
CD38 Inhibitor 78c Attenuates Pro-Inflammatory Cytokine Expression and Osteoclastogenesis in Macrophages


 , ,
, , 
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Reagents
2.2. Generation of L929 Conditioned Media
2.3. Generation of Bone Marrow-Derived Monocytes and Macrophages (BMMs)
2.4. Bacterial Culture
2.5. Generation of shRNA Lentivirus
2.6. NAD+ Assay
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Osteoclastogenesis Assay and Bone Resorption Assay
2.9. RNA Extraction and Real-Time PCR
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
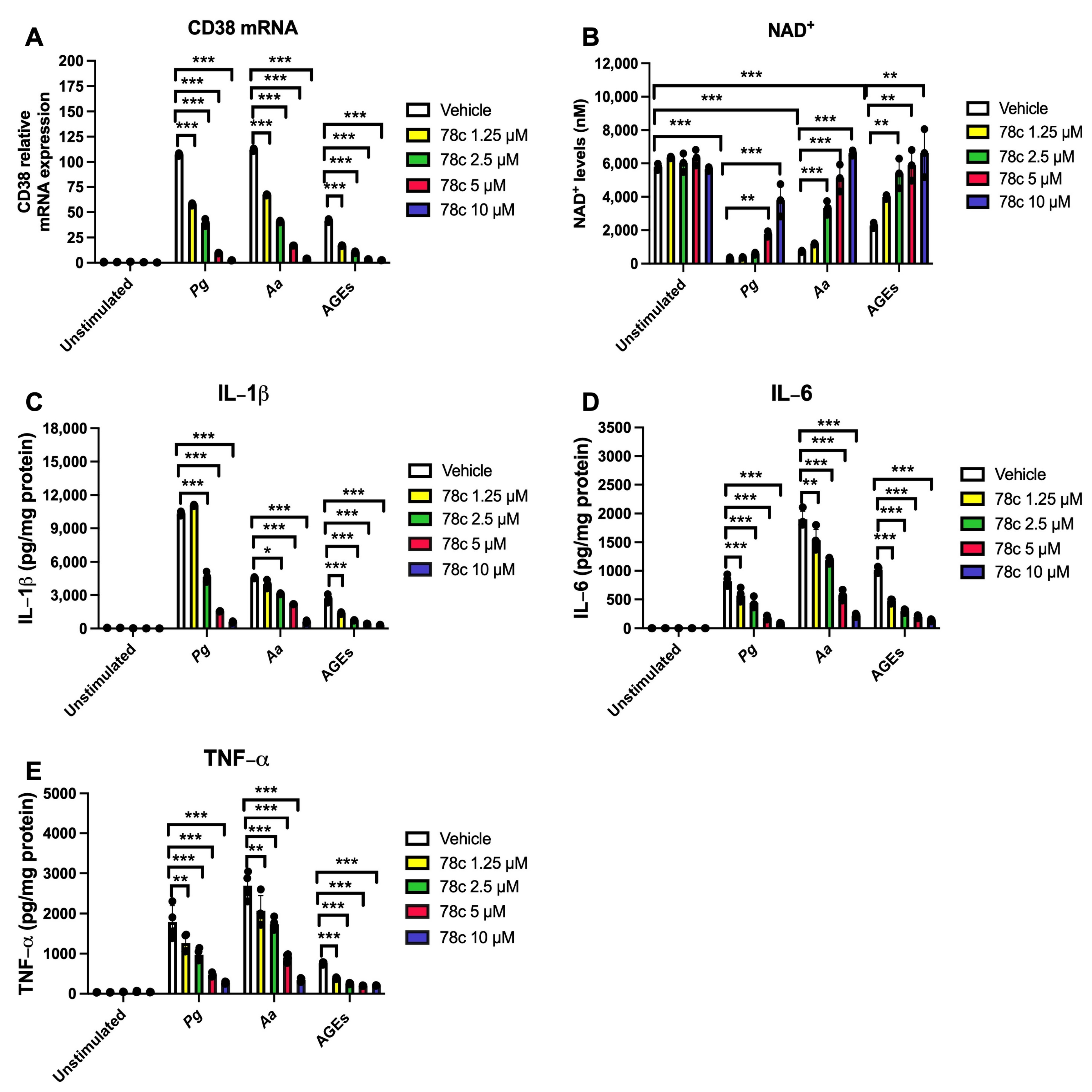
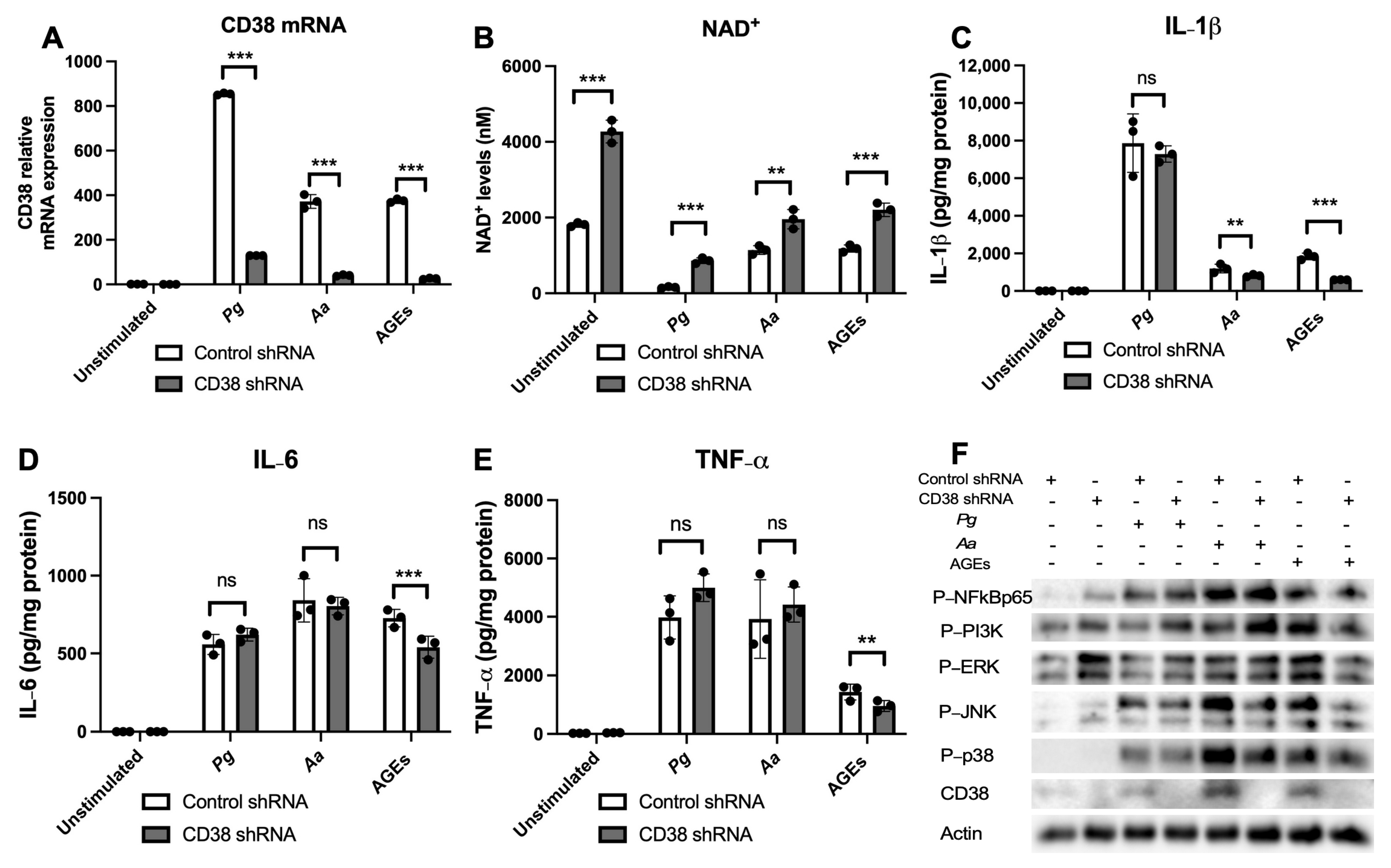
3.1. Inhibition of CD38 by 78c Reduced CD38, Reversed the Decline of NAD+, and Suppressed IL-1β, IL-6, and TNF-α Proinflammatory Cytokines in Macrophages Infected by Oral Pathogens or Stimulated by AGEs
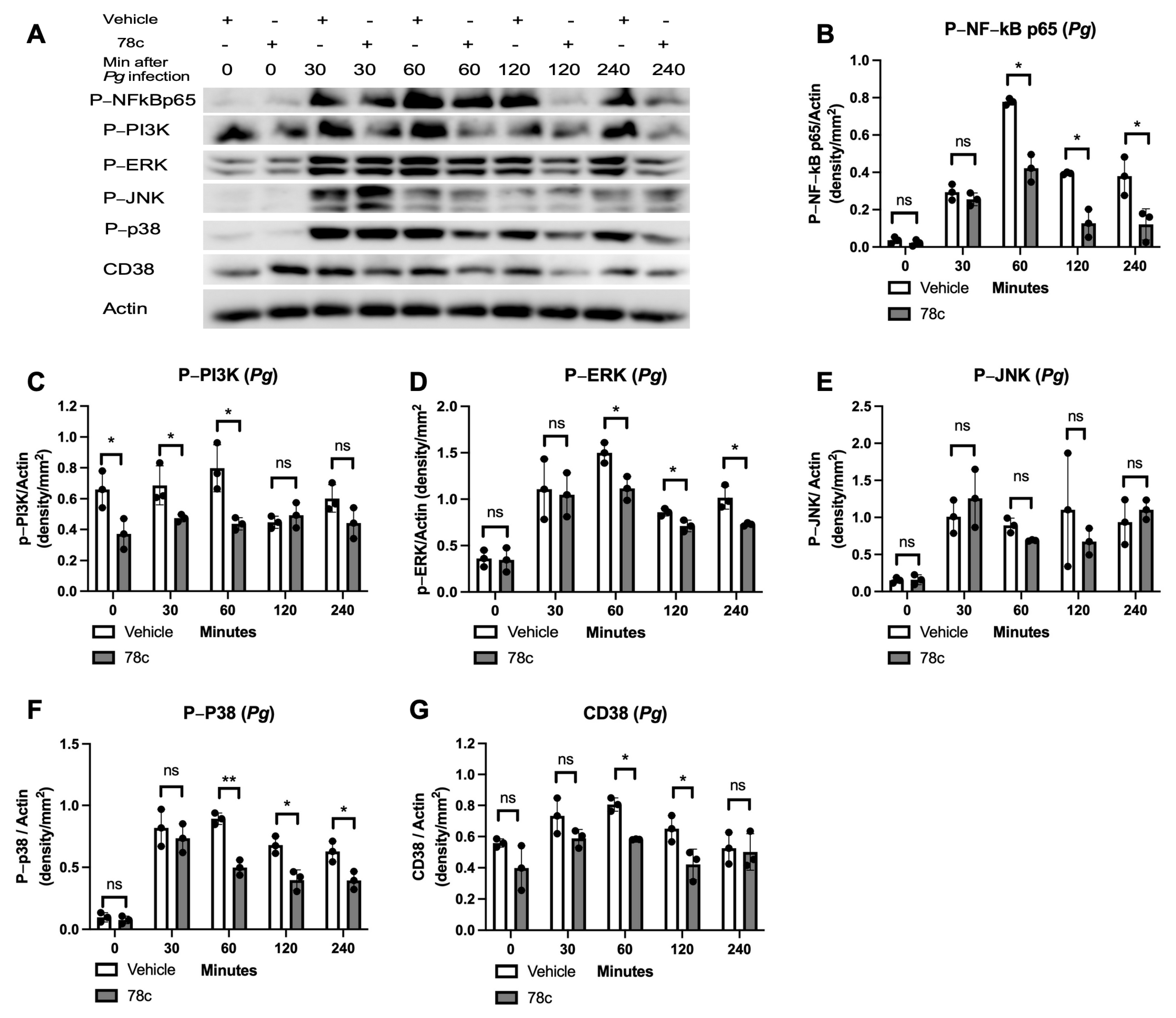
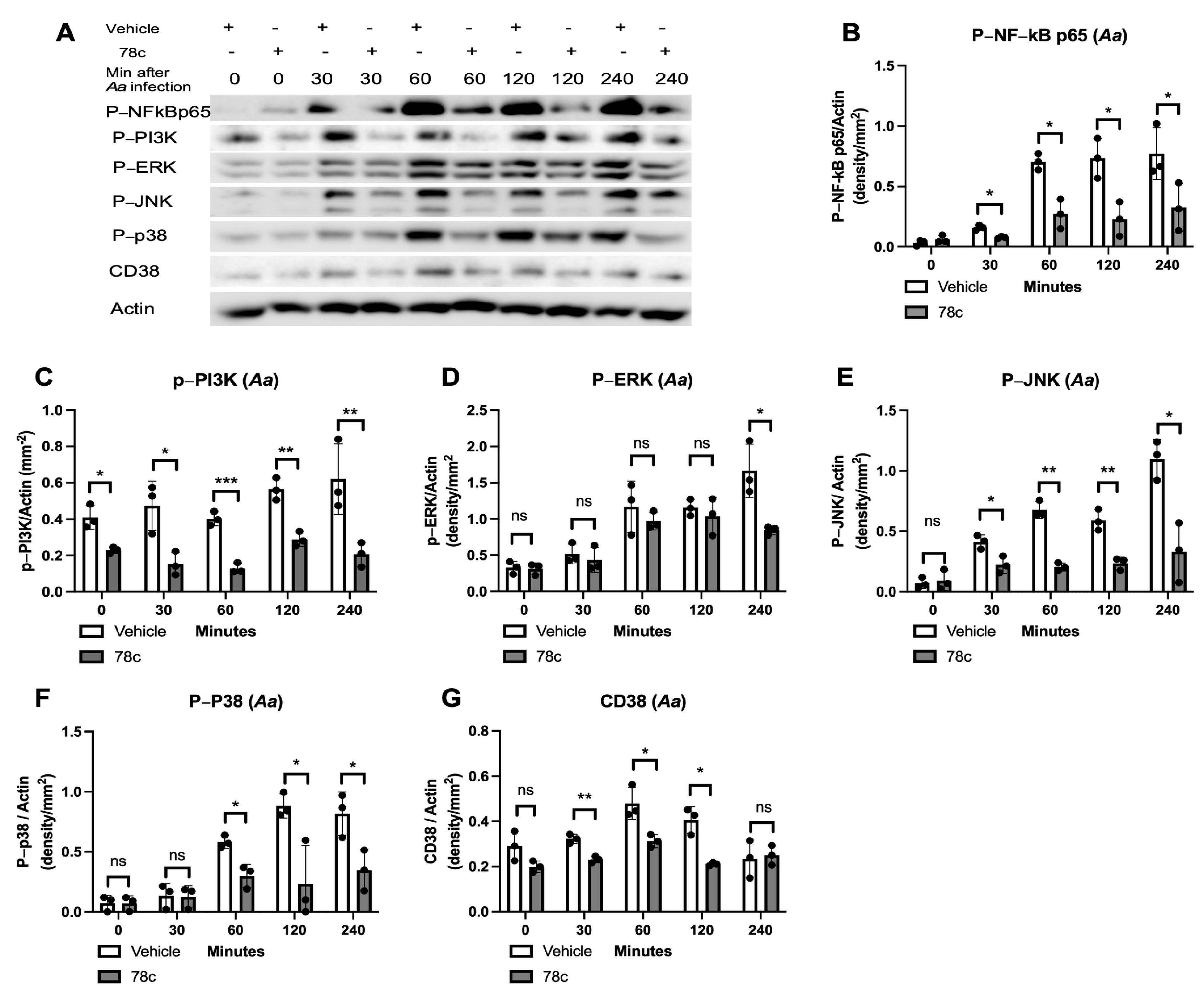
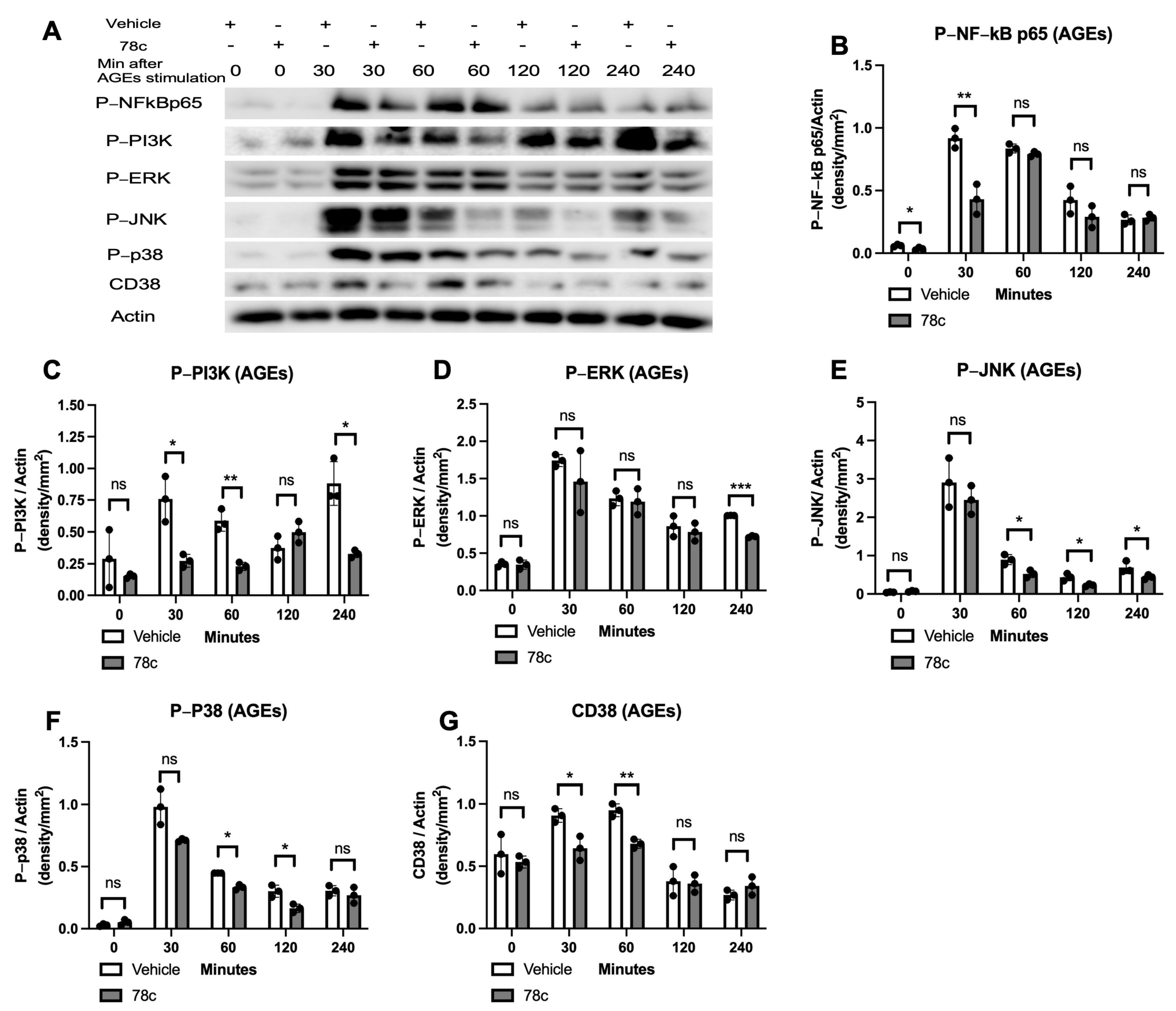
3.2. Inhibition of CD38 by 78c Reduced the Expressions of NF-kB, PI3K, MAPK, and CD38 Induced by Oral Pathogens or AGEs in Murine BMMs
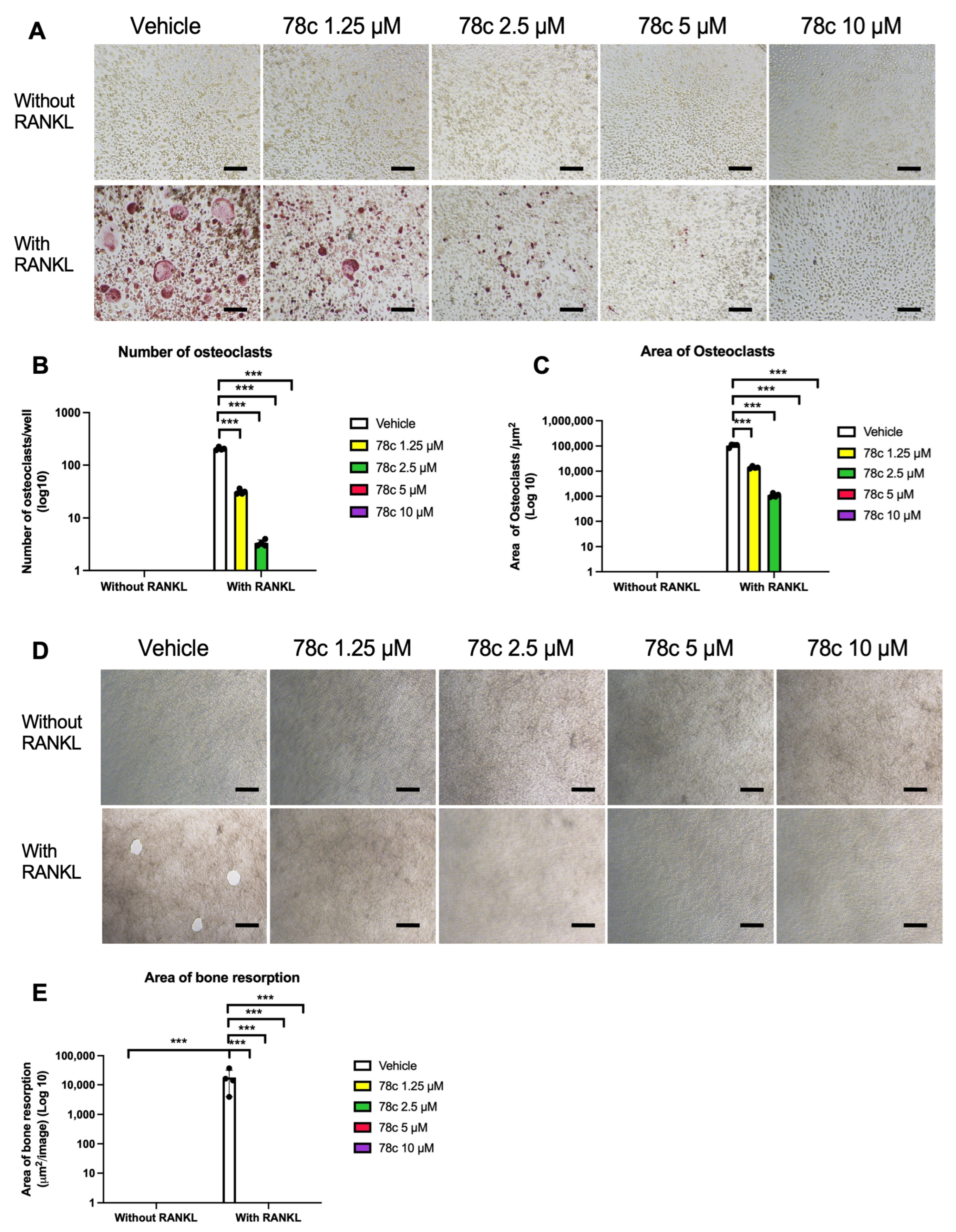
3.3. Inhibition of CD38 by 78c Suppressed Osteoclastogenesis and Bone Resorption Induced by RANKL
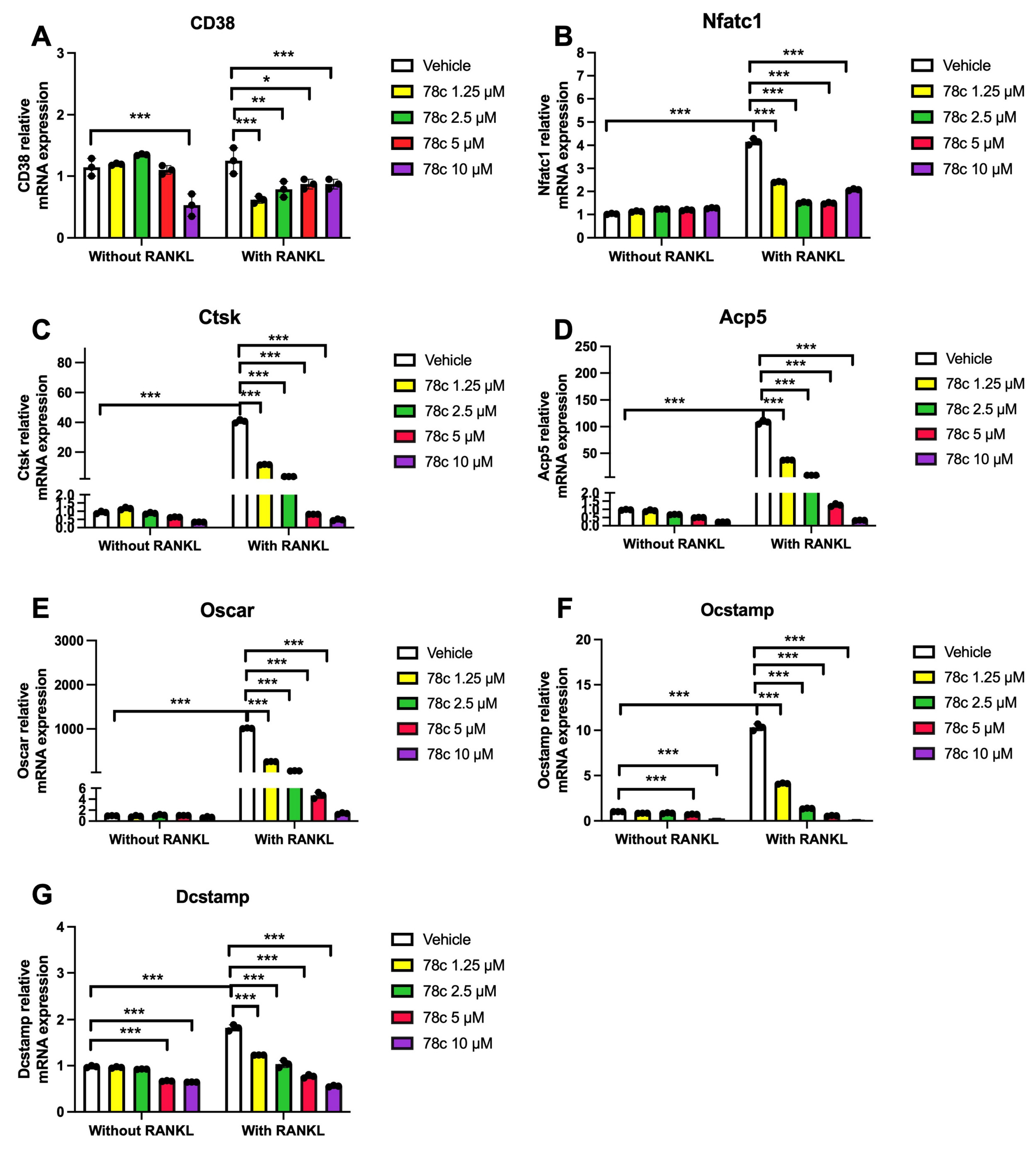
3.4. Inhibition of CD38 by 78c Reduced the mRNA Levels of Osteoclastogenic Factors, Including Nfatc1, Ctsk, Acp5, Oscar, Ocstamp, and Dcstamp, Induced by RANKL
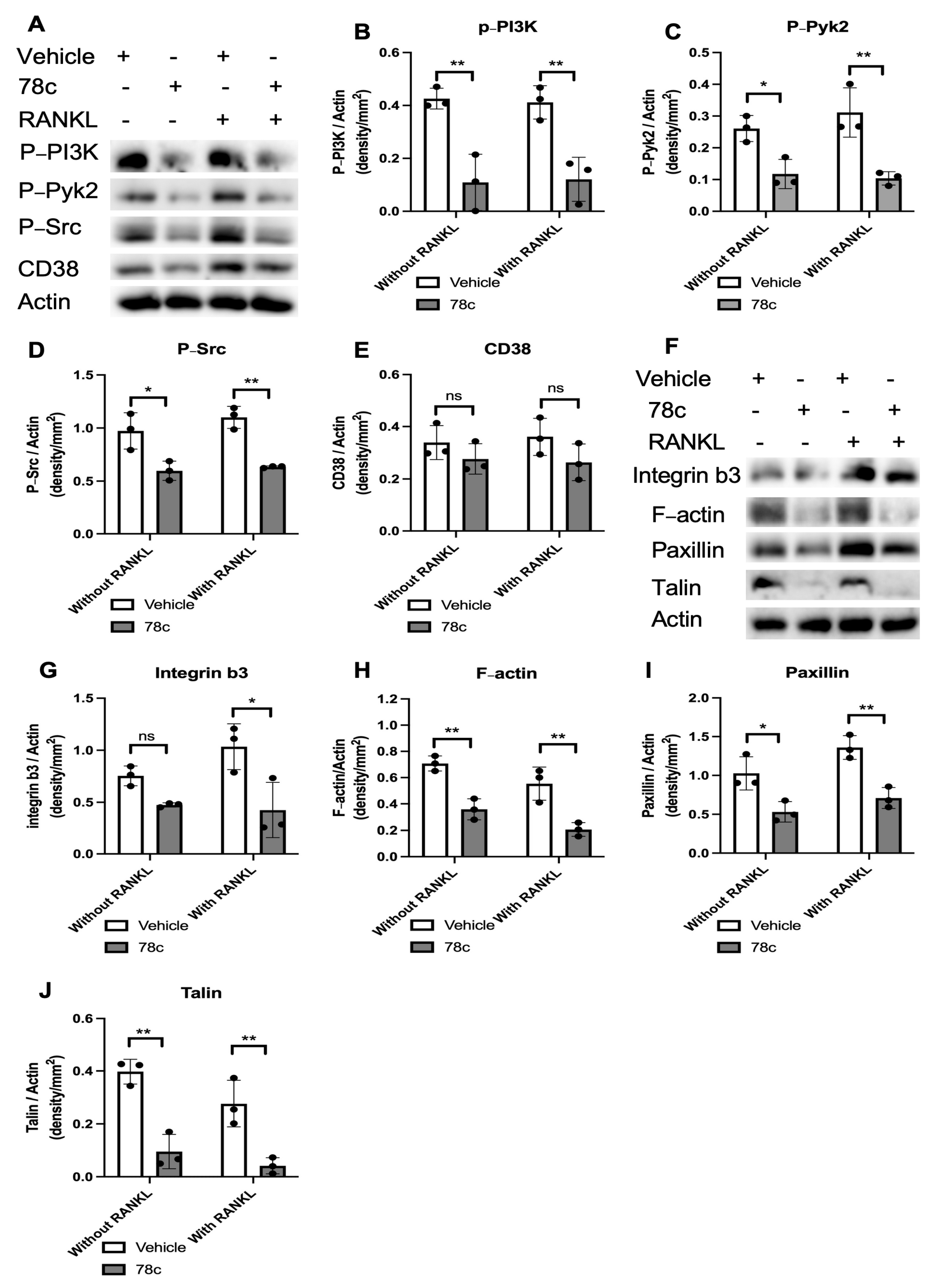
3.5. Inhibition of CD38 by 78c Decreased Podosome-Associated Protein Kinase and Adhesion Protein Levels Induced by RANKL
3.6. 78c Displays Some Off-Target Effects in Inhibiting IL-1β, IL-6, and TNF-α Cytokine Levels Induced by Oral Pathogens
3.7. 78c Has Some Off-Target Effects in Suppressing Osteoclastogenesis and Bone Resorption Induced by RANKL
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9, 1716. [Google Scholar] [CrossRef] [PubMed]
- Benzi, A.; Grozio, A.; Spinelli, S.; Sturla, L.; Guse, A.H.; De Flora, A.; Zocchi, E.; Heeren, J.; Bruzzone, S. Role of CD38 in Adipose Tissue: Tuning Coenzyme Availability? Nutrients 2021, 13, 3734. [Google Scholar] [CrossRef]
- Guse, A.H. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005, 272, 4590–4597. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Amici, S.A.; Young, N.A.; Narvaez-Miranda, J.; Jablonski, K.A.; Arcos, J.; Rosas, L.; Papenfuss, T.L.; Torrelles, J.B.; Jarjour, W.N.; Guerau-de-Arellano, M. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front. Immunol. 2018, 9, 1593. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Cacicedo, J.M.; Ido, Y. Impaired nicotinamide adenine dinucleotide (NAD(+)) metabolism in diabetes and diabetic tissues: Implications for nicotinamide-related compound treatment. J. Diabetes Investig. 2020, 11, 1403–1419. [Google Scholar] [CrossRef]
- Perez-Sanchez, C.; Escudero-Contreras, A.; Cerdó, T.; Sánchez-Mendoza, L.M.; Llamas-Urbano, A.; la Rosa, I.A.; Pérez-Rodriguez, M.; Muñoz-Barrera, L.; Del Carmen Abalos-Aguilera, M.; Barbarroja, N.; et al. Preclinical Characterization of Pharmacologic NAD(+) Boosting as a Promising Therapeutic Approach in Rheumatoid Arthritis. Arthritis Rheumatol. 2023, 75, 1749–1761. [Google Scholar] [CrossRef]
- Chini, E.N.; Chini, C.C.S.; Espindola Netto, J.M.; de Oliveira, G.C.; van Schooten, W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol. Sci. 2018, 39, 424–436. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Peclat, T.R.; Shi, B.; Varga, J.; Chini, E.N. The NADase enzyme CD38: An emerging pharmacological target for systemic sclerosis, systemic lupus erythematosus and rheumatoid arthritis. Curr. Opin. Rheumatol. 2020, 32, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.W.; Borgnakke, W.S. Periodontal disease: Associations with diabetes, glycemic control and complications. Oral Dis. 2008, 14, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Preshaw, P.M.; Bissett, S.M. Periodontitis: Oral complication of diabetes. Endocrinol. Metab. Clin. N. Am. 2013, 42, 849–867. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, H.; Ren, H.; Hu, X.; Wang, X.; Han, C. AGEs and chronic subclinical inflammation in diabetes: Disorders of immune system. Diabetes/Metab. Res. Rev. 2015, 31, 127–137. [Google Scholar] [CrossRef]
- Passarelli, M.; Machado, U.F.F. AGEs-Induced and Endoplasmic Reticulum Stress/Inflammation-Mediated Regulation of GLUT4 Expression and Atherogenesis in Diabetes Mellitus. Cells 2021, 11, 104. [Google Scholar] [CrossRef]
- Roberts, J.S.; Atanasova, K.R.; Lee, J.; Diamond, G.; Deguzman, J.; Hee Choi, C.; Yilmaz, Ö. Opportunistic Pathogen Porphyromonas gingivalis Modulates Danger Signal ATP-Mediated Antibacterial NOX2 Pathways in Primary Epithelial Cells. Front. Cell. Infect. Microbiol. 2017, 7, 291. [Google Scholar] [CrossRef]
- Lee, J.S.; Spooner, R.; Chowdhury, N.; Pandey, V.; Wellslager, B.; Atanasova, K.R.; Evans, Z.; Yilmaz, Ö. In Situ Intraepithelial Localizations of Opportunistic Pathogens, Porphyromonas gingivalis and Filifactor alocis, in Human Gingiva. Curr. Res. Microb. Sci. 2020, 1, 7–17. [Google Scholar] [CrossRef]
- Spooner, R.; Weigel, K.M.; Harrison, P.L.; Lee, K.; Cangelosi, G.A.; Yilmaz, Ö. In Situ Anabolic Activity of Periodontal Pathogens Porphyromonas gingivalis and Filifactor alocis in Chronic Periodontitis. Sci. Rep. 2016, 6, 33638. [Google Scholar] [CrossRef]
- Raja, M.; Ummer, F.; Dhivakar, C.P. Aggregatibacter actinomycetemcomitans—A tooth killer? J. Clin. Diagn Res. 2014, 8, ZE13–ZE16. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, J.; Guo, H.; Pan, Y.; Zhang, Y.; Zhao, W.; Li, X.; Li, Y. Recombinant fimbriae protein of Porphyromonas gingivalis induces an inflammatory response via the TLR4/NF-κB signaling pathway in human peripheral blood mononuclear cells. Int. J. Mol. Med. 2019, 43, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, C.P.; Laxton, R.C.; Patel, K.; Ye, S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2275–2281. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, S.; Nemoto, E.; Ogawa, T.; Shimauchi, H. Porphyromonas gingivalis fimbriae induce unique dendritic cell subsets via Toll-like receptor 2. J. Periodontal Res. 2009, 44, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, Y.; Cui, Q.; Xu, J.; Tang, Z.; Wang, Y.; He, C.; Wang, X. Toll-like receptor 4 plays a key role in advanced glycation end products-induced M1 macrophage polarization. Biochem. Biophys. Res. Commun. 2020, 531, 602–608. [Google Scholar] [CrossRef]
- Lima, H.R.; Gelani, V.; Fernandes, A.P.; Gasparoto, T.H.; Torres, S.A.; Santos, C.F.; Garlet, G.P.; da Silva, J.S.; Campanelli, A.P. The essential role of toll like receptor-4 in the control of Aggregatibacter actinomycetemcomitans infection in mice. J. Clin. Periodontol. 2010, 37, 248–254. [Google Scholar] [CrossRef]
- Vaananen, H.K.; Laitala-Leinonen, T. Osteoclast lineage and function. Arch. Biochem. Biophys. 2008, 473, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar]
- Nemeth, K.; Schoppet, M.; Al-Fakhri, N.; Helas, S.; Jessberger, R.; Hofbauer, L.C.; Goettsch, C. The role of osteoclast-associated receptor in osteoimmunology. J. Immunol. 2011, 186, 13–18. [Google Scholar] [CrossRef]
- Matsuo, K.; Irie, N. Transcription factors in osteoclast differentiation. Nihon Rinsho. Jpn. J. Clin. Med. 2005, 63, 1541–1546. [Google Scholar]
- Miyamoto, T. Regulators of osteoclast differentiation and cell-cell fusion. Keio J. Med. 2011, 60, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Yu, H. Sphingosine-1-Phosphate Receptor 2 Regulates Proinflammatory Cytokine Production and Osteoclastogenesis. PLoS ONE 2016, 11, e0156303. [Google Scholar] [CrossRef] [PubMed]
- Georgess, D.; Machuca-Gayet, I.; Blangy, A.; Jurdic, P. Podosome organization drives osteoclast-mediated bone resorption. Cell Adh. Migr. 2014, 8, 191–204. [Google Scholar] [CrossRef]
- Hsu, L.C.; Reddy, S.V.; Yilmaz, O.; Yu, H. Sphingosine-1-Phosphate Receptor 2 Controls Podosome Components Induced by RANKL Affecting Osteoclastogenesis and Bone Resorption. Cells 2019, 8, 17. [Google Scholar] [CrossRef]
- Duong, L.T.; Lakkakorpi, P.T.; Nakamura, I.; Machwate, M.; Nagy, R.M.; Rodan, G.A. PYK2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of alpha(v)beta3 integrin, and phosphorylated by src kinase. J. Clin. Investig. 1998, 102, 881–892. [Google Scholar] [CrossRef]
- Kim, J.H.; Stewart, T.P.; Soltani-Bejnood, M.; Wang, L.; Fortuna, J.M.; Mostafa, O.A.; Moustaid-Moussa, N.; Shoieb, A.M.; McEntee, M.F.; Wang, Y.; et al. Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J. Endocrinol. 2006, 191, 437–446. [Google Scholar] [CrossRef]
- Leiter, E.H.; Strobel, M.; O’Neill, A.; Schultz, D.; Schile, A.; Reifsnyder, P.C. Comparison of Two New Mouse Models of Polygenic Type 2 Diabetes at the Jackson Laboratory, NONcNZO10Lt/J and TALLYHO/JngJ. J. Diabetes Res. 2013, 2013, 165327. [Google Scholar] [CrossRef] [PubMed]
- Manzanero, S. Generation of mouse bone marrow-derived macrophages. Methods Mol. Biol. 2012, 844, 177–181. [Google Scholar] [PubMed]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 2008, 83, 14.1.1–14.1.14. [Google Scholar]
- Spooner, R.; DeGuzman, J.; Lee, K.L.; Yilmaz, O. Danger signal adenosine via adenosine 2a receptor stimulates growth of Porphyromonas gingivalis in primary gingival epithelial cells. Mol. Oral Microbiol. 2014, 29, 67–78. [Google Scholar] [CrossRef]
- Wellslager, B.; Roberts, J.; Chowdhury, N.; Madan, L.; Orellana, E.; Yilmaz, Ö. Porphyromonas gingivalis activates Heat-Shock-Protein 27 to drive a LC3C-specific probacterial form of select autophagy that is redox sensitive for intracellular bacterial survival in human gingival mucosa. bioRxiv 2024. [Google Scholar] [CrossRef]
- Roboon, J.; Hattori, T.; Ishii, H.; Takarada-Iemata, M.; Nguyen, D.T.; Heer, C.D.; O’Meally, D.; Brenner, C.; Yamamoto, Y.; Okamoto, H.; et al. Inhibition of CD38 and supplementation of nicotinamide riboside ameliorate lipopolysaccharide-induced microglial and astrocytic neuroinflammation by increasing NAD. J. Neurochem. 2021, 158, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Burns, E.; Bachrach, G.; Shapira, L.; Nussbaum, G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J. Immunol. 2006, 177, 8296–8300. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Kaufman, J.L.; Hofmeister, C.C.; Joseph, N.S.; Heffner, T.L.; Gupta, V.A.; Sullivan, H.C.; Neish, A.S.; Dhodapkar, M.V.; Lonial, S. Daratumumab in multiple myeloma. Cancer 2019, 125, 2364–2382. [Google Scholar] [CrossRef]
- Costa, F.; Toscani, D.; Chillemi, A.; Quarona, V.; Bolzoni, M.; Marchica, V.; Vescovini, R.; Mancini, C.; Martella, E.; Campanini, N.; et al. Expression of CD38 in myeloma bone niche: A rational basis for the use of anti-CD38 immunotherapy to inhibit osteoclast formation. Oncotarget 2017, 8, 56598–56611. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Iqbal, J.; Dolgilevich, S.; Yuen, T.; Wu, X.B.; Moonga, B.S.; Adebanjo, O.A.; Bevis, P.J.; Lund, F.; Huang, C.L.; et al. Disordered osteoclast formation and function in a CD38 (ADP-ribosyl cyclase)-deficient mouse establishes an essential role for CD38 in bone resorption. FASEB J. 2003, 17, 369–375. [Google Scholar] [CrossRef]
- Ilchovska, D.D.; Barrow, D.M. An Overview of the NF-kB mechanism of pathophysiology in rheumatoid arthritis, investigation of the NF-kB ligand RANKL and related nutritional interventions. Autoimmun. Rev. 2021, 20, 102741. [Google Scholar] [CrossRef]
- Lee, K.; Chung, Y.H.; Ahn, H.; Kim, H.; Rho, J.; Jeong, D. Selective Regulation of MAPK Signaling Mediates RANKL-dependent Osteoclast Differentiation. Int. J. Biol. Sci. 2016, 12, 235–245. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Verkleij, C.P.M.; Bosman, P.W.C.; Zweegman, S.; Otten, H.; van de Donk, N. CD38-targeted therapy with daratumumab reduces autoantibody levels in multiple myeloma patients. J. Transl. Autoimmun. 2019, 2, 100022. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Song, X.; Song, Y.; He, G.; Fang, K.; Chang, X. Compound 78c exerts a therapeutic effect on collagen-induced arthritis and rheumatoid arthritis. Clin. Exp. Rheumatol. 2023, 41, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018, 27, 1081–1095.e1010. [Google Scholar] [CrossRef] [PubMed]
- Peclat, T.R.; Thompson, K.L.; Warner, G.M.; Chini, C.C.S.; Tarragó, M.G.; Mazdeh, D.Z.; Zhang, C.; Zavala-Solorio, J.; Kolumam, G.; Liang Wong, Y.; et al. CD38 inhibitor 78c increases mice lifespan and healthspan in a model of chronological aging. Aging Cell 2022, 21, e13589. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lory, W.; Chowdhury, N.; Wellslager, B.; Pandruvada, S.; Huang, Y.; Yilmaz, Ö.; Yu, H. CD38 Inhibitor 78c Attenuates Pro-Inflammatory Cytokine Expression and Osteoclastogenesis in Macrophages. Cells 2024, 13, 1971. https://doi.org/10.3390/cells13231971
Lory W, Chowdhury N, Wellslager B, Pandruvada S, Huang Y, Yilmaz Ö, Yu H. CD38 Inhibitor 78c Attenuates Pro-Inflammatory Cytokine Expression and Osteoclastogenesis in Macrophages. Cells. 2024; 13(23):1971. https://doi.org/10.3390/cells13231971
Chicago/Turabian StyleLory, William, Nityananda Chowdhury, Bridgette Wellslager, Subramanya Pandruvada, Yan Huang, Özlem Yilmaz, and Hong Yu. 2024. "CD38 Inhibitor 78c Attenuates Pro-Inflammatory Cytokine Expression and Osteoclastogenesis in Macrophages" Cells 13, no. 23: 1971. https://doi.org/10.3390/cells13231971
APA StyleLory, W., Chowdhury, N., Wellslager, B., Pandruvada, S., Huang, Y., Yilmaz, Ö., & Yu, H. (2024). CD38 Inhibitor 78c Attenuates Pro-Inflammatory Cytokine Expression and Osteoclastogenesis in Macrophages. Cells, 13(23), 1971. https://doi.org/10.3390/cells13231971