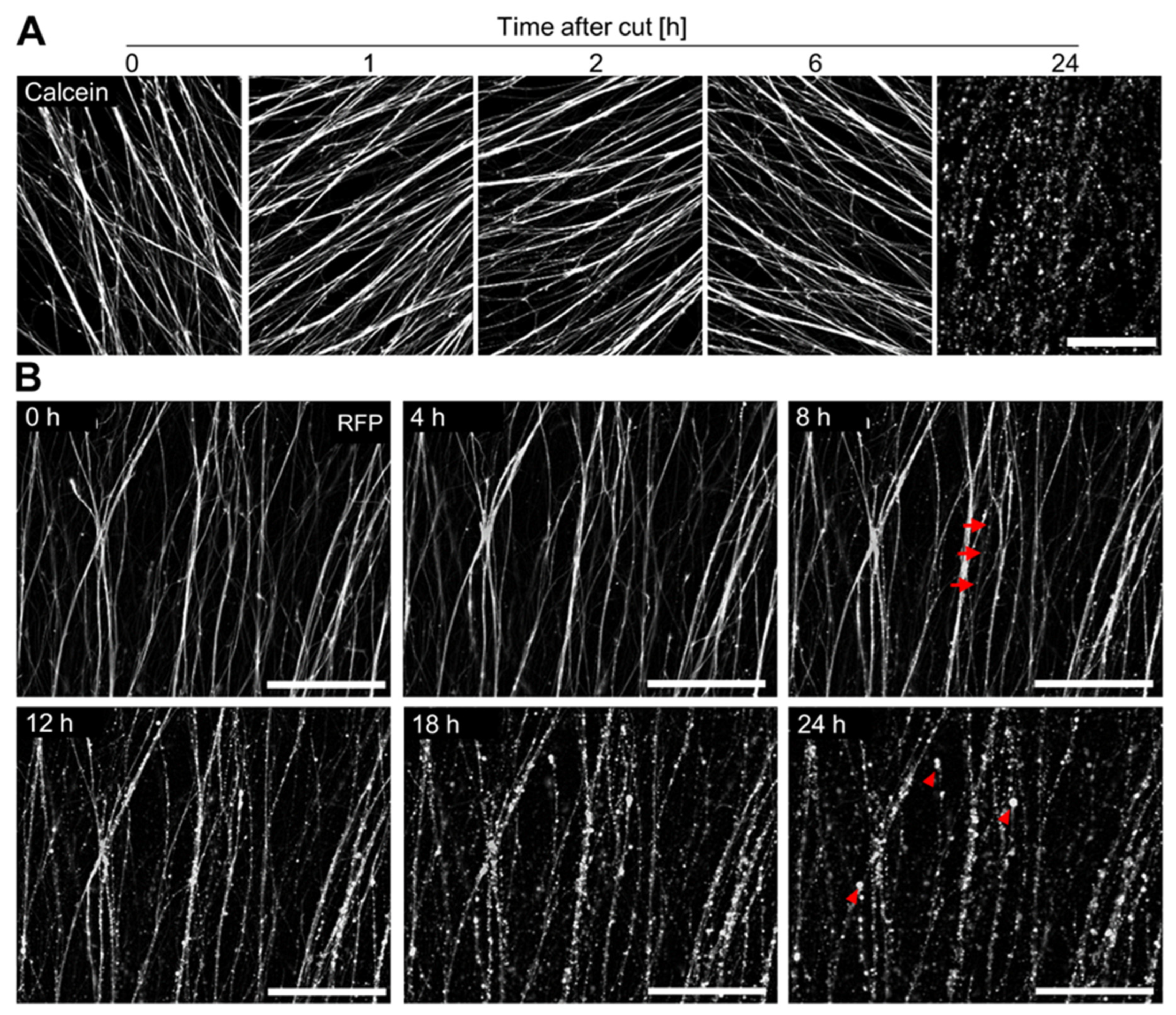
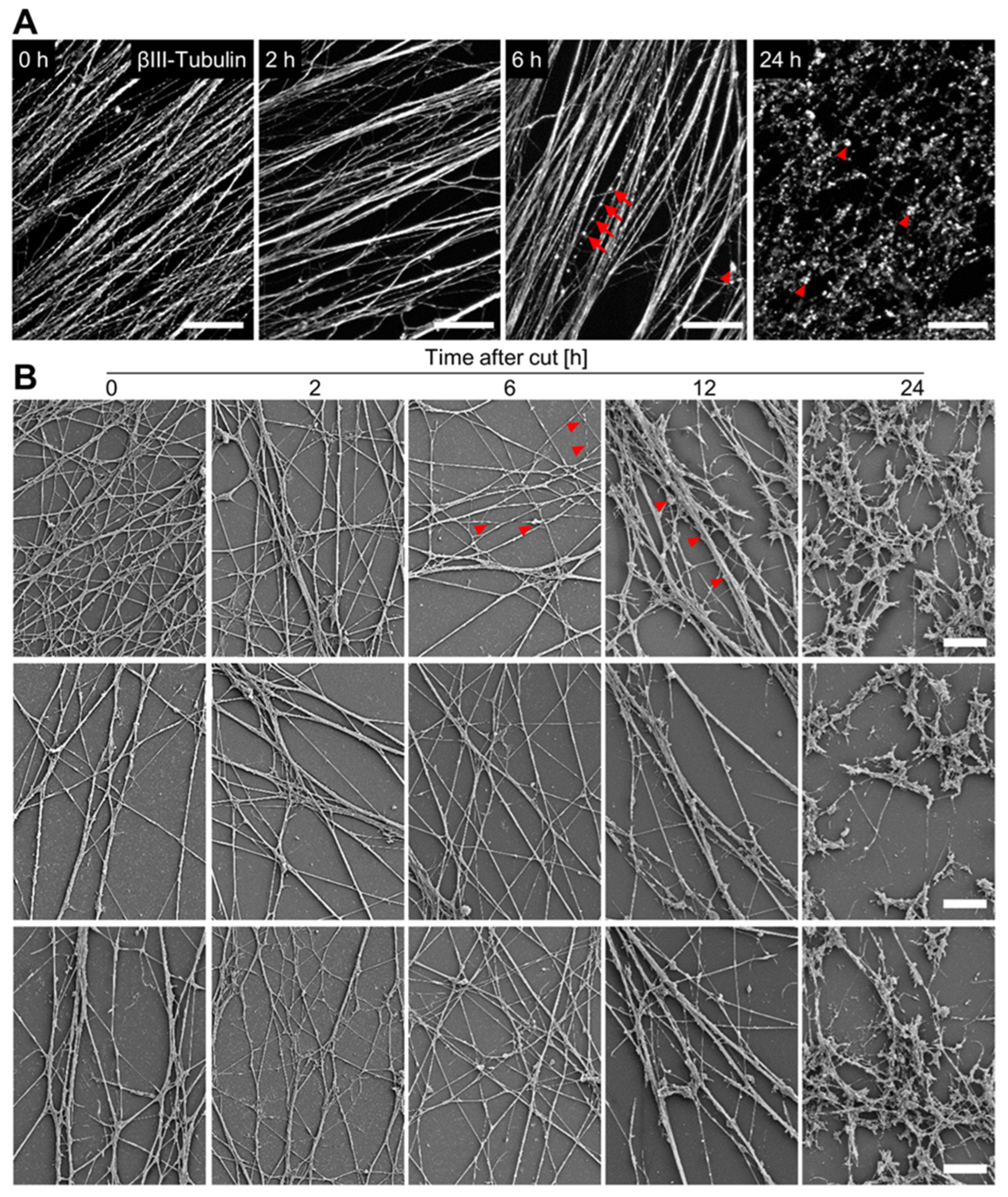
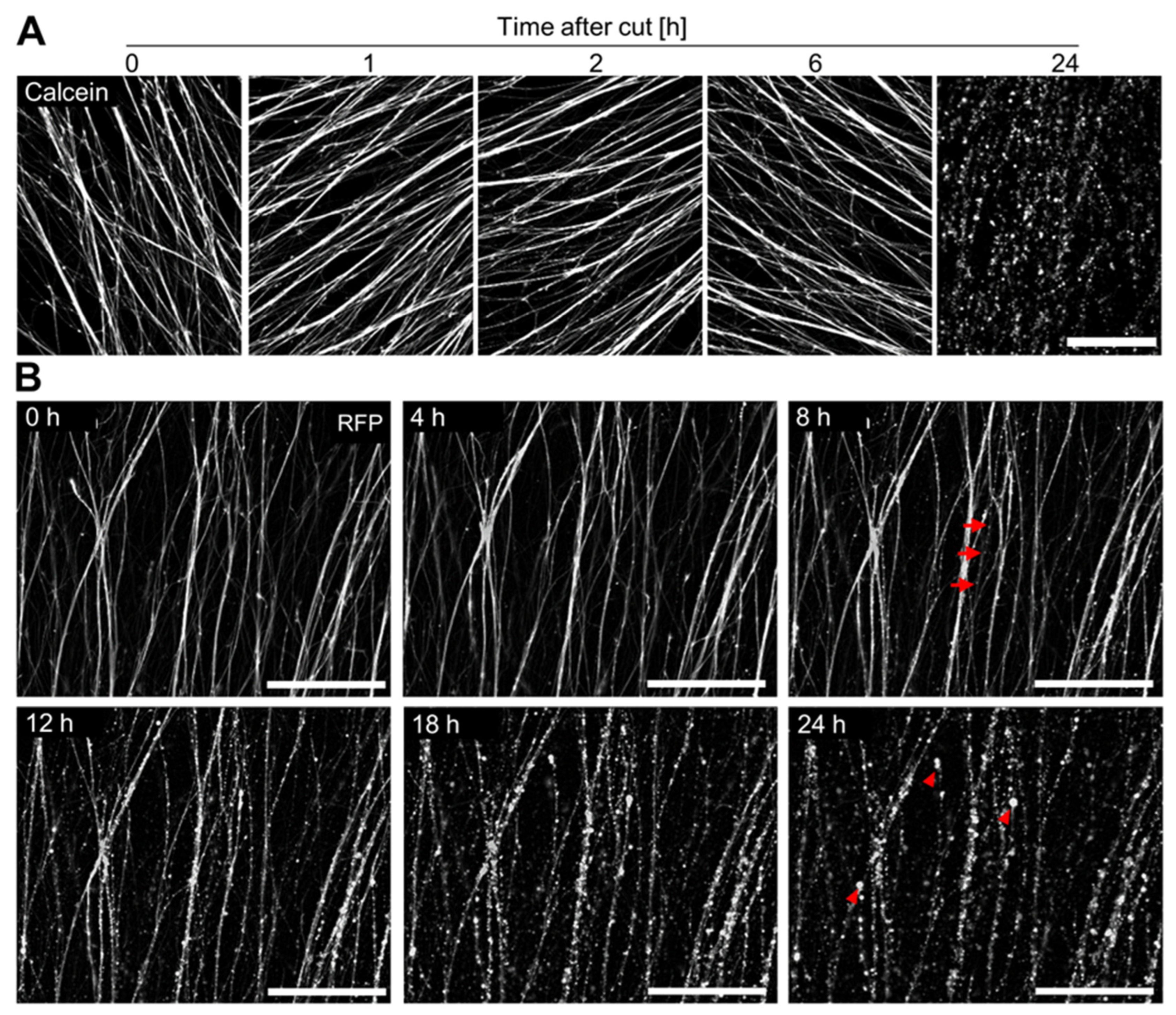
3.4. Time Course of Axotomy-Induced Neurite Degeneration
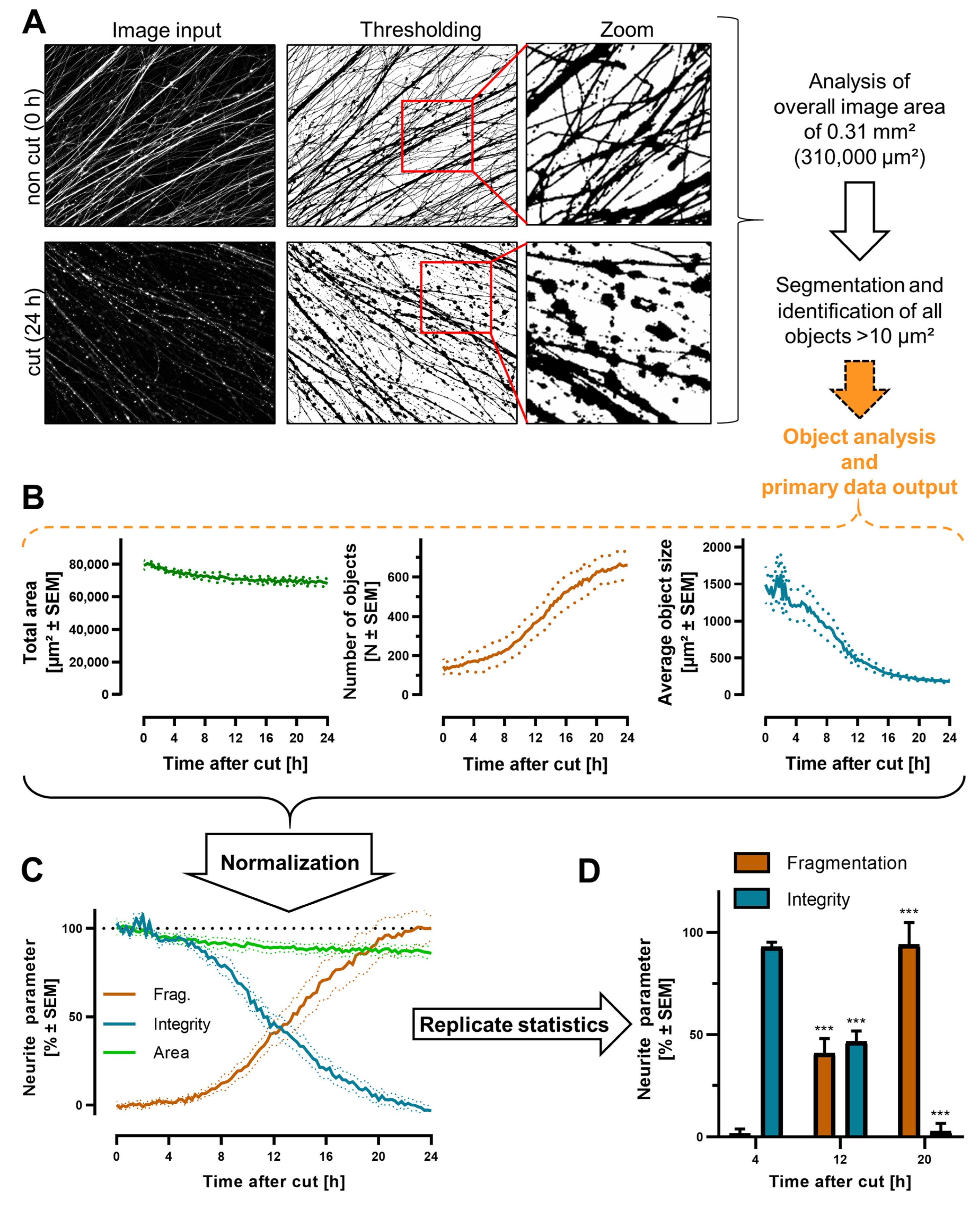
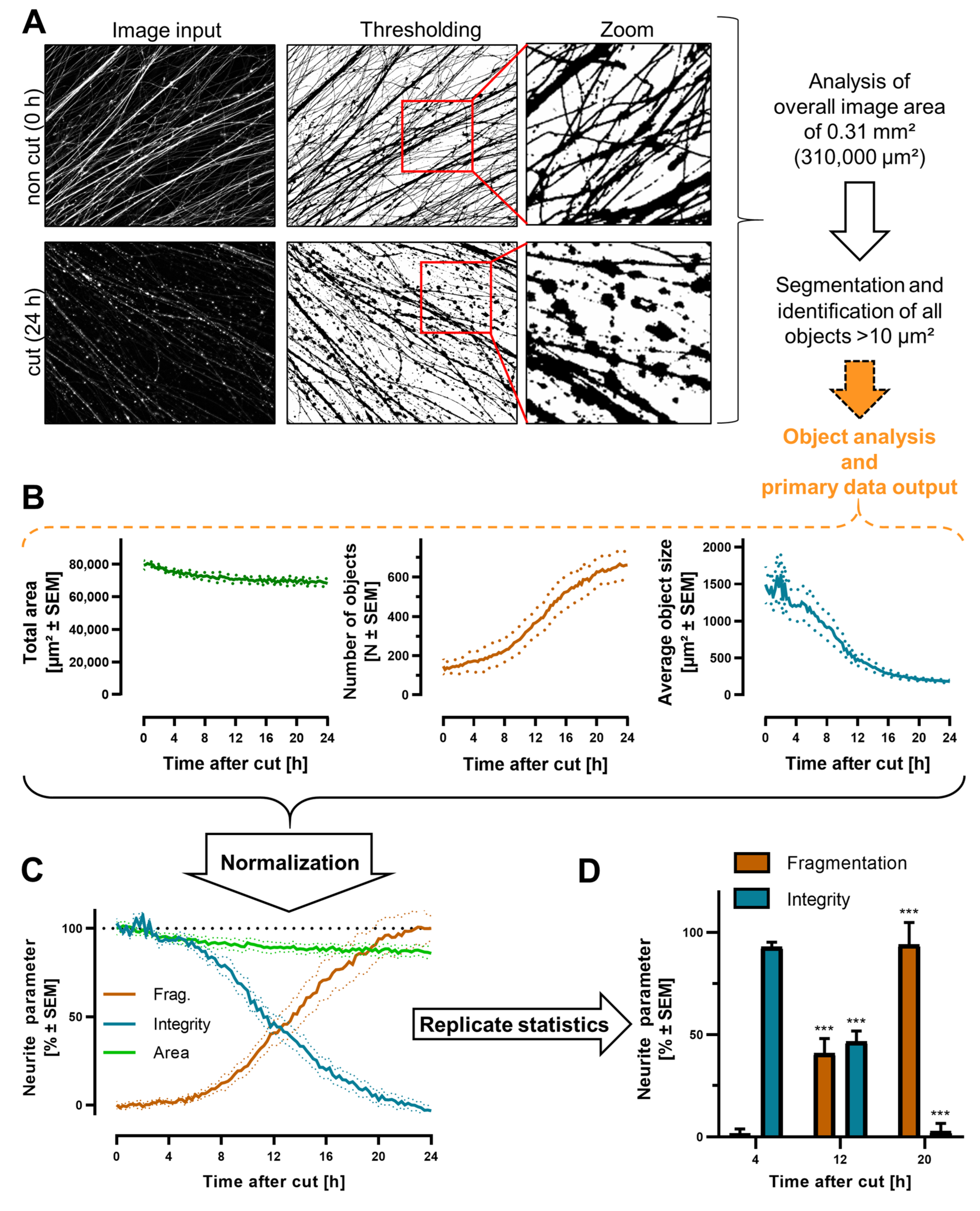
3.5. Quantification of Neurite Fragmentation
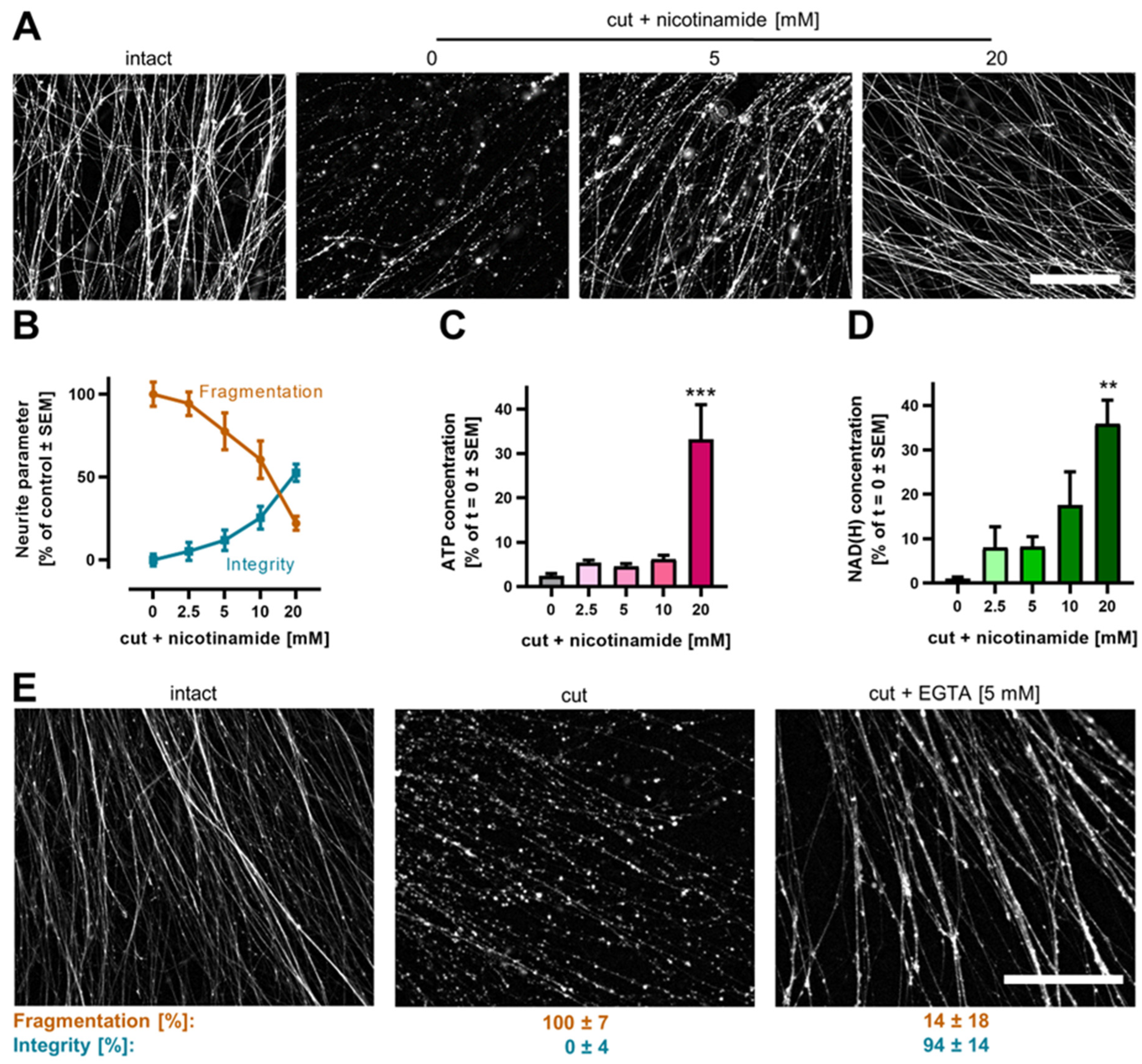
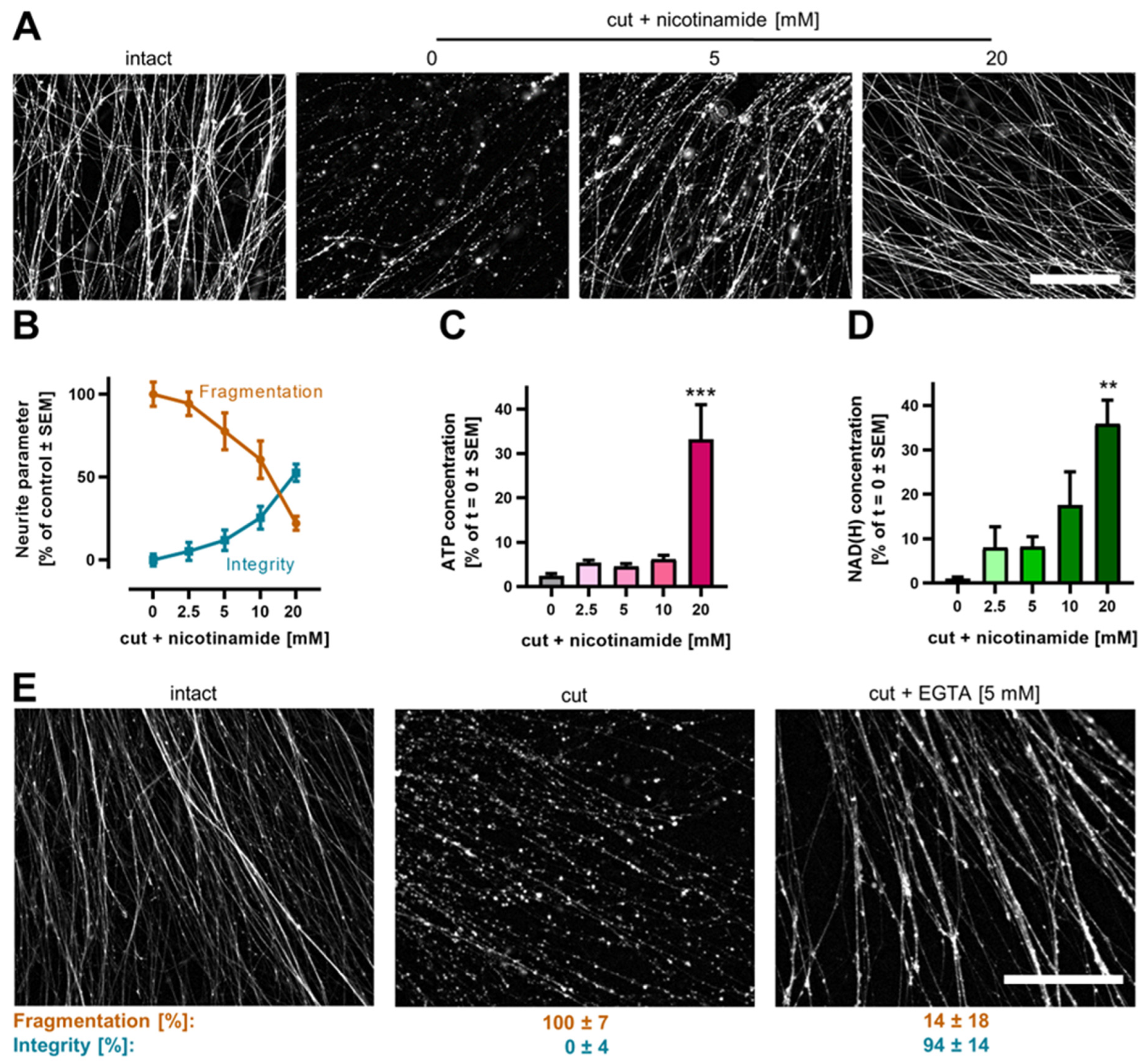
3.6. Pharmacological Intervention against Neurite Degeneration
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carelli, V.; La Morgia, C.; Ross-Cisneros, F.N.; Sadun, A.A. Optic neuropathies: The tip of the neurodegeneration iceberg. Hum. Mol. Genet. 2017, 26, R139–R150. [Google Scholar] [CrossRef]
- Newman, N.J.; Yu-Wai-Man, P.; Biousse, V.; Carelli, V. Understanding the molecular basis and pathogenesis of hereditary optic neuropathies: Towards improved diagnosis and management. Lancet Neurol. 2023, 22, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef]
- Krauss, R.; Bosanac, T.; Devraj, R.; Engber, T.; Hughes, R.O. Axons Matter: The Promise of Treating Neurodegenerative Disorders by Targeting SARM1-Mediated Axonal Degeneration. Trends Pharmacol. Sci. 2020, 41, 281–293. [Google Scholar] [CrossRef]
- Lee, B.; Cho, Y. Experimental Model Systems for Understanding Human Axonal Injury Responses. Int. J. Mol. Sci. 2021, 22, 474. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Loedige, I.; Baranovskii, A.; Mendonsa, S.; Dantsuji, S.; Popitsch, N.; Breimann, L.; Zerna, N.; Cherepanov, V.; Milek, M.; Ameres, S.; et al. mRNA stability and m6A are major determinants of subcellular mRNA localization in neurons. Mol. Cell 2023, 83, 2709–2725.e2710. [Google Scholar] [CrossRef]
- Dorrbaum, A.R.; Kochen, L.; Langer, J.D.; Schuman, E.M. Local and global influences on protein turnover in neurons and glia. Elife 2018, 7, e34202. [Google Scholar] [CrossRef]
- Hagenston, A.M.; Bading, H. Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004564. [Google Scholar] [CrossRef]
- Berth, S.H.; Lloyd, T.E. Disruption of axonal transport in neurodegeneration. J. Clin. Investig. 2023, 133, e168554. [Google Scholar] [CrossRef]
- Roy, S.; Zhang, B.; Lee, V.M.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef]
- Adalbert, R.; Coleman, M.P. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2013, 39, 90–108. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Pigino, G.F.; Brady, S.T.; Lazarov, O.; Binder, L.I.; Morfini, G.A. Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp. Neurol. 2013, 246, 44–53. [Google Scholar] [CrossRef]
- Li, H.; Li, S.H.; Yu, Z.X.; Shelbourne, P.; Li, X.J. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 2001, 21, 8473–8481. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef]
- Cajigas, I.J.; Tushev, G.; Will, T.J.; tom Dieck, S.; Fuerst, N.; Schuman, E.M. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 2012, 74, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Dieterich, D.C.; Ito, H.T.; Kim, S.A.; Schuman, E.M. Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 2010, 66, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Rhee, S.W.; Tu, C.H.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. Microfluidic Multicompartment Device for Neuroscience Research. Langmuir 2003, 19, 1551–1556. [Google Scholar] [CrossRef]
- Campenot, R.B. Local control of neurite development by nerve growth factor. Proc. Natl. Acad. Sci. USA 1977, 74, 4516–4519. [Google Scholar] [CrossRef]
- Ciolli Mattioli, C.; Rom, A.; Franke, V.; Imami, K.; Arrey, G.; Terne, M.; Woehler, A.; Akalin, A.; Ulitsky, I.; Chekulaeva, M. Alternative 3’ UTRs direct localization of functionally diverse protein isoforms in neuronal compartments. Nucleic Acids Res. 2019, 47, 2560–2573. [Google Scholar] [CrossRef]
- Unsain, N.; Heard, K.N.; Higgins, J.M.; Barker, P.A. Production and isolation of axons from sensory neurons for biochemical analysis using porous filters. J. Vis. Exp. 2014, 89, e51795. [Google Scholar] [CrossRef]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef]
- Vargas, M.E.; Yamagishi, Y.; Tessier-Lavigne, M.; Sagasti, A. Live Imaging of Calcium Dynamics during Axon Degeneration Reveals Two Functionally Distinct Phases of Calcium Influx. J. Neurosci. 2015, 35, 15026–15038. [Google Scholar] [CrossRef]
- Wang, X.; Yang, C.; Wang, X.; Miao, J.; Chen, W.; Zhou, Y.; Xu, Y.; An, Y.; Cheng, A.; Ye, W.; et al. Driving axon regeneration by orchestrating neuronal and non-neuronal innate immune responses via the IFNgamma-cGAS-STING axis. Neuron 2023, 111, 236–255.e237. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.R.; Herrmann, J.E.; Woo, M.J.; Tansey, K.E.; Doan, N.B.; Sofroniew, M.V. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 2004, 24, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Ghezzi, P.; Grasso, G.; Bianchi, R.; Villa, P.; Fratelli, M.; Savino, C.; Bianchi, M.; Nielsen, J.; Gerwien, J.; et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Haque, N.S.; Hlavin, M.L.; Fawcett, J.W.; Dunnett, S.B. The neurotrophin NT4/5, but not NT3, enhances the efficacy of nigral grafts in a rat model of Parkinson’s disease. Brain Res. 1996, 712, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Wilby, M.J.; Sinclair, S.R.; Muir, E.M.; Zietlow, R.; Adcock, K.H.; Horellou, P.; Rogers, J.H.; Dunnett, S.B.; Fawcett, J.W. A glial cell line-derived neurotrophic factor-secreting clone of the Schwann cell line SCTM41 enhances survival and fiber outgrowth from embryonic nigral neurons grafted to the striatum and to the lesioned substantia nigra. J. Neurosci. 1999, 19, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Al-Ali, H.; Beckerman, S.R.; Bixby, J.L.; Lemmon, V.P. In vitro models of axon regeneration. Exp. Neurol. 2017, 287, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 1984, 103, 285–293. [Google Scholar] [CrossRef]
- Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Neural transplantation of human neuroteratocarcinoma (hNT) neurons into ischemic rats. A quantitative dose-response analysis of cell survival and behavioral recovery. Neuroscience 1999, 91, 519–525. [Google Scholar] [CrossRef]
- Roloff, F.; Scheiblich, H.; Dewitz, C.; Dempewolf, S.; Stern, M.; Bicker, G. Enhanced neurite outgrowth of human model (NT2) neurons by small-molecule inhibitors of Rho/ROCK signaling. PLoS ONE 2015, 10, e0118536. [Google Scholar] [CrossRef]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar] [PubMed]
- Dravid, A.; Raos, B.; Svirskis, D.; O’Carroll, S.J. Optimised techniques for high-throughput screening of differentiated SH-SY5Y cells and application for neurite outgrowth assays. Sci. Rep. 2021, 11, 23935. [Google Scholar] [CrossRef]
- Culbreth, M.; Nyffeler, J.; Willis, C.; Harrill, J.A. Optimization of Human Neural Progenitor Cells for an Imaging-Based High-Throughput Phenotypic Profiling Assay for Developmental Neurotoxicity Screening. Front. Toxicol. 2021, 3, 803987. [Google Scholar] [CrossRef]
- Carstens, K.E.; Carpenter, A.F.; Martin, M.M.; Harrill, J.A.; Shafer, T.J.; Paul Friedman, K. Integrating Data From In Vitro New Approach Methodologies for Developmental Neurotoxicity. Toxicol. Sci. 2022, 187, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.S.; Bucelli, R.; Jett, D.A.; Bruun, D.; Yang, D.; Lein, P.J. Chlorpyrifos exerts opposing effects on axonal and dendritic growth in primary neuronal cultures. Toxicol. Appl. Pharmacol. 2005, 207, 112–124. [Google Scholar] [CrossRef]
- Pin, S.; Chen, H.; Lein, P.J.; Wang, M.M. Nucleic acid binding agents exert local toxic effects on neurites via a non-nuclear mechanism. J. Neurochem. 2006, 96, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Lunn, E.R.; Perry, V.H.; Brown, M.C.; Rosen, H.; Gordon, S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur. J. Neurosci. 1989, 1, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Bosanac, T.; Hughes, R.O.; Engber, T.; Devraj, R.; Brearley, A.; Danker, K.; Young, K.; Kopatz, J.; Hermann, M.; Berthemy, A.; et al. Pharmacological SARM1 inhibition protects axon structure and function in paclitaxel-induced peripheral neuropathy. Brain 2021, 144, 3226–3238. [Google Scholar] [CrossRef]
- Wang, J.; Zhai, Q.; Chen, Y.; Lin, E.; Gu, W.; McBurney, M.W.; He, Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol. 2005, 170, 349–355. [Google Scholar] [CrossRef]
- Di Stefano, M.; Nascimento-Ferreira, I.; Orsomando, G.; Mori, V.; Gilley, J.; Brown, R.; Janeckova, L.; Vargas, M.E.; Worrell, L.A.; Loreto, A.; et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015, 22, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e1335. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.P.; Hoke, A. Programmed axon degeneration: From mouse to mechanism to medicine. Nat. Rev. Neurosci. 2020, 21, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Gcwensa, N.Z.; Russell, D.L.; Cowell, R.M.; Volpicelli-Daley, L.A. Molecular Mechanisms Underlying Synaptic and Axon Degeneration in Parkinson’s Disease. Front. Cell Neurosci. 2021, 15, 626128. [Google Scholar] [CrossRef] [PubMed]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef]
- Gutbier, S.; May, P.; Berthelot, S.; Krishna, A.; Trefzer, T.; Behbehani, M.; Efremova, L.; Delp, J.; Gstraunthaler, G.; Waldmann, T.; et al. Major changes of cell function and toxicant sensitivity in cultured cells undergoing mild, quasi-natural genetic drift. Arch. Toxicol. 2018, 92, 3487–3503. [Google Scholar] [CrossRef]
- Scholz, D.; Poltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef]
- Delp, J.; Cediel-Ulloa, A.; Suciu, I.; Kranaster, P.; van Vugt-Lussenburg, B.M.; Munic Kos, V.; van der Stel, W.; Carta, G.; Bennekou, S.H.; Jennings, P.; et al. Neurotoxicity and underlying cellular changes of 21 mitochondrial respiratory chain inhibitors. Arch. Toxicol. 2021, 95, 591–615. [Google Scholar] [CrossRef]
- Delp, J.; Gutbier, S.; Klima, S.; Hoelting, L.; Pinto-Gil, K.; Hsieh, J.H.; Aichem, M.; Klein, K.; Schreiber, F.; Tice, R.R.; et al. A high-throughput approach to identify specific neurotoxicants/developmental toxicants in human neuronal cell function assays. ALTEX 2018, 35, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.K.; Balmer, N.V.; Matt, F.; Schonenberger, F.; Merhof, D.; Leist, M. Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch. Toxicol. 2013, 87, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Brull, M.; Spreng, A.S.; Gutbier, S.; Loser, D.; Krebs, A.; Reich, M.; Kraushaar, U.; Britschgi, M.; Patsch, C.; Leist, M. Incorporation of stem cell-derived astrocytes into neuronal organoids to allow neuro-glial interactions in toxicological studies. ALTEX 2020, 37, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Harris, G.; Eschment, M.; Orozco, S.P.; McCaffery, J.M.; Maclennan, R.; Severin, D.; Leist, M.; Kleensang, A.; Pamies, D.; Maertens, A.; et al. Toxicity, recovery, and resilience in a 3D dopaminergic neuronal in vitro model exposed to rotenone. Arch. Toxicol. 2018, 92, 2587–2606. [Google Scholar] [CrossRef] [PubMed]
- Schildknecht, S.; Karreman, C.; Poltl, D.; Efremova, L.; Kullmann, C.; Gutbier, S.; Krug, A.; Scholz, D.; Gerding, H.R.; Leist, M. Generation of genetically-modified human differentiated cells for toxicological tests and the study of neurodegenerative diseases. ALTEX 2013, 30, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.S.; Donlin-Asp, P.G.; Leitch, B.; Herzog, E.; Schuman, E.M. Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 2019, 364, eaau3644. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.E.; Martin, K.C.; Schuman, E.M. Local translation in neurons: Visualization and function. Nat. Struct. Mol. Biol. 2019, 26, 557–566. [Google Scholar] [CrossRef]
- Shigeoka, T.; Jung, H.; Jung, J.; Turner-Bridger, B.; Ohk, J.; Lin, J.Q.; Amieux, P.S.; Holt, C.E. Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell 2016, 166, 181–192. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Kaneko, S.; Wang, J.; Kaneko, M.; Yiu, G.; Hurrell, J.M.; Chitnis, T.; Khoury, S.J.; He, Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J. Neurosci. 2006, 26, 9794–9804. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Ainscow, E.K.; Mirshamsi, S.; Tang, T.; Ashford, M.L.; Rutter, G.A. Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: Evidence for ATP-independent control of ATP-sensitive K+ channels. J. Physiol. 2002, 544, 429–445. [Google Scholar] [CrossRef]
- Fukuda, J.; Fujita, Y.; Ohsawa, K. ATP content in isolated mammalian nerve cells assayed by a modified luciferin-luciferase method. J. Neurosci. Methods 1983, 8, 295–302. [Google Scholar] [CrossRef]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef]
- Toloe, J.; Mollajew, R.; Kugler, S.; Mironov, S.L. Metabolic differences in hippocampal ‘Rett’ neurons revealed by ATP imaging. Mol. Cell Neurosci. 2014, 59, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e1311. [Google Scholar] [CrossRef] [PubMed]
- Nicotera, P.; Leist, M.; Fava, E.; Berliocchi, L.; Volbracht, C. Energy requirement for caspase activation and neuronal cell death. Brain Pathol. 2000, 10, 276–282. [Google Scholar] [CrossRef]
- Poltl, D.; Schildknecht, S.; Karreman, C.; Leist, M. Uncoupling of ATP-depletion and cell death in human dopaminergic neurons. Neurotoxicology 2012, 33, 769–779. [Google Scholar] [CrossRef]
- Volbracht, C.; Leist, M.; Nicotera, P. ATP controls neuronal apoptosis triggered by microtubule breakdown or potassium deprivation. Mol. Med. 1999, 5, 477–489. [Google Scholar] [CrossRef]
- Le Masson, G.; Przedborski, S.; Abbott, L.F. A computational model of motor neuron degeneration. Neuron 2014, 83, 975–988. [Google Scholar] [CrossRef]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; An, R.; Umanah, G.K.; Park, H.; Nambiar, K.; Eacker, S.M.; Kim, B.; Bao, L.; Harraz, M.M.; Chang, C.; et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 2016, 354, aad6872. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Mitochondrial Mechanisms of Neuronal Cell Death: Potential Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Costa, A.C.; Celardo, I.; Loh, S.H.; Martins, L.M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 2016, 7, e2166. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fedele, G.; Celardo, I.; Loh, S.H.Y.; Martins, L.M. Parp mutations protect from mitochondrial toxicity in Alzheimer’s disease. Cell Death Dis. 2021, 12, 651. [Google Scholar] [CrossRef]
- Schlaepfer, W.W.; Bunge, R.P. Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J. Cell Biol. 1973, 59, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Loreto, A.; Di Stefano, M.; Gering, M.; Conforti, L. Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca2+ Influx but Only Modestly Influenced by Mitochondria. Cell Rep. 2015, 13, 2539–2552. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Ferguson, T.A.; Schoch, K.M.; Li, J.; Qian, Y.; Shofer, F.S.; Saatman, K.E.; Neumar, R.W. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 2013, 56, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Volbracht, C.; Fava, E.; Nicotera, P. 1-Methyl-4-phenylpyridinium induces autocrine excitotoxicity, protease activation, and neuronal apoptosis. Mol. Pharmacol. 1998, 54, 789–801. [Google Scholar] [CrossRef]
- Volbracht, C.; Fava, E.; Leist, M.; Nicotera, P. Calpain inhibitors prevent nitric oxide-triggered excitotoxic apoptosis. Neuroreport 2001, 12, 3645–3648. [Google Scholar] [CrossRef]
- Baudry, M.; Luo, Y.L.; Bi, X. Calpain-2 Inhibitors as Therapy for Traumatic Brain Injury. Neurotherapeutics 2023, 20, 1592–1602. [Google Scholar] [CrossRef]
- Harbauer, A.B.; Hees, J.T.; Wanderoy, S.; Segura, I.; Gibbs, W.; Cheng, Y.; Ordonez, M.; Cai, Z.; Cartoni, R.; Ashrafi, G.; et al. Neuronal mitochondria transport Pink1 mRNA via synaptojanin 2 to support local mitophagy. Neuron 2022, 110, 1516–1531.e1519. [Google Scholar] [CrossRef]
- Hu, Q.; Wu, D.; Walker, M.; Wang, P.; Tian, R.; Wang, W. Genetically encoded biosensors for evaluating NAD+/NADH ratio in cytosolic and mitochondrial compartments. Cell Rep. Methods 2021, 1, 100116. [Google Scholar] [CrossRef]
- Imamura, H.; Nhat, K.P.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef]
- Trevisiol, A.; Saab, A.S.; Winkler, U.; Marx, G.; Imamura, H.; Mobius, W.; Kusch, K.; Nave, K.A.; Hirrlinger, J. Monitoring ATP dynamics in electrically active white matter tracts. Elife 2017, 6, e24241. [Google Scholar] [CrossRef]
- von Kugelgen, N.; Chekulaeva, M. Conservation of a core neurite transcriptome across neuronal types and species. Wiley Interdiscip. Rev. RNA 2020, 11, e1590. [Google Scholar] [CrossRef]
- Schierle, G.S.; Hansson, O.; Leist, M.; Nicotera, P.; Widner, H.; Brundin, P. Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat. Med. 1999, 5, 97–100. [Google Scholar] [CrossRef]
- Schierle, G.S.; Leist, M.; Martinou, J.C.; Widner, H.; Nicotera, P.; Brundin, P. Differential effects of Bcl-2 overexpression on fibre outgrowth and survival of embryonic dopaminergic neurons in intracerebral transplants. Eur. J. Neurosci. 1999, 11, 3073–3081. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Castilho, R.F.; Kaminski Schierle, G.S.; Karlsson, J.; Nicotera, P.; Leist, M.; Brundin, P. Additive effects of caspase inhibitor and lazaroid on the survival of transplanted rat and human embryonic dopamine neurons. Exp. Neurol. 2000, 164, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Kirkeby, A.; Nelander, J.; Hoban, D.B.; Rogelius, N.; Bjartmarz, H.; Novo Nordisk Cell Therapy R&D; Storm, P.; Fiorenzano, A.; Adler, A.F.; Vale, S.; et al. Preclinical quality, safety, and efficacy of a human embryonic stem cell-derived product for the treatment of Parkinson’s disease, STEM-PD. Cell Stem Cell 2023, 30, 1299–1314.e1299. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.Z.; Lehmann, M.; Gutbier, S.; Nembo, E.; Noel, S.; Smirnova, L.; Forsby, A.; Hescheler, J.; Avci, H.X.; Hartung, T.; et al. In vitro acute and developmental neurotoxicity screening: An overview of cellular platforms and high-throughput technical possibilities. Arch. Toxicol. 2017, 91, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.B.; Kim, H.; El Touny, L.; Simeonov, A.; Gerhold, D. LUHMES Dopaminergic Neurons Are Uniquely Susceptible to Ferroptosis. Neurotox. Res. 2022, 40, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.B.; Hogberg, H.; Kuo, D.; Sakamuru, S.; Xia, M.; Smirnova, L.; Hartung, T.; Gerhold, D. Characterization of three human cell line models for high-throughput neuronal cytotoxicity screening. J. Appl. Toxicol. 2017, 37, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Tuszynski, M.H. Regulation of axonal regeneration after mammalian spinal cord injury. Nat. Rev. Mol. Cell Biol. 2023, 24, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Dohm, C.P.; Kermer, P.; Bahr, M. Aggregopathy in neurodegenerative diseases: Mechanisms and therapeutic implication. Neurodegener. Dis. 2008, 5, 321–338. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brüll, M.; Geese, N.; Celardo, I.; Laumann, M.; Leist, M. Preparation of Viable Human Neurites for Neurobiological and Neurodegeneration Studies. Cells 2024, 13, 242. https://doi.org/10.3390/cells13030242
Brüll M, Geese N, Celardo I, Laumann M, Leist M. Preparation of Viable Human Neurites for Neurobiological and Neurodegeneration Studies. Cells. 2024; 13(3):242. https://doi.org/10.3390/cells13030242
Chicago/Turabian StyleBrüll, Markus, Nils Geese, Ivana Celardo, Michael Laumann, and Marcel Leist. 2024. "Preparation of Viable Human Neurites for Neurobiological and Neurodegeneration Studies" Cells 13, no. 3: 242. https://doi.org/10.3390/cells13030242
APA StyleBrüll, M., Geese, N., Celardo, I., Laumann, M., & Leist, M. (2024). Preparation of Viable Human Neurites for Neurobiological and Neurodegeneration Studies. Cells, 13(3), 242. https://doi.org/10.3390/cells13030242