



Aberrant MET Receptor Tyrosine Kinase Signaling in Glioblastoma: Targeted Therapy and Future Directions

Abstract
1. Introduction
2. Met Structure and Function
3. MET/HGF Dysregulation and Oncogenic Paradigms in GBM
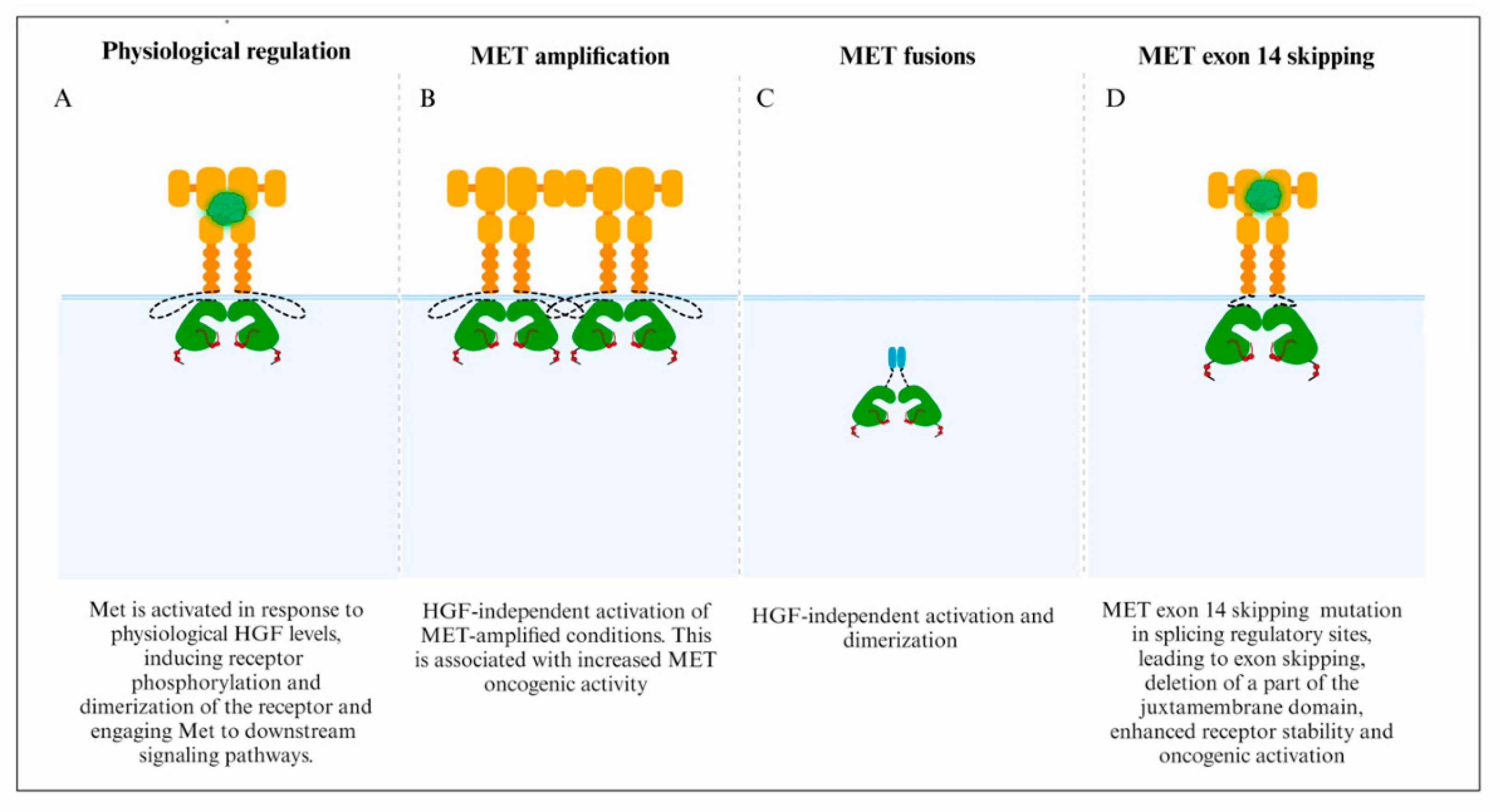
3.1. MET Focal Amplification
3.2. Fusion Genes
3.3. MET Exon 14 Skipping
4. MET-Targeted Therapies in GBMs
5. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Wu, F.; Chai, R.C.; Wang, Z.; Liu, Y.Q.; Zhao, Z.; Li, G.Z.; Jiang, H.Y. Molecular classification of IDH-mutant glioblastomas based on gene expression profiles. Carcinogenesis 2019, 40, 853–860. [Google Scholar] [CrossRef]
- Bausart, M.; Preat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef]
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular origin of glioblastoma and its implication in precision therapy. Cell. Mol. Immunol. 2018, 15, 737–739. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.; Odia, Y.; Khosla, A.A.; Ahluwalia, M.S. Key Clinical Principles in the Management of Glioblastoma. JCO Oncol. Pract. 2023, 19, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Skiriute, D.; Vaitkiene, P.; Saferis, V.; Asmoniene, V.; Skauminas, K.; Deltuva, V.P.; Tamasauskas, A. MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in glioblastoma. BMC Cancer 2012, 12, 218. [Google Scholar] [CrossRef]
- Li, E.; Hristova, K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry 2006, 45, 6241–6251. [Google Scholar] [CrossRef]
- Wintheiser, G.A.; Silberstein, P. Physiology, Tyrosine Kinase Receptors. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Stern, Y.E.; Al-Ghabkari, A.; Monast, A.; Fiset, B.; Aboualizadeh, F.; Yao, Z.; Stagljar, I.; Walsh, L.A.; Duhamel, S.; Park, M. Met-HER3 crosstalk supports proliferation via MPZL3 in MET-amplified cancer cells. Cell. Mol. Life Sci. 2022, 79, 178. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Eckerich, C.; Zapf, S.; Fillbrandt, R.; Loges, S.; Westphal, M.; Lamszus, K. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int. J. Cancer 2007, 121, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cazares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar] [CrossRef]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef]
- Park, M.; Dean, M.; Cooper, C.S.; Schmidt, M.; O’Brien, S.J.; Blair, D.G.; Vande Woude, G.F. Mechanism of met oncogene activation. Cell 1986, 45, 895–904. [Google Scholar] [CrossRef]
- Rodrigues, G.A.; Park, M. Dimerization mediated through a leucine zipper activates the oncogenic potential of the met receptor tyrosine kinase. Mol. Cell Biol. 1993, 13, 6711–6722. [Google Scholar] [CrossRef]
- Dean, M.; Park, M.; Vande Woude, G.F. Characterization of the rearranged tpr-met oncogene breakpoint. Mol. Cell Biol. 1987, 7, 921–924. [Google Scholar] [CrossRef]
- Cai, K.; Zhang, X.; Bai, X.C. Cryo-electron Microscopic Analysis of Single-Pass Transmembrane Receptors. Chem. Rev. 2022, 122, 13952–13988. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. The Hepatocyte Growth Factor Receptor: Structure, Function and Pharmacological Targeting in Cancer. Curr. Signal Transduct. Ther. 2011, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Maulik, G.; Shrikhande, A.; Kijima, T.; Ma, P.C.; Morrison, P.T.; Salgia, R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002, 13, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Ma, P.C.; Salgia, R. Therapeutic targeting of the receptor tyrosine kinase Met. Cancer Treat. Res. 2004, 119, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor: Molecular structure, roles in liver regeneration, and other biological functions. Crit. Rev. Oncog. 1992, 3, 27–54. [Google Scholar] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef]
- Toshikazu, N. Structure and function of hepatocyte growth factor. Prog. Growth Factor Res. 1991, 3, 67–85. [Google Scholar] [CrossRef]
- Uchikawa, E.; Chen, Z.; Xiao, G.-Y.; Zhang, X.; Bai, X.-C. Structural basis of the activation of c-MET receptor. Nat. Commun. 2021, 12, 4074. [Google Scholar] [CrossRef]
- Frigault, M.M.; Naujokas, M.A.; Park, M. Gab2 requires membrane targeting and the Met binding motif to promote lamellipodia, cell scatter, and epithelial morphogenesis downstream from the Met receptor. J. Cell Physiol. 2008, 214, 694–705. [Google Scholar] [CrossRef]
- Lock, L.S.; Maroun, C.R.; Naujokas, M.A.; Park, M. Distinct recruitment and function of Gab1 and Gab2 in Met receptor-mediated epithelial morphogenesis. Mol. Biol. Cell 2002, 13, 2132–2146. [Google Scholar] [CrossRef] [PubMed]
- Mood, K.; Saucier, C.; Bong, Y.S.; Lee, H.S.; Park, M.; Daar, I.O. Gab1 is required for cell cycle transition, cell proliferation, and transformation induced by an oncogenic met receptor. Mol. Biol. Cell 2006, 17, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Lock, L.S.; Frigault, M.M.; Saucier, C.; Park, M. Grb2-independent recruitment of Gab1 requires the C-terminal lobe and structural integrity of the Met receptor kinase domain. J. Biol. Chem. 2003, 278, 30083–30090. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Morrison, H.; Matzke, A.; Kastilan, T.; Pace, G.; Herrlich, P.; Ponta, H. Hepatocyte growth factor-induced Ras activation requires ERM proteins linked to both CD44v6 and F-actin. Mol. Biol. Cell 2007, 18, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Maroun, C.R.; Holgado-Madruga, M.; Royal, I.; Naujokas, M.A.; Fournier, T.M.; Wong, A.J.; Park, M. The Gab1 PH domain is required for localization of Gab1 at sites of cell-cell contact and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell Biol. 1999, 19, 1784–1799. [Google Scholar] [CrossRef]
- Rajadurai, C.V.; Havrylov, S.; Zaoui, K.; Vaillancourt, R.; Stuible, M.; Naujokas, M.; Zuo, D.; Tremblay, M.L.; Park, M. Met receptor tyrosine kinase signals through a cortactin-Gab1 scaffold complex, to mediate invadopodia. J. Cell Sci. 2012, 125, 2940–2953. [Google Scholar] [CrossRef]
- Lai, A.Z.; Abella, J.V.; Park, M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol. 2009, 19, 542–551. [Google Scholar] [CrossRef]
- Chen, S.Y.; Chen, H.C. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell Biol. 2006, 26, 5155–5167. [Google Scholar] [CrossRef]
- McCall-Culbreath, K.D.; Li, Z.; Zutter, M.M. Crosstalk between the alpha2beta1 integrin and c-met/HGF-R regulates innate immunity. Blood 2008, 111, 3562–3570. [Google Scholar] [CrossRef][Green Version]
- Yamamoto, N.; Mammadova, G.; Song, R.X.; Fukami, Y.; Sato, K. Tyrosine phosphorylation of p145met mediated by EGFR and Src is required for serum-independent survival of human bladder carcinoma cells. J. Cell Sci. 2006, 119, 4623–4633. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.; Naujokas, M.A.; Zuo, D.; Sangwan, V.; Frigault, M.M.; Petkiewicz, S.; Dankort, D.L.; Muller, W.J.; Park, M. HGF converts ErbB2/Neu epithelial morphogenesis to cell invasion. Mol. Biol. Cell 2005, 16, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.W.; Somcio, R.J.; Fan, F.; Liu, W.; Johnson, M.; Lesslie, D.P.; Evans, D.B.; Gallick, G.E.; Ellis, L.M. Regulatory role of c-Met in insulin-like growth factor-I receptor-mediated migration and invasion of human pancreatic carcinoma cells. Mol. Cancer Ther. 2006, 5, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Salokas, K.; Liu, X.; Ohman, T.; Chowdhury, I.; Gawriyski, L.; Keskitalo, S.; Varjosalo, M. Physical and functional interactome atlas of human receptor tyrosine kinases. EMBO Rep. 2022, 23, e54041. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, M.; Abounader, R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9, 87. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e520. [Google Scholar] [CrossRef]
- Vaubel, R.A.; Tian, S.; Remonde, D.; Schroeder, M.A.; Mladek, A.C.; Kitange, G.J.; Caron, A.; Kollmeyer, T.M.; Grove, R.; Peng, S.; et al. Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clin. Cancer Res. 2020, 26, 1094–1104. [Google Scholar] [CrossRef]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Jonsson, P.; Lin, A.L.; Young, R.J.; DiStefano, N.M.; Hyman, D.M.; Li, B.T.; Berger, M.F.; Zehir, A.; Ladanyi, M.; Solit, D.B.; et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin. Cancer Res. 2019, 25, 5537–5547. [Google Scholar] [CrossRef] [PubMed]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, V.; Luebeck, J.; Nguyen, N.D.; Bakhtiari, M.; Turner, K.M.; Schwab, R.; Carter, H.; Mischel, P.S.; Bafna, V. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat. Commun. 2019, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Joo, K.M.; Yoo, J.S.; Koh, J.S.; Dong, S.M.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Petterson, S.A.; Dahlrot, R.H.; Hermansen, S.K.; KA Munthe, S.; Gundesen, M.T.; Wohlleben, H.; Rasmussen, T.; Beier, C.P.; Hansen, S.; Kristensen, B.W. High levels of c-Met is associated with poor prognosis in glioblastoma. J. Neuro-Oncol. 2015, 122, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Lal, B.; Xia, S.; Abounader, R.; Laterra, J. Targeting the c-Met pathway potentiates glioblastoma responses to gamma-radiation. Clin. Cancer Res. 2005, 11, 4479–4486. [Google Scholar] [CrossRef]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 2012, 30, e30–e33. [Google Scholar] [CrossRef]
- Joo, K.M.; Jin, J.; Kim, E.; Ho Kim, K.; Kim, Y.; Gu Kang, B.; Kang, Y.J.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838. [Google Scholar] [CrossRef]
- De Bacco, F.; D’Ambrosio, A.; Casanova, E.; Orzan, F.; Neggia, R.; Albano, R.; Verginelli, F.; Cominelli, M.; Poliani, P.L.; Luraghi, P.; et al. MET inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol. Med. 2016, 8, 550–568. [Google Scholar] [CrossRef]
- Lolkema, M.P.; Bohets, H.H.; Arkenau, H.T.; Lampo, A.; Barale, E.; de Jonge, M.J.A.; van Doorn, L.; Hellemans, P.; de Bono, J.S.; Eskens, F. The c-Met Tyrosine Kinase Inhibitor JNJ-38877605 Causes Renal Toxicity through Species-Specific Insoluble Metabolite Formation. Clin. Cancer Res. 2015, 21, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.Y.C.; Knight, J.F.; Johnson, R.M.; Stern, Y.E.; Saleh, S.M.; Savage, P.; Monast, A.; Zuo, D.; Duhamel, S.; Park, M. Co-dependency for MET and FGFR1 in basal triple-negative breast cancers. NPJ Breast Cancer 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.L. Targeting gene fusions in glioma. Curr. Opin. Neurol. 2021, 34, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Charest, A.; Lane, K.; McMahon, K.; Park, J.; Preisinger, E.; Conroy, H.; Housman, D. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer 2003, 37, 58–71. [Google Scholar] [CrossRef] [PubMed]
- You, G.; Fan, X.; Hu, H.; Jiang, T.; Chen, C.C. Fusion Genes Altered in Adult Malignant Gliomas. Front. Neurol. 2021, 12, 715206. [Google Scholar] [CrossRef] [PubMed]
- Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [CrossRef]
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014, 24, 1765–1773. [Google Scholar] [CrossRef]
- Huang, R.; Liu, Y.; Wang, K.; Wang, Z.; Zhang, C.; Zhang, W.; Zhao, Z.; Li, G.; Huang, L.; Chang, Y.; et al. High-sensitive clinical diagnostic method for PTPRZ1-MET and the characteristic protein structure contributing to ligand-independent MET activation. CNS Neurosci. Ther. 2021, 27, 617–628. [Google Scholar] [CrossRef]
- Lai, A.Z.; Cory, S.; Zhao, H.; Gigoux, M.; Monast, A.; Guiot, M.C.; Huang, S.; Tofigh, A.; Thompson, C.; Naujokas, M.; et al. Dynamic reprogramming of signaling upon met inhibition reveals a mechanism of drug resistance in gastric cancer. Sci. Signal 2014, 7, ra38. [Google Scholar] [CrossRef]
- Zeng, A.L.; Yan, W.; Liu, Y.W.; Wang, Z.; Hu, Q.; Nie, E.; Zhou, X.; Li, R.; Wang, X.F.; Jiang, T.; et al. Tumour exosomes from cells harbouring PTPRZ1-MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 2017, 36, 5369–5381. [Google Scholar] [CrossRef]
- Hu, H.; Mu, Q.; Bao, Z.; Chen, Y.; Liu, Y.; Chen, J.; Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678.e1618. [Google Scholar] [CrossRef]
- Lamorte, L.; Park, M. The receptor tyrosine kinases: Role in cancer progression. Surg. Oncol. Clin. N. Am. 2001, 10, 271–288. [Google Scholar] [CrossRef]
- Peschard, P.; Park, M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene 2007, 26, 1276–1285. [Google Scholar] [CrossRef]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.-L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12, R6. [Google Scholar] [CrossRef]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Peschard, P.; Park, M. Escape from Cbl-mediated downregulation: A recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 2003, 3, 519–523. [Google Scholar] [CrossRef]
- Hammond, D.E.; Urbé, S.; Vande Woude, G.F.; Clague, M.J. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770. [Google Scholar] [CrossRef]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef]
- Fujino, T.; Suda, K.; Mitsudomi, T. Lung Cancer with MET exon 14 Skipping Mutation: Genetic Feature, Current Treatments, and Future Challenges. Lung Cancer 2021, 12, 35–50. [Google Scholar] [CrossRef]
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non-Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5, PO.20.00516. [Google Scholar] [CrossRef]
- Recondo, G.; Che, J.; Jänne, P.A.; Awad, M.M. Targeting MET Dysregulation in Cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Yamada, K.M. Identification of a novel type of alternative splicing of a tyrosine kinase receptor. Juxtamembrane deletion of the c-met protein kinase C serine phosphorylation regulatory site. J. Biol. Chem. 1994, 269, 19457–19461. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ou, S.H.I.; Lee, J.M.; Kim, H.C.; Hong, M.; Kim, S.Y.; Jang, J.; Ahn, S.; Kang, S.Y.; Lee, S.; et al. Gastrointestinal malignancies harbor actionable MET exon 14 deletions. Oncotarget 2015, 6, 28211–28222. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Sattler, M.; Scheele, J.; Stroh, C.; Felip, E. The promise of selective MET inhibitors in non-small cell lung cancer with MET exon 14 skipping. Cancer Treat. Rev. 2020, 87, 102022. [Google Scholar] [CrossRef]
- Sohn, S.-H.; Sul, H.J.; Kim, B.J.; Zang, D.Y. Responses to the Tepotinib in Gastric Cancers with MET Amplification or MET Exon 14 Skipping Mutations and High Expression of Both PD-L1 and CD44. Cancers 2022, 14, 3444. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Si-Yang, L.; Lan-Ying, G.; An-Na, L.; Na-Na, L.; Hong-Fei, G.; Jian, S.; Jin-Ji, Y.; Xu-Chao, Z.; Yang, S.; Zhong-Yi, D.; et al. The Unique Characteristics of MET Exon 14 Mutation in Chinese Patients with NSCLC. J. Thorac. Oncol. 2016, 11, 1503–1510. [Google Scholar] [CrossRef]
- Clotilde, D.; Frédéric, L.; Fabienne, E.; Zoulika, K.; Martin, F.; Shéhérazade, S.; Simon, B.; Valérie, G.; Philippe, J.; Marie-Christine, C.; et al. Optimization of Routine Testing for MET Exon 14 Splice Site Mutations in NSCLC Patients. J. Thorac. Oncol. 2018, 13, 1873–1883. [Google Scholar] [CrossRef]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping with the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.-W.; Hida, T.; De Jonge, M.J.; Orlov, S.V.; et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J. Clin. Oncol. 2019, 37, 9004. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Borghei, A.; Hakimian, B.; Ali, S.M.; Ou, S.I. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. J. Thorac. Oncol. 2017, 12, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Cierra, H.; Kristen, A.B.; Garland, A.; Luis, V.; Daniel, J.G.; Tian, Z. Control of renal cell carcinoma brain metastases with cabozantinib following progression on immune checkpoint inhibitor therapy. Curr. Probl. Cancer Case Rep. 2021, 3, 100060. [Google Scholar] [CrossRef]
- Zhang, Y.; Guessous, F.; Kofman, A.; Schiff, D.; Abounader, R. XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs 2010, 13, 112–121. [Google Scholar] [PubMed]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol. 2005, 7, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Laterra, J.; Rosen, E.; Nam, M.; Ranganathan, S.; Fielding, K.; Johnston, P. Scatter factor/hepatocyte growth factor expression enhances human glioblastoma tumorigenicity and growth. Biochem. Biophys. Res. Commun. 1997, 235, 743–747. [Google Scholar] [CrossRef]
- Lamszus, K.; Schmidt, N.O.; Jin, L.; Laterra, J.; Zagzag, D.; Way, D.; Witte, M.; Weinand, M.; Goldberg, I.D.; Westphal, M.; et al. Scatter factor promotes motility of human glioma and neuromicrovascular endothelial cells. Int. J. Cancer 1998, 75, 19–28. [Google Scholar] [CrossRef]
- Abounader, R.; Ranganathan, S.; Lal, B.; Fielding, K.; Book, A.; Dietz, H.; Burger, P.; Laterra, J. Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J. Natl. Cancer Inst. 1999, 91, 1548–1556. [Google Scholar] [CrossRef]
- Wen, P.Y.; Drappatz, J.; de Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Holland, J.; et al. Phase II study of cabozantinib in patients with progressive glioblastoma: Subset analysis of patients naive to antiangiogenic therapy. Neuro Oncol. 2018, 20, 249–258. [Google Scholar] [CrossRef]
- Baltschukat, S.; Engstler, B.S.; Huang, A.; Hao, H.X.; Tam, A.; Wang, H.Q.; Liang, J.; DiMare, M.T.; Bhang, H.C.; Wang, Y.; et al. Capmatinib (INC280) Is Active Against Models of Non-Small Cell Lung Cancer and Other Cancer Types with Defined Mechanisms of MET Activation. Clin. Cancer Res. 2019, 25, 3164–3175. [Google Scholar] [CrossRef]
- Bang, Y.J.; Su, W.C.; Schuler, M.; Nam, D.H.; Lim, W.T.; Bauer, T.M.; Azaro, A.; Poon, R.T.P.; Hong, D.; Lin, C.C.; et al. Phase 1 study of capmatinib in MET-positive solid tumor patients: Dose escalation and expansion of selected cohorts. Cancer Sci. 2020, 111, 536–547. [Google Scholar] [CrossRef]
- Esaki, T.; Hirai, F.; Makiyama, A.; Seto, T.; Bando, H.; Naito, Y.; Yoh, K.; Ishihara, K.; Kakizume, T.; Natsume, K.; et al. Phase I dose-escalation study of capmatinib (INC280) in Japanese patients with advanced solid tumors. Cancer Sci. 2019, 110, 1340–1351. [Google Scholar] [CrossRef]
- van den Bent, M.; Azaro, A.; De Vos, F.; Sepulveda, J.; Yung, W.K.A.; Wen, P.Y.; Lassman, A.B.; Joerger, M.; Tabatabai, G.; Rodon, J.; et al. A Phase Ib/II, open-label, multicenter study of INC280 (capmatinib) alone and in combination with buparlisib (BKM120) in adult patients with recurrent glioblastoma. J. Neuro-Oncol. 2020, 146, 79–89. [Google Scholar] [CrossRef]
- Dong, X.; Li, X.; Chen, J.; Ma, S.; Mu, D.; Hu, J.; Lu, S. Phase 1 Study of the Selective c-MET Inhibitor, HS-10241, in Patients With Advanced Solid Tumors. JTO Clin. Res. Rep. 2023, 4, 100449. [Google Scholar] [CrossRef]
- Gavine, P.R.; Ren, Y.; Han, L.; Lv, J.; Fan, S.; Zhang, W.; Xu, W.; Liu, Y.J.; Zhang, T.; Fu, H.; et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol. Oncol. 2015, 9, 323–333. [Google Scholar] [CrossRef]
- Lorente, M.; Torres, S.; Salazar, M.; Carracedo, A.; Hernández-Tiedra, S.; Rodríguez-Fornés, F.; García-Taboada, E.; Meléndez, B.; Mollejo, M.; Campos-Martín, Y.; et al. Stimulation of ALK by the growth factor midkine renders glioma cells resistant to autophagy-mediated cell death. Autophagy 2011, 7, 1071–1073. [Google Scholar] [CrossRef]
- Ma, J.; Lang, B.; Wang, X.; Wang, L.; Dong, Y.; Hu, H. Co-expression of midkine and pleiotrophin predicts poor survival in human glioma. J. Clin. Neurosci. 2014, 21, 1885–1890. [Google Scholar] [CrossRef]
- López-Valero, I.; Dávila, D.; González-Martínez, J.; Salvador-Tormo, N.; Lorente, M.; Saiz-Ladera, C.; Torres, S.; Gabicagogeascoa, E.; Hernández-Tiedra, S.; García-Taboada, E.; et al. Midkine signaling maintains the self-renewal and tumorigenic capacity of glioma initiating cells. Theranostics 2020, 10, 5120–5136. [Google Scholar] [CrossRef]
- Martínez-García, M.; Velasco, G.; Pineda, E.; Gil-Gil, M.; Alameda, F.; Capellades, J.; Martín-Soberón, M.C.; López-Valero, I.; Tovar Ambel, E.; Foro, P.; et al. Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial. Cancers 2022, 14, 2393. [Google Scholar] [CrossRef]
- Dymova, M.A.; Kuligina, E.V.; Richter, V.A. Molecular Mechanisms of Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6385. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, L.; Ye, M.; Kang, C.; You, H. Treatment Progress of Immune Checkpoint Blockade Therapy for Glioblastoma. Front. Immunol. 2020, 11, 592612. [Google Scholar] [CrossRef]
- Merchant, M.; Ma, X.; Maun, H.R.; Zheng, Z.; Peng, J.; Romero, M.; Huang, A.; Yang, N.Y.; Nishimura, M.; Greve, J.; et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl. Acad. Sci. USA 2013, 110, E2987–E2996. [Google Scholar] [CrossRef]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O(6)-Methylguanine-DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results from the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non-Small-Cell Lung Cancer: METLung. J. Clin. Oncol. 2017, 35, 412–420. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, J.Y.; Lim, H.; Lee, S.H.; Moon, Y.J.; Pyo, H.J.; Ryu, S.E.; Shin, W.; Heo, Y.-S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. [Google Scholar] [CrossRef]
- Nero, T.L.; Parker, M.W.; Morton, C.J. Protein structure and computational drug discovery. Biochem. Soc. Trans. 2018, 46, 1367–1379. [Google Scholar] [CrossRef]
- Casteran, N.; De Sepulveda, P.; Beslu, N.; Aoubala, M.; Letard, S.; Lecocq, E.; Rottapel, R.; Dubreuil, P. Signal transduction by several KIT juxtamembrane domain mutations. Oncogene 2003, 22, 4710–4722. [Google Scholar] [CrossRef]
- Wang, X.; Yennawar, N.; Hankey, P.A. Autoinhibition of the Ron receptor tyrosine kinase by the juxtamembrane domain. Cell Commun. Signal. 2014, 12, 28. [Google Scholar] [CrossRef]
- Estevam, G.O.; Linossi, E.M.; Macdonald, C.B.; Espinoza, C.A.; Michaud, J.M.; Coyote-Maestas, W.; Collisson, E.A.; Jura, N.; Fraser, J.S. Conserved regulatory motifs in the juxtamembrane domain and kinase N-lobe revealed through deep mutational scanning of the MET receptor tyrosine kinase domain. bioRxiv 2023. [Google Scholar] [CrossRef]
- Arnold, C. AlphaFold touted as next big thing for drug discovery—But is it? Nature 2023, 622, 15–17. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Surka, C.; Jin, L.; Mbong, N.; Lu, C.C.; Jang, I.S.; Rychak, E.; Mendy, D.; Clayton, T.; Tindall, E.; Hsu, C.; et al. CC-90009, a novel cereblon E3 ligase modulator, targets acute myeloid leukemia blasts and leukemia stem cells. Blood 2021, 137, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Mullard, A. First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. 2019, 18, 237–239. [Google Scholar] [CrossRef]
- Grinshpun, A. Clinician’s guide to targeted estrogen receptor degradation using PROTAC in patients with estrogen receptor-positive metastatic breast cancer. Curr. Opin. Oncol. 2023, 35, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-Y.; Wang, H.-P.; Mao, Y.-Z.; Zhang, H.; Xin, M.; Xi, X.-X.; Lei, H.; Mao, S.; Li, D.-H.; Zhang, S.-Q. Discovery of Potent PROTACs Targeting EGFR Mutants through the Optimization of Covalent EGFR Ligands. J. Med. Chem. 2022, 65, 4709–4726. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Kim, T.M.; Lee, Y.; Kim, S.; Park, S.; Ju, Y.S.; Kim, M.; Keam, B.; Jeon, Y.K.; Kim, D.W.; et al. Acquired resistance to third-generation EGFR tyrosine kinase inhibitors in patients with de novo EGFRT790M-mutant NSCLC. J. Thorac. Oncol. 2021, 16, 1859–1871. [Google Scholar] [CrossRef]
- Jang, J.; To, C.; De Clercq, D.J.H.; Park, E.; Ponthier, C.M.; Shin, B.H.; Mushajiang, M.; Nowak, R.P.; Fischer, E.S.; Eck, M.J.; et al. Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations. Angew. Chem. Int. Ed. 2020, 59, 14481–14489. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 2018, 25, 67–77.e3. [Google Scholar] [CrossRef]
- Wang, Z.; Tan, M.; Su, W.; Huang, W.; Zhang, J.; Jia, F.; Cao, G.; Liu, X.; Song, H.; Ran, H.; et al. Persistent Degradation of HER2 Protein by Hybrid nanoPROTAC for Programmed Cell Death. J. Med. Chem. 2023, 66, 6263–6273. [Google Scholar] [CrossRef]
- Maneiro, M.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody–PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2022, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Jia, C.; Fan, Z.; Hu, X.; Zhang, W.; Liu, K.; Sun, S.; Guo, H.; Yang, N.; Zhu, M.; et al. Discovery of novel exceptionally potent and orally active c-MET PROTACs for the treatment of tumors with MET alterations. Acta Pharm. Sin. B 2023, 13, 2715–2735. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jin, J.; Gu, W.; Zhao, Z.; Yuan, H.; Zhou, Y.; Nagle, D.G.; Xi, Q.; Zhang, X.; Sun, Q.; et al. Crizotinib-based proteolysis targeting chimera suppresses gastric cancer by promoting MET degradation. Cancer Sci. 2023, 114, 1958–1971. [Google Scholar] [CrossRef]
- Bollu, L.R.; Bommi, P.V.; Monsen, P.J.; Zhai, L.; Lauing, K.L.; Bell, A.; Kim, M.; Ladomersky, E.; Yang, X.; Platanias, L.C.; et al. Identification and Characterization of a Novel Indoleamine 2,3-Dioxygenase 1 Protein Degrader for Glioblastoma. J. Med. Chem. 2022, 65, 15642–15662. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, W.; Wang, Y.; Yao, D.; Ye, T.; Yao, Y.; Chen, B.; Liu, G.; Yang, X.; Wang, W.; et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm. Sin. B 2020, 10, 1943–1953. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study NCT Number | Summary | Conditions | Drug(s) | Mechanisms of Action | Phase |
|---|---|---|---|---|---|
| 00704288 | Evaluate XL184 (cabozantinib) for GBM | Glioblastoma multiforme | Drug: XL184 | VEGFR2, MET, and RET inhibitor | II [104] completed |
| 00960492 | Determine safe dose of XL184 (cabozantinib) | Glioblastoma | Drug: XL184, temozolomide, radiation therapy | VEGFR2, MET, and RET kinase inhibitor | I [111] completed |
| 01068782 | Evaluate XL184 (cabozantinib) in astrocytic tumors | Grrade IV astrocytic tumors | Drug: XL184 | VEGFR2, MET, and RET kinase inhibitor | II completed |
| 01324479 | Evaluate INC280 (capmatinib) for solid tumors | Solid tumors | Drug: INC280 | MET inhibitor | I [114] completed |
| 01870726 | Evaluate INC280 (capmatinib) and buparlisib for recurrent glioblastoma | c-MET inhibitor, PI3K inhibitor, PTEN mutations, homozygous del. of PTEN or PTEN neg. by IHC, c-Met amplification by FISH, INC280, BKM120, buparlisib, recurrent GBM | DrugRUG: INC280, buparlisib | MET and PI3K inhibitor | I/II [115] active |
| 02386826 | Evaluate INC280 (capmatinib) and bevacizumab for recurrent glioblastoma | Glioblastoma gliosarcoma | Drug: INC280, bevacizumab | MET and VEGFR inhibitor | I completed |
| 01441388 | A study of crizotinib plus VEGF inhibitor combinations | Advanced solid tumors | Drug: crizotinib plus VEGF inhibitor combinations | MET and VEGFR inhibitor | I completed |
| 02270034 | Assess crizotinib for newly diagnosed glioblastoma | Glioblastoma multiforme (grade IV) of cerebellum | Drug: crizotinib | MET inhibitor | I completed |
| 01632228 | Assess onartuzumab and bevacizumab for recurrent glioblastoma | Glioblastoma | Drug: bevacizumab, onartuzumab, placebo | MET and VEGFR inhibitor | I [127] completed |
| 02885324 | Study cabozantinib for CNS tumors in children | Glioblastoma multiforme, anaplastic astrocytoma, malignant brain tumor, high-grade glioma | Drug: cabozantinib | VEGFR2, MET, and RET inhibitor | II completed |
| 03175224 | Study APL-101 for advanced solid tumors | Solid tumor, advanced cancer, renal cancer, gastric cancer, gastroesophageal junction adenocarcinoma, NSCLC, lung cancer, brain tumor, glioblastoma multiforme | D: APL-101 oral capsules | MET inhibitor | I/II active |
| 03598244 | Assess volitinib (savolitinib) for recurrent primary CNS tumors | Central nervous system (CNS) tumors | Drug: volitinib (savolitinib) | MET inhibitor | I active |
| 05039281 | Study atezolizumab and cabozantinib for recurrent glioblastoma | Recurrent glioblastoma, recurrent gliosarcoma | Biological: atezolizumab, Drug: cabozantinib | Atezolizumab: targets PD-L1, cabozantinib: VEGFR2, MET, and RET kinase inhibitor | I/II active |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Ghabkari, A.; Huang, B.; Park, M. Aberrant MET Receptor Tyrosine Kinase Signaling in Glioblastoma: Targeted Therapy and Future Directions. Cells 2024, 13, 218. https://doi.org/10.3390/cells13030218
Al-Ghabkari A, Huang B, Park M. Aberrant MET Receptor Tyrosine Kinase Signaling in Glioblastoma: Targeted Therapy and Future Directions. Cells. 2024; 13(3):218. https://doi.org/10.3390/cells13030218
Chicago/Turabian StyleAl-Ghabkari, Abdulhameed, Bruce Huang, and Morag Park. 2024. "Aberrant MET Receptor Tyrosine Kinase Signaling in Glioblastoma: Targeted Therapy and Future Directions" Cells 13, no. 3: 218. https://doi.org/10.3390/cells13030218
APA StyleAl-Ghabkari, A., Huang, B., & Park, M. (2024). Aberrant MET Receptor Tyrosine Kinase Signaling in Glioblastoma: Targeted Therapy and Future Directions. Cells, 13(3), 218. https://doi.org/10.3390/cells13030218