
Metabolic Reprogramming Induced by Aging Modifies the Tumor Microenvironment

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Immune Infiltration Analysis by ssGSEA
2.3. DNAm Age Calculation
2.4. RNA Age Calculation
2.5. Trajectory Analysis Based on Bulk Sequencing
2.6. Single-Cell RNA-Seq Analysis
2.6.1. Data Preprocessing, Cell Clustering, and Annotation
2.6.2. Integration of Multiple Single-Cell Transcriptome Data Cohorts across Samples
2.6.3. Trajectory Analysis to Infer T Cell Fates in the Aging Process
2.6.4. Cell Type Enrichment of Various Age-Based Subgroups
2.6.5. Detecting Malignant Cell Based on Genomic Copy Number Inferring
2.7. Proteomics Analysis
2.8. Statistical Analysis
3. Results
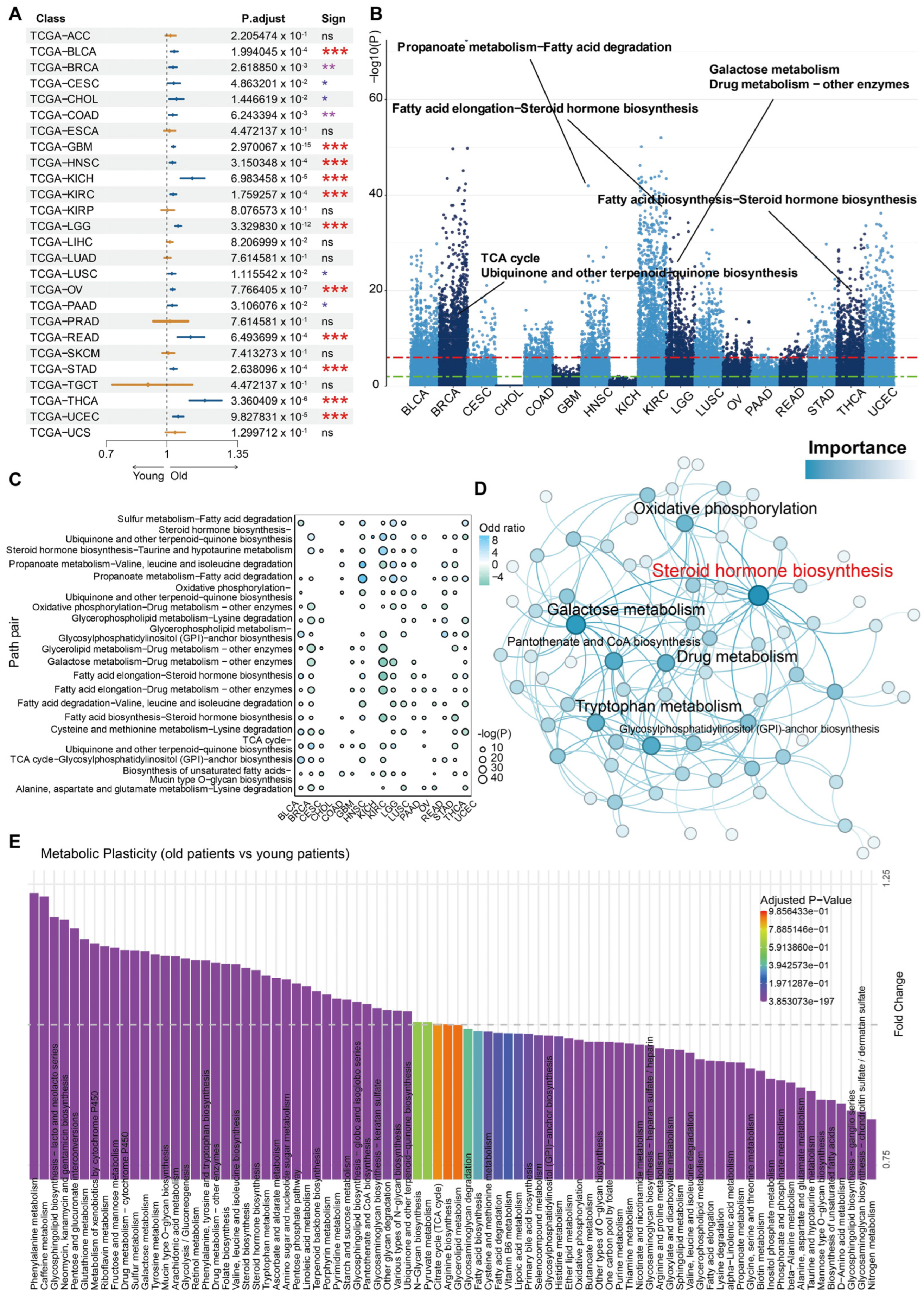
3.1. Aging-Related Metabolic Alterations: Insights from Pan-Cancer Bulk Sequencing Transcriptome Analysis
3.2. The Metabolic Switch Correlates to Molecular Features of Senescence in Aging-Related Cancers
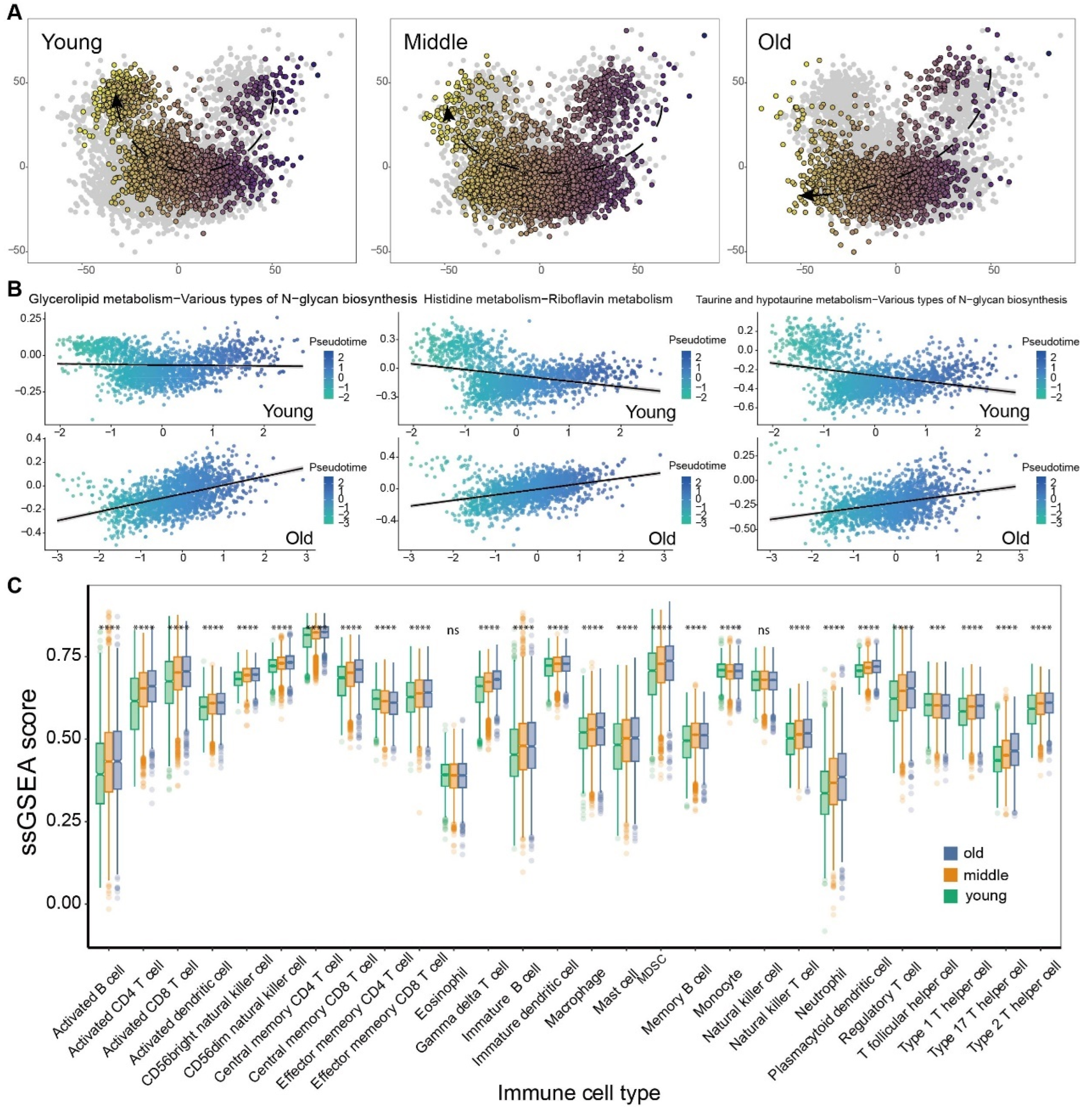
3.3. The Landscape of Tumor Microenvironment Showed Distinct Discrepancy among Aged-Based Subgroups
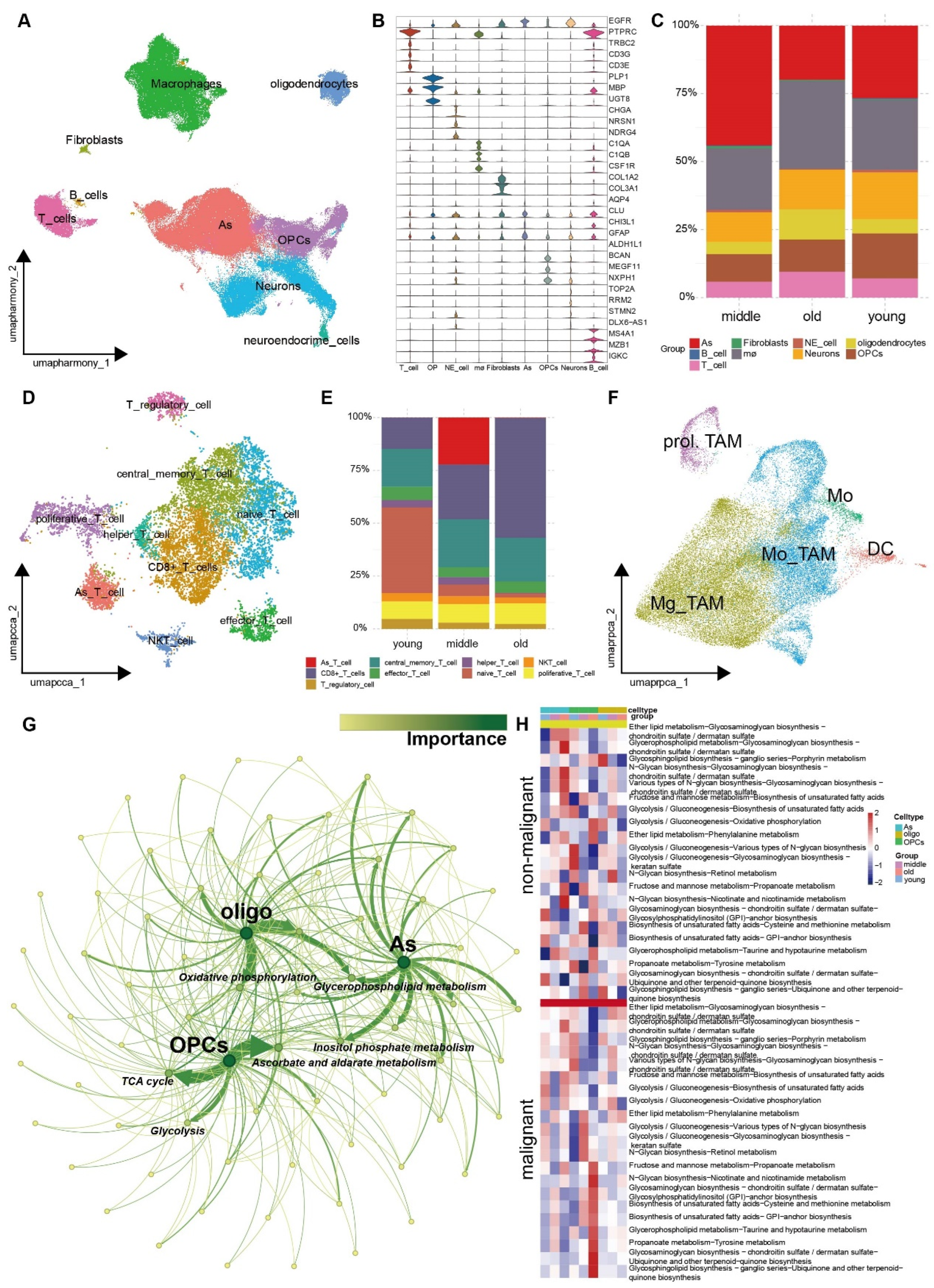
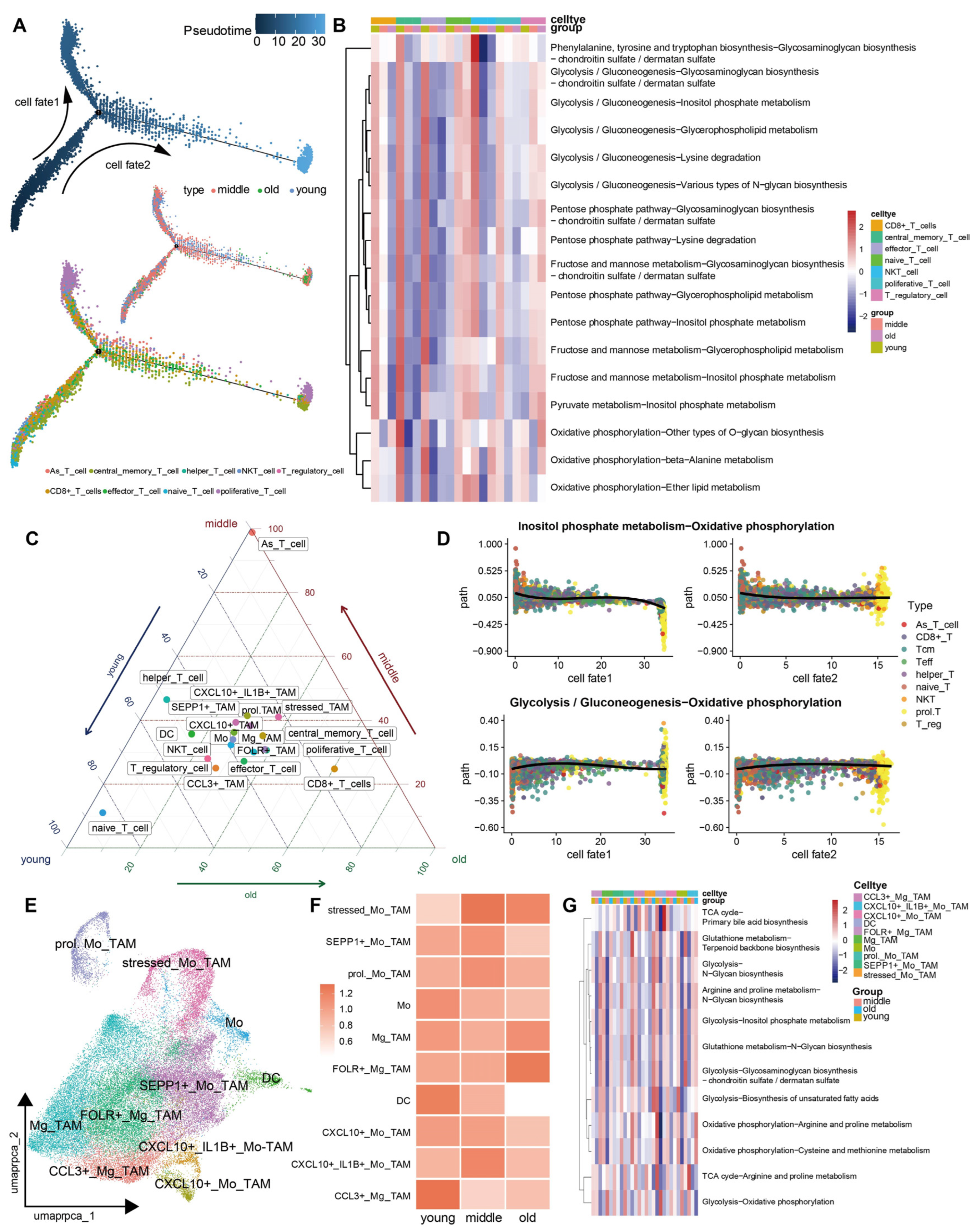
3.4. The Metabolic Reprogramming of Various Cell Types within the TME Exhibits Increased Heterogeneity as the Aging Progresses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- White, M.C.; Holman, D.M.; Goodman, R.A.; Richardson, L.C. Cancer Risk Among Older Adults: Time for Cancer Prevention to Go Silver. Gerontologist 2019, 59, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Lauper, J.M.; Krause, A.; Vaughan, T.L.; Monnat, R.J. Spectrum and Risk of Neoplasia in Werner Syndrome: A Systematic Review. PLoS ONE 2013, 8, e59709. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Georgiev, P.; Ringel, A.E.; Sharpe, A.H.; Haigis, M.C. Age-associated remodeling of T cell immunity and metabolism. Cell Metab. 2023, 35, 36–55. [Google Scholar] [CrossRef]
- Weistuch, C.; Mujica-Parodi, L.R.; Razban, R.M.; Antal, B.; van Nieuwenhuizen, H.; Amgalan, A.; Dill, K.A. Metabolism modulates network synchrony in the aging brain. Proc. Natl. Acad. Sci. USA 2021, 118, e2025727118. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Drapela, S.; Ilter, D.; Gomes, A.P. Metabolic reprogramming: A bridge between aging and tumorigenesis. Mol. Oncol. 2022, 16, 3295–3318. [Google Scholar] [CrossRef]
- Nakajima, Y.; Chamoto, K.; Oura, T.; Honjo, T. Critical role of the CD44 (low) CD62L (low) CD8 (+) T cell subset in restoring antitumor immunity in aged mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2103730118. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Notarangelo, G.; Ghergurovich, J.M.; Paulo, J.A.; Sage, P.T.; Santos, D.; Satterstrom, F.K.; Gygi, S.P.; Rabinowitz, J.D.; Sharpe, A.H.; et al. Defective respiration and one-carbon metabolism contribute to impaired naive T cell activation in aged mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13347–13352. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Deng, M.; Wang, Z.; Huang, C. MMP3C: An in-silico framework to depict cancer metabolic plasticity using gene expression profiles. Brief. Bioinform. 2023, 25, bbad471. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Deng, M.; Leng, D.; Sun, B.; Zheng, P.; Zhang, X.D. MIRS: An AI scoring system for predicting the prognosis and therapy of breast cancer. iScience 2023, 26, 108322. [Google Scholar] [CrossRef]
- Feng, Y.W.; Chen, X.Y.; Zhang, X.D.; Huang, C. Metabolic Pathway Pairwise-Based Signature as a Potential Non-Invasive Diagnostic Marker in Alzheimer’s Disease Patients. Genes 2023, 14, 1285. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Pelegí-Sisó, D.; de Prado, P.; Ronkainen, J.; Bustamante, M.; González, J.R. A Bioconductor package to estimate DNA methylation age. Bioinformatics 2021, 37, 1759–1760. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2015, 16, 3156. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Kuan, P.F. RNAAgeCalc: A multi-tissue transcriptional age calculator. PLoS ONE 2020, 15, e0237006. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.R.; Yau, C. Uncovering pseudotemporal trajectories with covariates from single cell and bulk expression data. Nat. Commun. 2018, 9, 2442. [Google Scholar] [CrossRef]
- R Core Team, R. R: A Language and Environment for Statistical Computing, Version 2.7.0; The R Development Core Team: Vienna, Austria, 2013. [Google Scholar]
- Hao, Y.H.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.W.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Germain, P.L.; Lun, A.; Garcia Meixide, C.; Macnair, W.; Robinson, M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Research 2021, 10, 979. [Google Scholar] [CrossRef]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.A.; Trapnell, C. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.; Verma, A.; Marderstein, A.R.; White, J.; Bhinder, B.; Medina, J.S.G.; Elemento, O. Pan-cancer analysis reveals molecular patterns associated with age. Cell Rep. 2021, 37, 110100. [Google Scholar] [CrossRef]
- Vuononvirta, J.; Marelli-Berg, F.M.; Poobalasingam, T. Metabolic regulation of T lymphocyte motility and migration. Mol. Asp. Med. 2021, 77, 100888. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Bell, A.; Lauing, K.L.; Ladomersky, E.; Zhai, L.J.; Penco-Campillo, M.; Shah, Y.J.; Mauer, E.; Xiu, J.; Nicolaides, T.; et al. Advanced Age in Humans and Mouse Models of Glioblastoma Show Decreased Survival from Extratumoral Influence. Clin. Cancer Res. 2023, 29, 4973–4989. [Google Scholar] [CrossRef]
- Zhao, T.; Zeng, J.; Xu, Y.; Su, Z.; Chong, Y.; Ling, T.; Xu, H.; Shi, H.; Zhu, M.; Mo, Q.; et al. Chitinase-3 like-protein-1 promotes glioma progression via the NF-kappaB signaling pathway and tumor microenvironment reprogramming. Theranostics 2022, 12, 6989–7008. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.X.; Li, T.Y.; Xu, Y.Q.; Zhang, X.X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.Q.; Yang, K.Y.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Whitaker-Menezes, D.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Witkiewicz, A.K.; Vander Heiden, M.G.; Migneco, G.; Chiavarina, B.; et al. The reverse Warburg effect: Glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 2010, 9, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Kang, C. Stressing the Role of CCL3 in Reversing the Immunosuppressive Microenvironment in Gliomas. Cancer Immunol. Res. 2024, 12, 514. [Google Scholar] [CrossRef]
- Kodama, M.; Nakayama, K.I. A second Warburg-like effect in cancer metabolism: The metabolic shift of glutamine-derived nitrogen A shift in glutamine-derived nitrogen metabolism from glutaminolysis to de novo nucleotide biosynthesis contributes to malignant evolution of cancer. Bioessays 2020, 42, e2000169. [Google Scholar] [CrossRef]
- Feng, Z.; Hanson, R.W.; Berger, N.A.; Trubitsyn, A. Reprogramming of energy metabolism as a driver of aging. Oncotarget 2016, 7, 15410–15420. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Wang, Z.; Zhu, B.; Deng, M.; Qiu, J.; Feng, Y.; Ding, N.; Huang, C. Metabolic Reprogramming Induced by Aging Modifies the Tumor Microenvironment. Cells 2024, 13, 1721. https://doi.org/10.3390/cells13201721
Chen X, Wang Z, Zhu B, Deng M, Qiu J, Feng Y, Ding N, Huang C. Metabolic Reprogramming Induced by Aging Modifies the Tumor Microenvironment. Cells. 2024; 13(20):1721. https://doi.org/10.3390/cells13201721
Chicago/Turabian StyleChen, Xingyu, Zihan Wang, Bo Zhu, Min Deng, Jiayue Qiu, Yunwen Feng, Ning Ding, and Chen Huang. 2024. "Metabolic Reprogramming Induced by Aging Modifies the Tumor Microenvironment" Cells 13, no. 20: 1721. https://doi.org/10.3390/cells13201721
APA StyleChen, X., Wang, Z., Zhu, B., Deng, M., Qiu, J., Feng, Y., Ding, N., & Huang, C. (2024). Metabolic Reprogramming Induced by Aging Modifies the Tumor Microenvironment. Cells, 13(20), 1721. https://doi.org/10.3390/cells13201721