
Differential Infiltration of Key Immune T-Cell Populations Across Malignancies Varying by Immunogenic Potential and the Likelihood of Response to Immunotherapy

 , , , , , , , , , , , ,
, , , , , , , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Data Compilation
2.2. RNA Sequencing and Data Retrieval
2.3. Quantification Analysis of RNA Gene Expression
2.4. Generation of Gene Expression Signature Scores
2.5. Enrichment Analysis of Gene Sets
2.6. Study Outcomes
2.7. Validation
2.8. Statistical Analysis
3. Results
3.1. Patients Characteristics
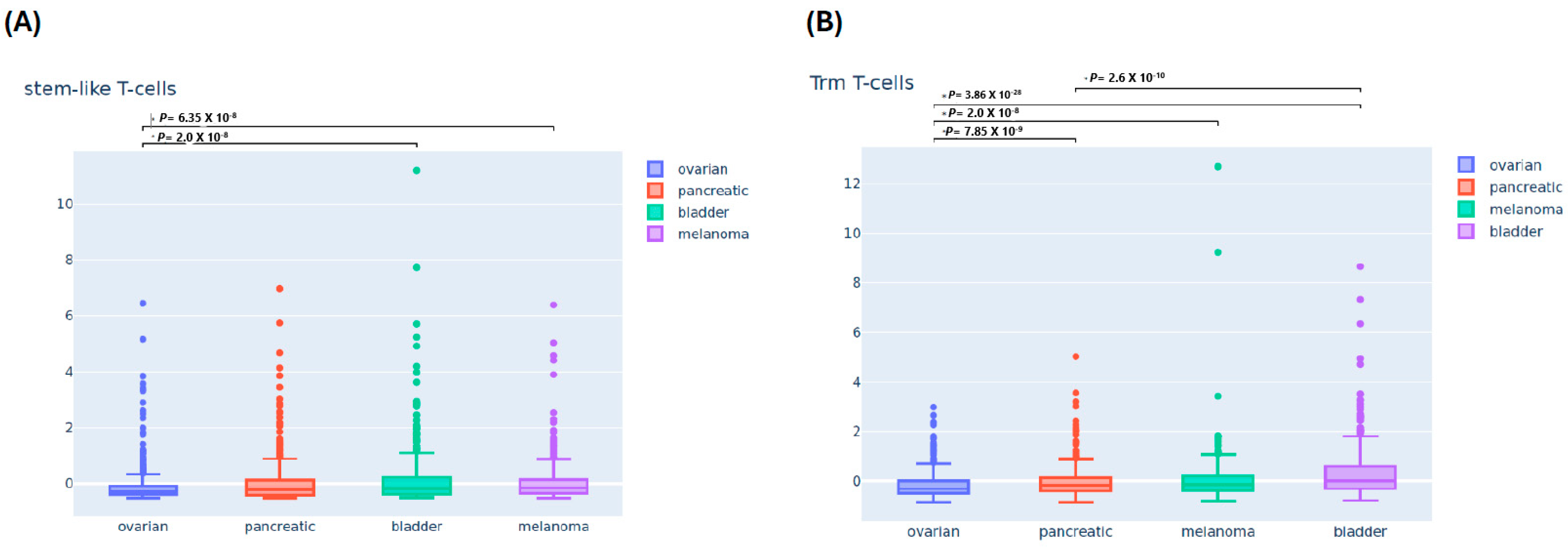
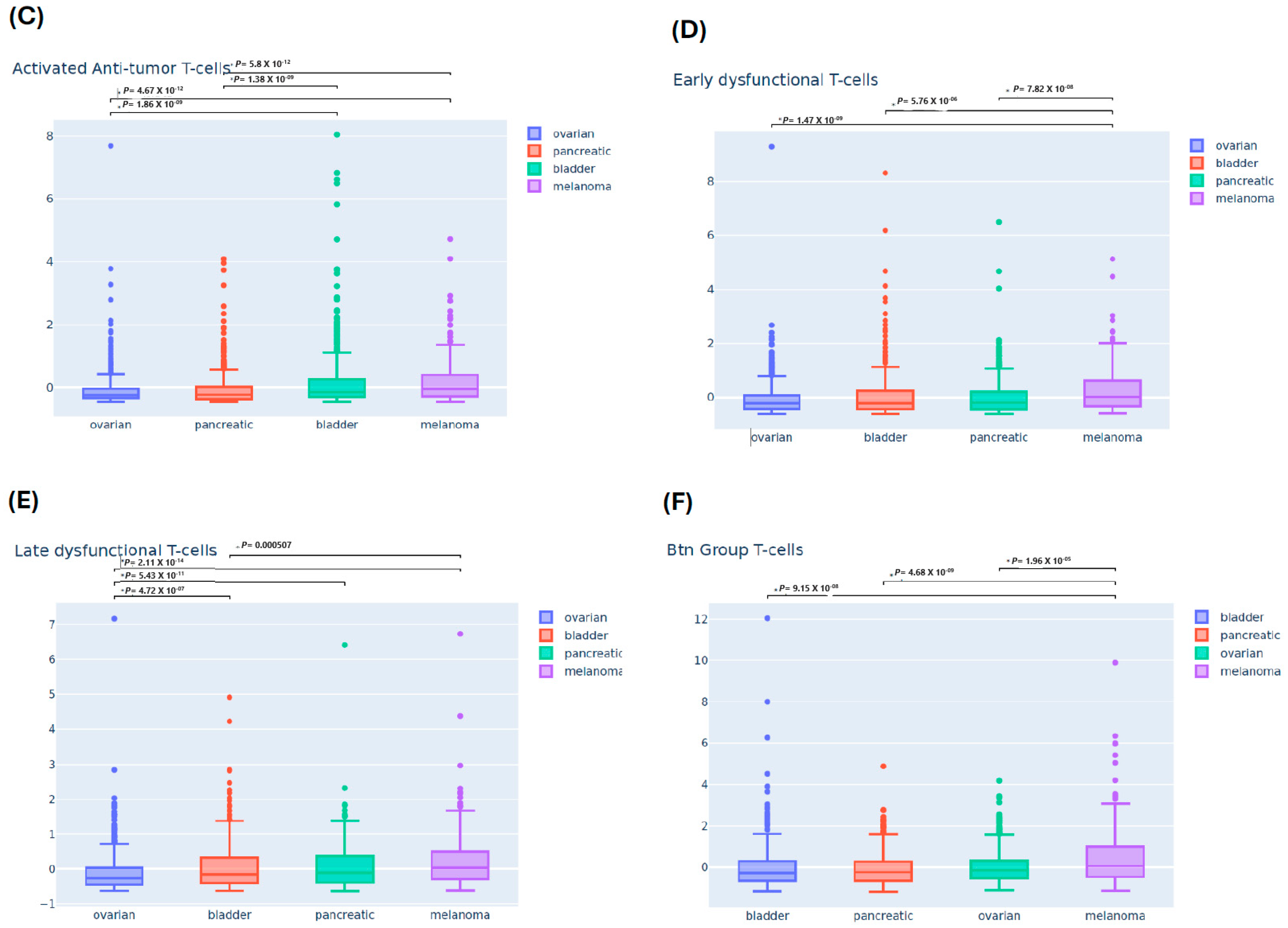
3.2. Differential Immune Infiltrating T-Cells Across Four Cancers
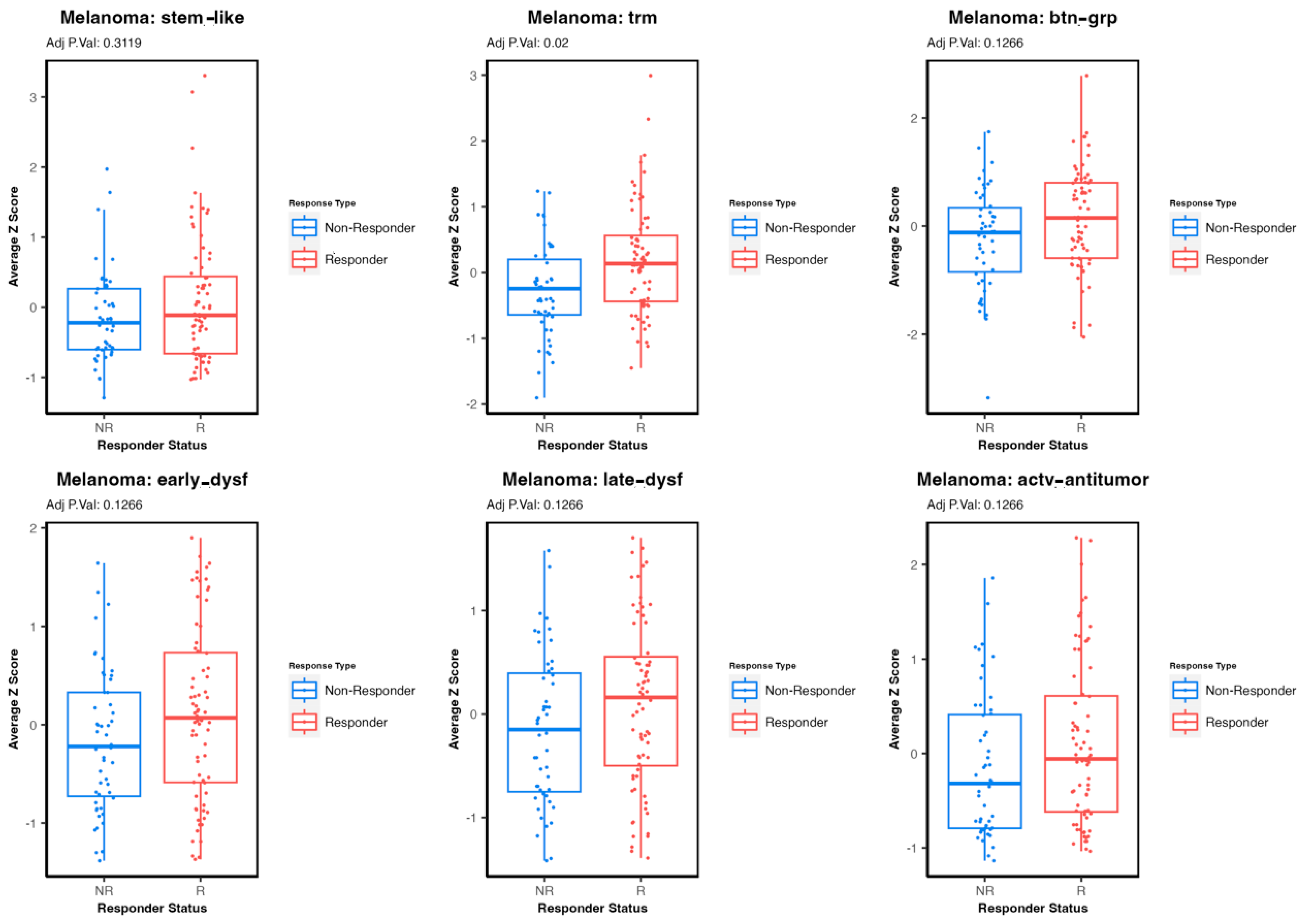
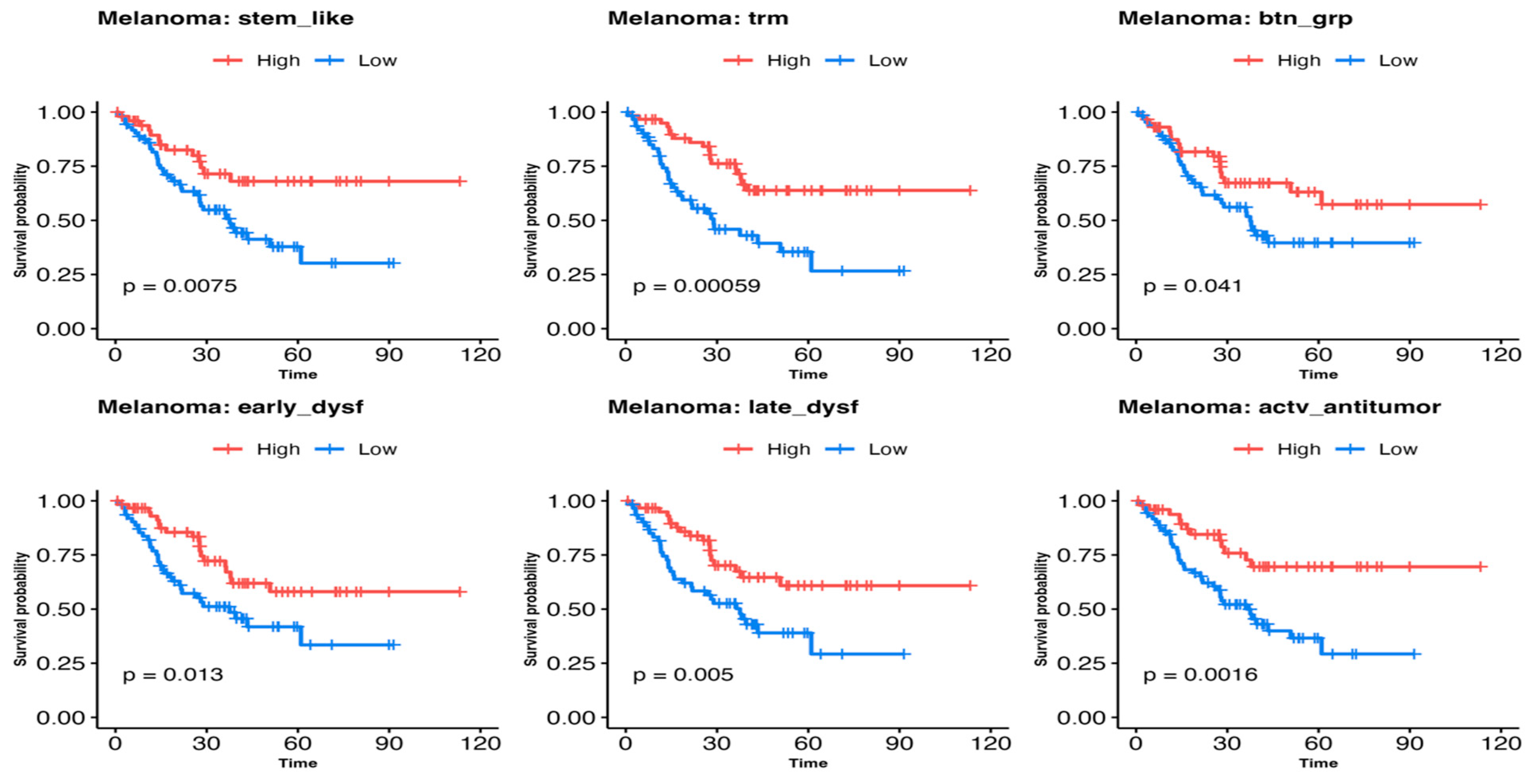
3.3. Comparing Immune T-Cell Subtype Infiltration in Patients with Melanoma Treated with Immunetherapy and Testing Association with Survival Outcomes
3.4. Immune Cell Infiltration Gene Expression Signature Validation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, D.C.; Theocharopoulos, C.; Lialios, P.P.; Foteinou, D.; Koumprentziotis, I.A.; Xynos, G.; Gogas, H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment. Cancers 2023, 15, 2718. [Google Scholar] [CrossRef]
- Bondhopadhyay, B.; Sisodiya, S.; Chikara, A.; Khan, A.; Tanwar, P.; Afroze, D.; Singh, N.; Agrawal, U.; Mehrotra, R.; Hussain, S. Cancer immunotherapy: A promising dawn in cancer research. Am. J. Blood Res. 2020, 10, 375–385. [Google Scholar] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Finotello, F.; Trajanoski, Z. New strategies for cancer immunotherapy: Targeting regulatory T cells. Genome Med. 2017, 9, 10. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Fenstermacher, D.A.; Wenham, R.M.; Rollison, D.E.; Dalton, W.S. Implementing personalized medicine in a cancer center. Cancer J. 2011, 17, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.A.; Dalton, W.S.; Rodriguez, L.; Sellers, T.; Willman, C.L. Orien. Oncol. Issues 2016, 31, 62–66. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- DeLuca, D.S.; Levin, J.Z.; Sivachenko, A.; Fennell, T.; Nazaire, M.D.; Williams, C.; Reich, M.; Winckler, W.; Getz, G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 2012, 28, 1530–1532. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Chuang, H.Y.; Kim, J.W.; Ideker, T.; Lee, D. Inferring pathway activity toward precise disease classification. PLoS Comput. Biol. 2008, 4, e1000217. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Xie, M.; Lee, K.; Lockhart, J.H.; Cukras, S.D.; Carvajal, R.; Beg, A.A.; Flores, E.R.; Teng, M.; Chung, C.H.; Tan, A.C. TIMEx: Tumor-immune microenvironment deconvolution web-portal for bulk transcriptomics using pan-cancer scRNA-seq signatures. Bioinformatics 2021, 37, 3681–3683. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Wei, S.; Wei, Z.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Sugarman, E.; Gabrilovich, D.I.; Sullivan, R.J.; et al. Pathway signatures derived from on-treatment tumor specimens predict response to anti-PD1 blockade in metastatic melanoma. Nat. Commun. 2021, 12, 6023. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e236. [Google Scholar] [CrossRef]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 blockade drives loss of T(reg) stability in glycolysis-low tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Algazi, A.; Ascierto, P.A.; Butler, M.O.; Chandra, S.; Gordon, M.; Hernandez-Aya, L.; Lawrence, D.; Lutzky, J.; Miller, W.H., Jr.; et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nat. Commun. 2020, 11, 6262. [Google Scholar] [CrossRef]
- Freeman, S.S.; Sade-Feldman, M.; Kim, J.; Stewart, C.; Gonye, A.L.K.; Ravi, A.; Arniella, M.B.; Gushterova, I.; LaSalle, T.J.; Blaum, E.M.; et al. Combined tumor and immune signals from genomes or transcriptomes predict outcomes of checkpoint inhibition in melanoma. Cell Rep. Med. 2022, 3, 100500. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Torngren, T.; Kvist, A.; et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1738. [Google Scholar] [CrossRef]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1897. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Maffuid, K.; Cao, Y. Decoding the Complexity of Immune-Cancer Cell Interactions: Empowering the Future of Cancer Immunotherapy. Cancers 2023, 15, 4188. [Google Scholar] [CrossRef] [PubMed]
- Blessin, N.C.; Li, W.; Mandelkow, T.; Jansen, H.L.; Yang, C.; Raedler, J.B.; Simon, R.; Buscheck, F.; Dum, D.; Luebke, A.M.; et al. Prognostic role of proliferating CD8(+) cytotoxic Tcells in human cancers. Cell Oncol. 2021, 44, 793–803. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Wang, S.; Dong, J.; Wei, J. Pan-Cancer Analysis of Immune Cell Infiltration Identifies a Prognostic Immune-Cell Characteristic Score (ICCS) in Lung Adenocarcinoma. Front. Immunol. 2020, 11, 1218. [Google Scholar] [CrossRef]
- Yi, L.; Yang, L. Stem-like T cells and niches: Implications in human health and disease. Front. Immunol. 2022, 13, 907172. [Google Scholar] [CrossRef]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Choo, D.; Vezys, V.; Wherry, E.J.; Duraiswamy, J.; Akondy, R.; Wang, J.; Casey, K.A.; Barber, D.L.; Kawamura, K.S.; et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010, 207, 553–564. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8(+) T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef]
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigne, C.M.; Monkkonen, H.; Monkkonen, J.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.A.; Stammers, M.; Malcherek, G.; Beck, S.; Trowsdale, J. The cluster of BTN genes in the extended major histocompatibility complex. Genomics 2001, 71, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Yang, B.; Li, X.; Zhang, W.; Fan, J.; Zhou, Y.; Li, W.; Yin, J.; Yang, X.; Guo, E.; Li, X.; et al. Spatial heterogeneity of infiltrating T cells in high-grade serous ovarian cancer revealed by multi-omics analysis. Cell Rep. Med. 2022, 3, 100856. [Google Scholar] [CrossRef]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef]
- Nesseler, J.P.; Lee, M.H.; Nguyen, C.; Kalbasi, A.; Sayre, J.W.; Romero, T.; Nickers, P.; McBride, W.H.; Schaue, D. Tumor Size Matters-Understanding Concomitant Tumor Immunity in the Context of Hypofractionated Radiotherapy with Immunotherapy. Cancers 2020, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]
- Wang, R.C.; Wang, Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers 2023, 15, 3837. [Google Scholar] [CrossRef]
- Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martínez-Cano, S.; Mejías-Pérez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat. Commun. 2017, 8, 16073. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.T.; Byrne, K.T.; Vella, J.L.; Zhang, P.; Shabaneh, T.B.; Steinberg, S.M.; Molodtsov, A.K.; Bowers, J.S.; Angeles, C.V.; Paulos, C.M.; et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci. Immunol. 2017, 2, eaam6346. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.; Chiffelle, J.; Genolet, R.; Navarro Rodrigo, B.; Perez, M.A.S.; Huber, F.; Magnin, M.; Nguyen-Ngoc, T.; Guillaume, P.; Baumgaertner, P.; et al. Sensitive identification of neoantigens and cognate TCRs in human solid tumors. Nat. Biotechnol. 2022, 40, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.A.; Miron, M.; Farber, D.L. Location, location, location: Tissue resident memory T cells in mice and humans. Sci. Immunol. 2019, 4, eaas9673. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Cohort | Treatment(s) | Pre /On/Post Treatment | Patients (N) | REF |
|---|---|---|---|---|---|
| Du | Du | Ipilimumab/Nivolumab/Pembrolizumab | Pre/On | 50 | [23] |
| Gide | Gide_Pre_PD-1_CTLA4 | Ipilimumab/Nivolumab/Pembrolizumab | Pre/On | 41 | [24] |
| Gide_Pre_PD-1 | Pembrolizumab/Nivolumab | Pre/On | 50 | ||
| GSE | GSE165278 | Ipilimumab | Pre/Post | 22 | [25] |
| GSE158403 | Durvalumab | Pre/On | 81 | [26] | |
| Freeman | Freeman | N/A | Pre/Post | 38 | [27] |
| Hugo | HugoLo_IPRES_2016 | Pembrolizumab | Pre/On | 26 | [28] |
| Lauss | Lauss | Adoptive T-cell therapy | Pre | 25 | [29] |
| Lee | Lee | Pembrolizumab/Nivolumab | Pre/On | 78 | [30] |
| Liu | Liu | Pembrolizumab/Nivolumab | Pre/On | 122 | [31] |
| Riaz | Riaz | Nivolumab | Pre/On | 98 | [32] |
| Van Allen | VanAllen_anti-CTLA4_2015 | Ipilimumab | Pre | 41 | [33] |
| Characteristic | Total (N = 1892) | Cutaneous Melanoma (N = 232) | Ovarian Cancer (N = 664) | Pancreatic Adenocarcinoma (N = 647) | Bladder Urothelial Carcinoma (N = 349) |
|---|---|---|---|---|---|
| Age in years Mean ± SD | 62 ± 13 | 59 ± 14 | 59 ± 13 | 63 ± 13 | 68 ± 11 |
| Sex, n (%) Female Male | 1141 (60.3) 751 (39.7) | 89 (38.4) 143 (61.6) | 664 (100) 0 (0) | 301 (46.5) 346 (53.5) | 87 (24.9) 262 (75.1) |
| Ethnicity, n (%) Hispanic Non-Hispanic Unknown | 94 (5.0) 1749 (92.4) 49 (2.6) | 9 (3.9) 217 (93.5) 6 (2.6) | 39 (5.9) 618 (93.1) 7 (1.1) | 32 (4.9) 603 (93.2) 12 (1.9) | 14 (4.0) 311 (89.1) 24 (6.9) |
| Race, n (%) African American American Indian or Alaska Native Asian Native Hawaiian or Other Pacific Islander White Other Unknown | 55 (2.9) 11 (0.6) 19 (1.0) 2 (0.1) 1757 (92.9) 24 (1.3) 24 (1.3) | 1 (0.4) 1 (0.4) 0 (0) 0 (0) 226 (97.4) 1 (0.4) 3 (1.3) | 21 (3.2) 8 (1.2) 9 (1.4) 2 (0.3) 610 (91.9) 6 (0.9) 8 (1.2) | 23 (3.6) 2 (0.3) 7 (1.1) 0 (0) 601 (92.9) 7 (1.1) 7 (1.1) | 10 (2.9) 0 (0) 3 (0.9) 0 (0) 320 (91.7) 10 (2.9) 6 (1.7) |
| Cancer stage at initial diagnosis, n (%) Stage I Stage II Stage III Stage IV Unknown | 282 (14.9) 527 (27.9) 526 (27.8) 324 (17.1 193 (10.2) | 25 (10.8) 48 (20.7) 73 (31.5) 43 (18.5) 43 (18.5) | 97 (14.6) 74 (11.1) 292 (44.0) 129 (19.4) 72 (10.8) | 117 (18.1) 350 (54.1) 47 (7.3) 77 (11.9) 36 (5.6) | 43 (12.3) 55 (15.8) 114 (32.7) 75 (21.5) 42 (12.0) |
| Performance status (ECOG), n (%) 0 1 2 3 Unknown | 386 (20.4) 282 (14.9) 48 (2.5) 8 (0.4) 1168 (61.7) | 39 (16.8) 11 (4.7) 4 (1.7) 1 (0.4) 177 (76.3 | 140 (21.1) 117 (17.6) 22 (3.3) 4 (0.6) 381 (57.4) | 141 (21.8) 114 (17.6) 12 (1.9) 1 (0.2) 379 (58.6) | 66 (18.9) 40 (11.5) 10 (2.9) 2 (0.6) 231 (66.2) |
| T-Cells Populations | Stem-like TILs | TRM T-Cells | Activated-Potentially Anti-Tumor T-Cells | Early Dysfunction T-Cells | Late Dysfunction T-Cells | BTN3A Group-Related T-Cells |
|---|---|---|---|---|---|---|
| OREIN dataset | 0.554 | 0.655 | 0.586 | 0.593 | 0.588 | 0.594 |
| Public datasets * | 0.633 | 0.605 | 0.633 | 0.619 | 0.638 | 0.625 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eljilany, I.; Coleman, S.; Tan, A.C.; McCarter, M.D.; Carpten, J.; Colman, H.; Naqash, A.R.; Puzanov, I.; Arnold, S.M.; Churchman, M.L.; et al. Differential Infiltration of Key Immune T-Cell Populations Across Malignancies Varying by Immunogenic Potential and the Likelihood of Response to Immunotherapy. Cells 2024, 13, 1993. https://doi.org/10.3390/cells13231993
Eljilany I, Coleman S, Tan AC, McCarter MD, Carpten J, Colman H, Naqash AR, Puzanov I, Arnold SM, Churchman ML, et al. Differential Infiltration of Key Immune T-Cell Populations Across Malignancies Varying by Immunogenic Potential and the Likelihood of Response to Immunotherapy. Cells. 2024; 13(23):1993. https://doi.org/10.3390/cells13231993
Chicago/Turabian StyleEljilany, Islam, Sam Coleman, Aik Choon Tan, Martin D. McCarter, John Carpten, Howard Colman, Abdul Rafeh Naqash, Igor Puzanov, Susanne M. Arnold, Michelle L. Churchman, and et al. 2024. "Differential Infiltration of Key Immune T-Cell Populations Across Malignancies Varying by Immunogenic Potential and the Likelihood of Response to Immunotherapy" Cells 13, no. 23: 1993. https://doi.org/10.3390/cells13231993
APA StyleEljilany, I., Coleman, S., Tan, A. C., McCarter, M. D., Carpten, J., Colman, H., Naqash, A. R., Puzanov, I., Arnold, S. M., Churchman, M. L., Spakowicz, D., Salhia, B., Marin-Acevedo, J. A., Ganesan, S., Ratan, A., Shriver, C., Hwu, P., Dalton, W. S., Weiner, G. J., ... Tarhini, A. A. (2024). Differential Infiltration of Key Immune T-Cell Populations Across Malignancies Varying by Immunogenic Potential and the Likelihood of Response to Immunotherapy. Cells, 13(23), 1993. https://doi.org/10.3390/cells13231993