

Synergistic Protection of Retinal Ganglion Cells (RGCs) by SARM1 Inactivation with CNTF in a Rodent Model of Nonarteritic Anterior Ischemic Optic Neuropathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Loss of SARM1 Activity Inhibits Early RGC Axonal Degeneration
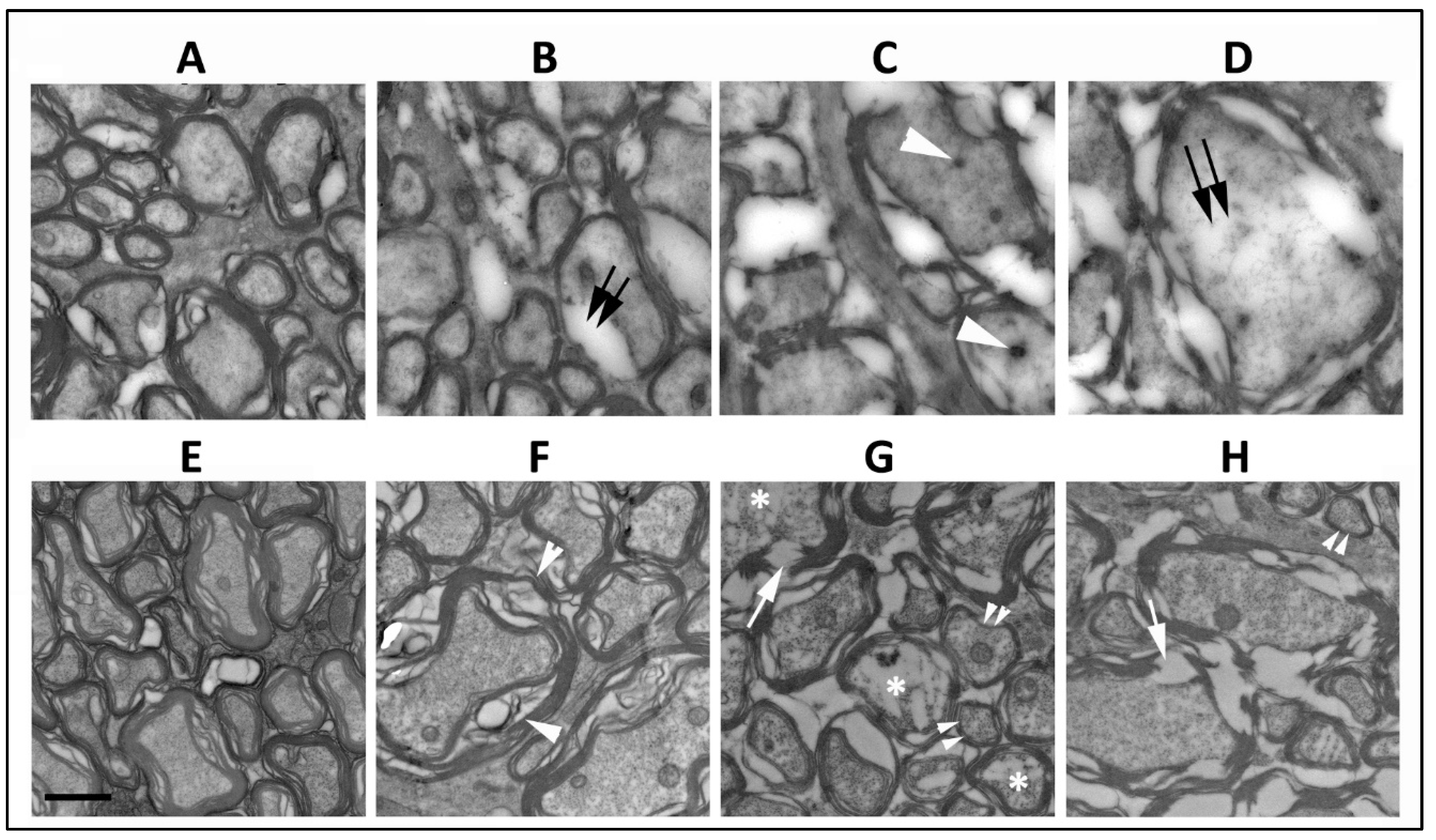
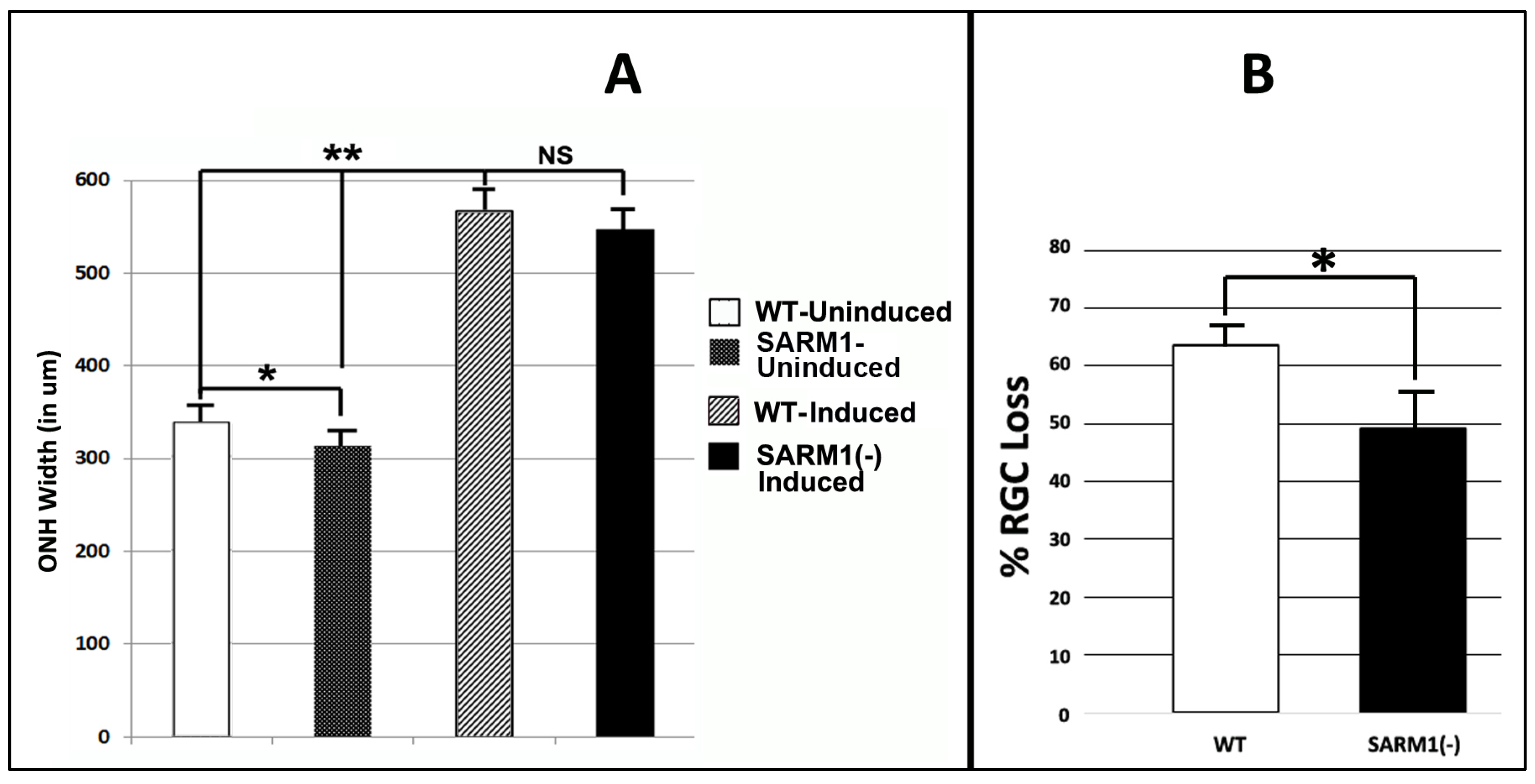
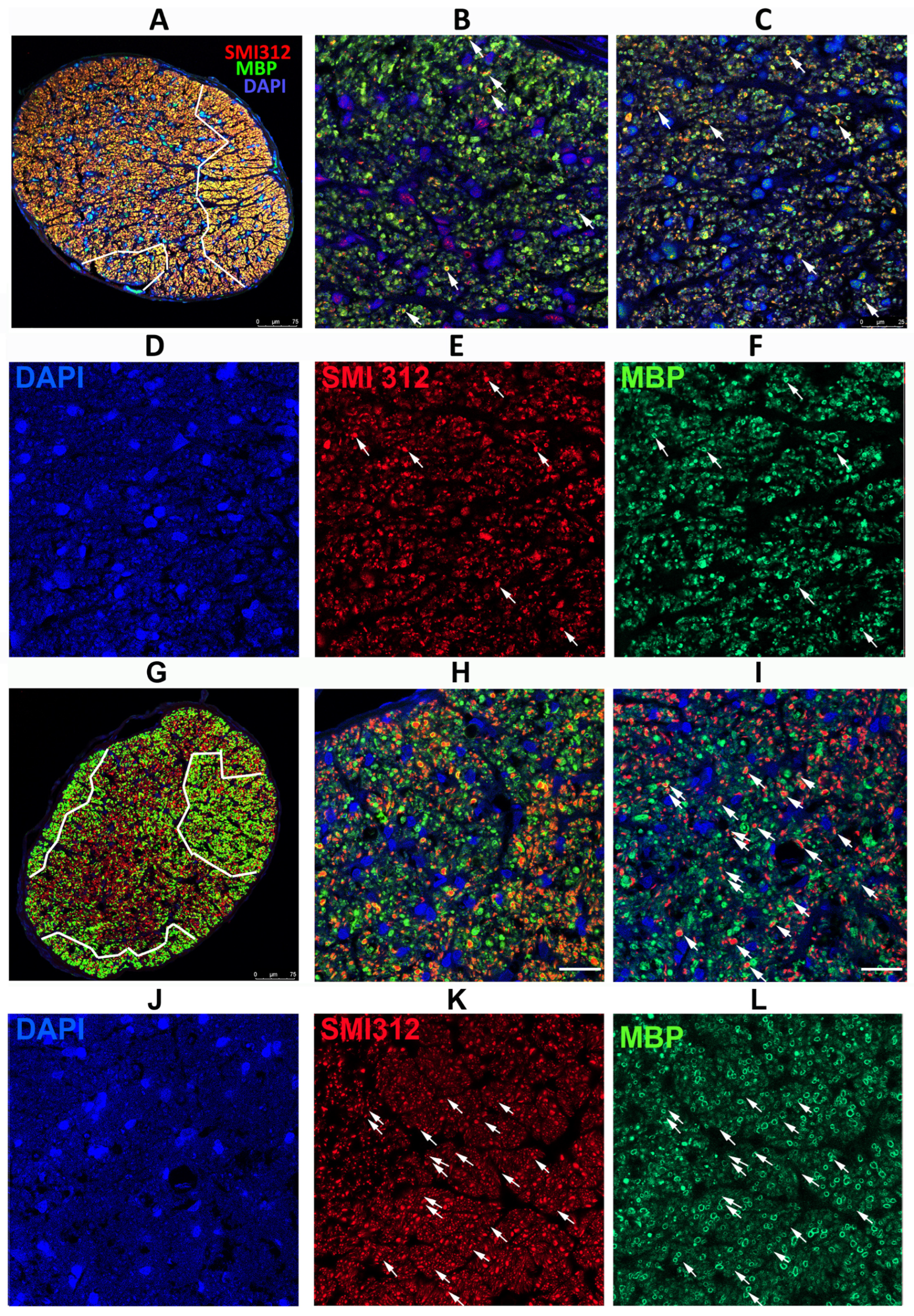
3.2. SARM1 Loss Inhibits Early ON Axonal and Myelin Degeneration
3.3. Eliminating SARM1 Activity Enhances RGC Somatic Survival after rNAION
3.4. Eliminating SARM1 Activity Enhances Long-Term RGC Axonal Survival after rNAION
3.5. Eliminating SARM1 Activity Synergizes with CNTF-Growth Factor-Mediated, Long-Term Neuroprotection
4. Discussion
4.1. Eliminating SARM1 Activity Results in Profound Protection of ON Axons from Ischemia
4.2. Combining SARM1 Suppression and CNTF Generates Synergistic Neuroprotection
4.3. Mechanisms Responsible for Synergistic Neuroprotection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, N.M.; Chew, S.S.; Danesh-Meyer, H.V. Non-arteritic anterior ischaemic optic neuropathy: A review and update. J. Clin. Neurosci. 2009, 16, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.R.; Arnold, A.C. Current concepts in the diagnosis, pathogenesis and management of nonarteritic anterior ischaemic optic neuropathy. Eye 2014, 29, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.; Cheng, A.C.; Leung, C.K.; Cheung, C.Y.; Yung, A.Y.; Gong, B.; Lam, D.S. Quantitative assessment of optic nerve head morphology and retinal nerve fibre layer in non-arteritic anterior ischaemic optic neuropathy with optical coherence tomography and confocal scanning laser ophthalmoloscopy. Br. J. Ophthalmol. 2009, 93, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.C. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J. Neuroophthalmol. 2003, 23, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Behbehani, R.; Mathews, M.K.; Sergott, R.C.; Savino, P.J. Nonarteritic anterior ischemic optic neuropathy in patients with sleep apnea while being treated with continuous positive airway pressure. Am. J. Ophthalmol. 2005, 139, 518–521. [Google Scholar] [CrossRef] [PubMed]
- McCulley, T.J.; Lam, B.L.; Feuer, W.J. A comparison of risk factors for postoperative and spontaneous nonarteritic anterior ischemic optic neuropathy. J. Neuroophthalmol. 2005, 25, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Tesser, R.A.; Niendorf, E.R.; Levin, L.A. The morphology of an infarct in nonarteritic anterior ischemic optic neuropathy. Ophthalmology 2003, 110, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.H.; Liao, Y.J. Structure-Function Analysis of Nonarteritic Anterior Ischemic Optic Neuropathy and Age-Related Differences in Outcome. J. Neuroophthalmol. 2017, 37, 258–264. [Google Scholar] [CrossRef]
- Mercado, J.L.; Purvin, V.A.; Kawasaki, A.; Wu, D.D. Bilateral sequential nonarteritic anterior ischemic optic neuropathy: A comparison of visual outcomes in fellow eyes using quantitative analysis of goldmann visual fields. Arch. Ophthalmol. 2012, 130, 863–867. [Google Scholar] [CrossRef]
- Salvetat, M.L.; Pellegrini, F.; Spadea, L.; Salati, C.; Zeppieri, M. Non-Arteritic Anterior Ischemic Optic Neuropathy (NA-AION): A Comprehensive Overview. Vision 2023, 7, 72. [Google Scholar] [CrossRef]
- Katz, D.M.; Trobe, J.D. Is there treatment for nonarteritic anterior ischemic optic neuropathy. Curr. Opin. Ophthalmol. 2015, 26, 458–463. [Google Scholar] [CrossRef]
- Ischemic Optic Neuropathy Decompression Trial Study Group. Characteristics of patients with nonarteritic anterior ischemic optic neuropathy eligible for the Ischemic Optic Neuropathy Decompression Trial. Arch. Ophthalmol. 1996, 114, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Lantos, K.; Dömötör, Z.R.; Farkas, N.; Kiss, S.; Szakács, Z.; Garami, A.; Varga, G.; Lujber, L.; Kanaan, R.; Hegyi, P.; et al. Efficacy of Treatments in Nonarteritic Ischemic Optic Neuropathy: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 2718. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.L.; Guo, Y.; Kelman, S.E.; Flower, R.W.; Johnson, M.A. Functional and cellular responses in a novel rodent model of anterior ischemic optic neuropathy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4153–4162. [Google Scholar] [CrossRef] [PubMed]
- Peeler, C.; Cestari, D.M. Non-Arteritic Anterior Ischemic Optic Neuropathy (NAION): A Review and Update on Animal Models. Semin. Ophthalmol. 2016, 31, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Huang, S.P.; Chang, C.H.; Lin, K.H.; Chang, S.W.; Tsai, R.K. Protective effects of systemic treatment with methylprednisolone in a rodent model of non-arteritic anterior ischemic optic neuropathy (rAION). Exp. Eye Res. 2015, 131, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Wen, Y.T.; Chang, C.H.; Chang, S.W.; Lin, K.H.; Tsai, R.K. Early Methylprednisolone Treatment Can Stabilize the Blood-Optic Nerve Barrier in a Rat Model of Anterior Ischemic Optic Neuropathy (rAION). Investig. Ophthalmol. Vis. Sci. 2017, 58, 1628–1636. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.T.; Huang, T.L.; Huang, S.P.; Chang, C.H.; Tsai, R.K. Early applications of granulocyte colony-stimulating factor (G-CSF) can stabilize the blood-optic-nerve barrier and ameliorate inflammation in a rat model of anterior ischemic optic neuropathy (rAION). Dis. Model Mech. 2016, 9, 1193–1202. [Google Scholar] [CrossRef]
- Nicholson, J.D.; Puche, A.C.; Guo, Y.; Weinreich, D.; Slater, B.J.; Bernstein, S.L. PGJ2 Provides Prolonged CNS Stroke Protection by Reducing White Matter Edema. PLoS ONE 2012, 7, e50021. [Google Scholar] [CrossRef]
- Yi, Z.; Chen, L.; Wang, X.; Chen, C.; Xing, Y. Protective Effects of Intravitreal Injection of the Rho-Kinase Inhibitor Y-27632 in a Rodent Model of Nonarteritic Anterior Ischemic Optic Neuropathy (rAION). J. Ophthalmol. 2020, 2020, 1485425. [Google Scholar] [CrossRef]
- Chang, C.-H.; Huang, T.-L.; Huang, S.-P.; Tsai, R.-K. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in a rat model of anterior ischemic optic neuropathy (rAION). Exp. Eye Res. 2014, 118, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, T.; Wen, Y.-T.; Chang, C.-H.; Kolovos, P.; Kalogerou, M.; Prokopiou, E.; Neokleous, A.; Huang, C.-T.; Tsai, R.-K. Neuroprotective Effects of Omega-3 Polyunsaturated Fatty Acids in a Rat Model of Anterior Ischemic Optic Neuropathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Mathews, M.K.; Guo, Y.; Langenberg, P.; Bernstein, S.L. Ciliary neurotrophic factor (CNTF)-mediated ganglion cell survival in a rodent model of non-arteritic anterior ischaemic optic neuropathy (NAION). Br. J. Ophthalmol. 2014, 99, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Di Polo, A.; Aigner, L.J.; Dunn, R.J.; Bray, G.M.; Aguayo, A.J. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Muller cells temporarily rescues injured retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3978–3983. [Google Scholar] [CrossRef] [PubMed]
- Ali Shariati, M.; Kumar, V.; Yang, T.; Chakraborty, C.; Barres, B.A.; Longo, F.M.; Liao, Y.J. A Small Molecule TrkB Neurotrophin Receptor Partial Agonist as Possible Treatment for Experimental Nonarteritic Anterior Ischemic Optic Neuropathy. Curr. Eye Res. 2018, 43, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Ngo Le, M.A.; Wen, Y.T.; Ho, Y.C.; Kapupara, K.; Tsai, R.K. Therapeutic Effects of Puerarin Against Anterior Ischemic Optic Neuropathy Through Antiapoptotic and Anti-Inflammatory Actions. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3481–3491. [Google Scholar] [CrossRef]
- Hoopfer, E.D.; McLaughlin, T.; Watts, R.J.; Schuldiner, O.; O’Leary, D.D.; Luo, L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron 2006, 50, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Soto, I.; Libby, R.T.; John, S.W.M. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp. Neurol. 2013, 246, 54–61. [Google Scholar] [CrossRef]
- Beirowski, B.; Babetto, E.; Coleman, M.P.; Martin, K.R. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur. J. Neurosci. 2008, 28, 1166–1179. [Google Scholar] [CrossRef]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343. [Google Scholar] [CrossRef]
- Czech, V.L.; O’Connor, L.C.; Philippon, B.; Norman, E.; Byrne, A.B. TIR-1/SARM1 inhibits axon regeneration and promotes axon degeneration. Elife 2023, 12, e80856. [Google Scholar] [CrossRef] [PubMed]
- Figley, M.D.; DiAntonio, A. The SARM1 axon degeneration pathway: Control of the NAD(+) metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol. 2020, 63, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, C.; Amici, A.; Gilley, J.; Loreto, A.; Trapanotto, A.G.; Antoniou, C.; Merlini, E.; Coleman, M.P.; Orsomando, G. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 2022, 25, 103812. [Google Scholar] [CrossRef] [PubMed]
- Gilley, J.; Mayer, P.R.; Yu, G.; Coleman, M.P. Low levels of NMNAT2 compromise axon development and survival. Hum. Mol. Genet. 2019, 28, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, W.; Jiang, H.; Huang, H.; Liu, L.; Fang, F.; Li, L.; Feng, X.; Liu, D.; Dalal, R.; et al. Differential effects of SARM1 inhibition in traumatic glaucoma and EAE optic neuropathies. Mol. Ther. Nucleic Acids 2023, 32, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.A.; Gilley, J.; Ling, K.; Jafar-Nejad, P.; Rigo, F.; Coleman, M. Sarm1 haploinsufficiency or low expression levels after antisense oligonucleotides delay programmed axon degeneration. Cell Rep. 2021, 37, 110108. [Google Scholar] [CrossRef] [PubMed]
- Turkiew, E.; Falconer, D.; Reed, N.; Hoke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171. [Google Scholar] [CrossRef]
- Sato-Yamada, Y.; Strickland, A.; Sasaki, Y.; Bloom, J.; DiAntonio, A.; Milbrandt, J. A SARM1-mitochondrial feedback loop drives neuropathogenesis in a Charcot-Marie-Tooth disease type 2A rat model. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Ko, K.W.; Milbrandt, J.; DiAntonio, A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 2020, 219, e201912047. [Google Scholar] [CrossRef]
- Fernandes, K.A.; Mitchell, K.L.; Patel, A.; Marola, O.J.; Shrager, P.; Zack, D.J.; Libby, R.T.; Welsbie, D.S. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp. Eye Res. 2018, 171, 54–61. [Google Scholar] [CrossRef]
- Krauss, R.; Bosanac, T.; Devraj, R.; Engber, T.; Hughes, R.O. Axons Matter: The Promise of Treating Neurodegenerative Disorders by Targeting SARM1-Mediated Axonal Degeneration. Trends Pharmacol. Sci. 2020, 41, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Bratkowski, M.; Burdett, T.C.; Danao, J.; Wang, X.; Mathur, P.; Gu, W.; Beckstead, J.A.; Talreja, S.; Yang, Y.S.; Danko, G.; et al. Uncompetitive, adduct-forming SARM1 inhibitors are neuroprotective in preclinical models of nerve injury and disease. Neuron 2022, 110, 3711–3726.e3716. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kerry, P.S.; Nanson, J.D.; Bosanac, T.; Sasaki, Y.; Krauss, R.; Saikot, F.K.; Adams, S.E.; Mosaiab, T.; Masic, V.; et al. Structural basis of SARM1 activation, substrate recognition, and inhibition by small molecules. Mol. Cell 2022, 82, 1643–1659.e1610. [Google Scholar] [CrossRef]
- Geisler, S.; Huang, S.X.; Strickland, A.; Doan, R.A.; Summers, D.W.; Mao, X.; Park, J.; DiAntonio, A.; Milbrandt, J. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J. Exp. Med. 2019, 216, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.A.; Stafford, B.K.; Nguyen, P.L.; Lien, B.V.; Wang, C.; Zukor, K.; He, Z.; Huberman, A.D. Neural activity promotes long-distance, target-specific regeneration of adult retinal axons. Nat. Neurosci. 2016, 19, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Qiao, M.; Bei, F.; Kim, I.-J.; He, Z.; Sanes, J.R. Subtype-Specific Regeneration of Retinal Ganglion Cells following Axotomy: Effects of Osteopontin and mTOR Signaling. Neuron 2015, 85, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Leaver, S.G.; Cui, Q.; Plant, G.W.; Arulpragasam, A.; Hisheh, S.; Verhaagen, J.; Harvey, A.R. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006, 13, 1328–1341. [Google Scholar] [CrossRef]
- Parrilla-Reverter, G.; Agudo, M.; Sobrado-Calvo, P.; Salinas-Navarro, M.; Villegas-Prez, M.A.P.; Vidal-Sanz, M. Effects of different neurotrophic factors on the survival of retinal ganglion cells after a complete intraorbital nerve crush injury: A quantitative in vivo study. Exp. Eye Res. 2009, 89, 32–41. [Google Scholar] [CrossRef]
- Ji, J.Z.; Elyaman, W.; Yip, H.K.; Lee, V.W.; Yick, L.W.; Hugon, J.; So, K.F. CNTF promotes survival of retinal ganglion cells after induction of ocular hypertension in rats: The possible involvement of STAT3 pathway. Eur. J. Neurosci. 2004, 19, 265–272. [Google Scholar] [CrossRef]
- Finnegan, L.K.; Chadderton, N.; Kenna, P.F.; Palfi, A.; Carty, M.; Bowie, A.G.; Millington-Ward, S.; Farrar, G.J. SARM1 Ablation Is Protective and Preserves Spatial Vision in an In Vivo Mouse Model of Retinal Ganglion Cell Degeneration. Int. J. Mol. Sci. 2022, 23, 1606. [Google Scholar] [CrossRef]
- Guo, Y.; Mehrabian, Z.; Johnson, M.A.; Miller, N.R.; Henderson, A.D.; Hamlyn, J.; Bernstein, S.L. Biomarkers of lesion severity in a rodent model of nonarteritic anterior ischemic optic neuropathy (rNAION). PLoS ONE 2021, 16, e0243186. [Google Scholar] [CrossRef]
- Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Nieto-Lopez, L.; Canovas-Martinez, I.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo, M. Brn3a as a marker of retinal ganglion cells: Qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3860–3868. [Google Scholar] [CrossRef] [PubMed]
- Crowe, M.J.; Bresnahan, J.C.; Shuman, S.L.; Masters, J.N.; Beattie, M.S. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 1997, 3, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Monavarfeshani, A.; Yan, W.; Pappas, C.; Odenigbo, K.A.; He, Z.; Segrè, A.V.; van Zyl, T.; Hageman, G.S.; Sanes, J.R. Transcriptomic analysis of the ocular posterior segment completes a cell atlas of the human eye. Proc. Natl. Acad. Sci. USA 2023, 120, e2306153120. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.O.; Bosanac, T.; Mao, X.; Engber, T.M.; DiAntonio, A.; Milbrandt, J.; Devraj, R.; Krauss, R. Small Molecule SARM1 Inhibitors Recapitulate the SARM1(-/-) Phenotype and Allow Recovery of a Metastable Pool of Axons Fated to Degenerate. Cell Rep. 2021, 34, 108588. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Franke, A.; Kaplan, M.R.; Pfrieger, F.W.; Barres, B.A. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron 1995, 15, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Van Adel, B.A.; Kostic, C.; Deglon, N.; Ball, A.K.; Arsenijevic, Y. Delivery of ciliary neurotrophic factor via lentiviral-mediated transfer protects axotomized retinal ganglion cells for an extended period of time. Hum. Gene Ther. 2003, 14, 103–115. [Google Scholar] [CrossRef]
- Ma, C.H.; Taylor, J.S. Trophic responsiveness of purified postnatal and adult rat retinal ganglion cells. Cell Tissue Res. 2010, 339, 297–310. [Google Scholar] [CrossRef]
- Sendtner, M.; Dittrich, F.; Hughes, R.A.; Thoenen, H. Actions of CNTF and neurotrophins on degenerating motoneurons: Preclinical studies and clinical implications. J. Neurol. Sci. 1994, 124, 77–83. [Google Scholar] [CrossRef]
- Sánchez-Migallón, M.C.; Nadal-Nicolás, F.M.; Jiménez-López, M.; Sobrado-Calvo, P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Brain derived neurotrophic factor maintains Brn3a expression in axotomized rat retinal ganglion cells. Exp. Eye Res. 2011, 92, 260–267. [Google Scholar] [CrossRef]
- Lee, K.; Choi, J.O.; Hwang, A.; Bae, H.W.; Kim, C.Y. Ciliary Neurotrophic Factor Derived From Astrocytes Protects Retinal Ganglion Cells Through PI3K/AKT, JAK/STAT, and MAPK/ERK Pathways. Investig. Ophthalmol. Vis. Sci. 2022, 63, 4. [Google Scholar] [CrossRef] [PubMed]
- Fantone, S.; Tossetta, G.; Montironi, R.; Senzacqua, M.; Marzioni, D.; Mazzucchelli, R. Ciliary neurotrophic factor (CNTF) and its receptor (CNTFRα) signal through MAPK/ERK pathway in human prostate tissues: A morphological and biomolecular study. Eur. J. Histochem. 2020, 64, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Mey, J.; Thanos, S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993, 602, 304–317. [Google Scholar] [CrossRef]
- Jo, S.A.; Wang, E.; Benowitz, L.I. Ciliary neurotrophic factor is an axogenesis factor for retinal ganglion cells. Neuroscience 1999, 89, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Chan, P.M.; So, K.F.; Yip, H.K.; Chung, S.K. Ciliary neurotrophic factor promotes the regrowth capacity but not the survival of intraorbitally axotomized retinal ganglion cells in adult hamsters. Neuroscience 1999, 94, 623–628. [Google Scholar] [CrossRef]
- Krol, J.; Roska, B. Treatment synergy in axon regeneration. Nat. Neurosci. 2016, 19, 983–984. [Google Scholar] [CrossRef]
- Shen, C.; Vohra, M.; Zhang, P.; Mao, X.; Figley, M.D.; Zhu, J.; Sasaki, Y.; Wu, H.; DiAntonio, A.; Milbrandt, J. Multiple domain interfaces mediate SARM1 autoinhibition. Proc. Natl. Acad. Sci. USA 2021, 118, e2023151118. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Mehrabian, Z.; Milbrandt, J.; DiAntonio, A.; Bernstein, S.L. Synergistic Protection of Retinal Ganglion Cells (RGCs) by SARM1 Inactivation with CNTF in a Rodent Model of Nonarteritic Anterior Ischemic Optic Neuropathy. Cells 2024, 13, 202. https://doi.org/10.3390/cells13030202
Guo Y, Mehrabian Z, Milbrandt J, DiAntonio A, Bernstein SL. Synergistic Protection of Retinal Ganglion Cells (RGCs) by SARM1 Inactivation with CNTF in a Rodent Model of Nonarteritic Anterior Ischemic Optic Neuropathy. Cells. 2024; 13(3):202. https://doi.org/10.3390/cells13030202
Chicago/Turabian StyleGuo, Yan, Zara Mehrabian, Jeffrey Milbrandt, Aaron DiAntonio, and Steven L. Bernstein. 2024. "Synergistic Protection of Retinal Ganglion Cells (RGCs) by SARM1 Inactivation with CNTF in a Rodent Model of Nonarteritic Anterior Ischemic Optic Neuropathy" Cells 13, no. 3: 202. https://doi.org/10.3390/cells13030202
APA StyleGuo, Y., Mehrabian, Z., Milbrandt, J., DiAntonio, A., & Bernstein, S. L. (2024). Synergistic Protection of Retinal Ganglion Cells (RGCs) by SARM1 Inactivation with CNTF in a Rodent Model of Nonarteritic Anterior Ischemic Optic Neuropathy. Cells, 13(3), 202. https://doi.org/10.3390/cells13030202