
CTRP13-Mediated Effects on Endothelial Cell Function and Their Potential Role in Obesity


Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Model
2.2. Rat Model
2.3. Patient Samples
2.4. Cell Isolation and Culture
2.5. RNA Isolation and qPCR
2.6. Western Blotting
2.7. Isolation of Recombinant CTRP13
2.8. Immunocytochemistry and Confocal Microscopy
2.9. Serum Analyses
2.10. Migration Assay
2.11. Sprouting Assay
2.12. Cell Cycle Analysis
2.13. Adenovirus Generation and Utilization
2.14. BrdU Incorporation
2.15. Antibodies and Chemicals
2.16. Statistical Analysis
3. Results
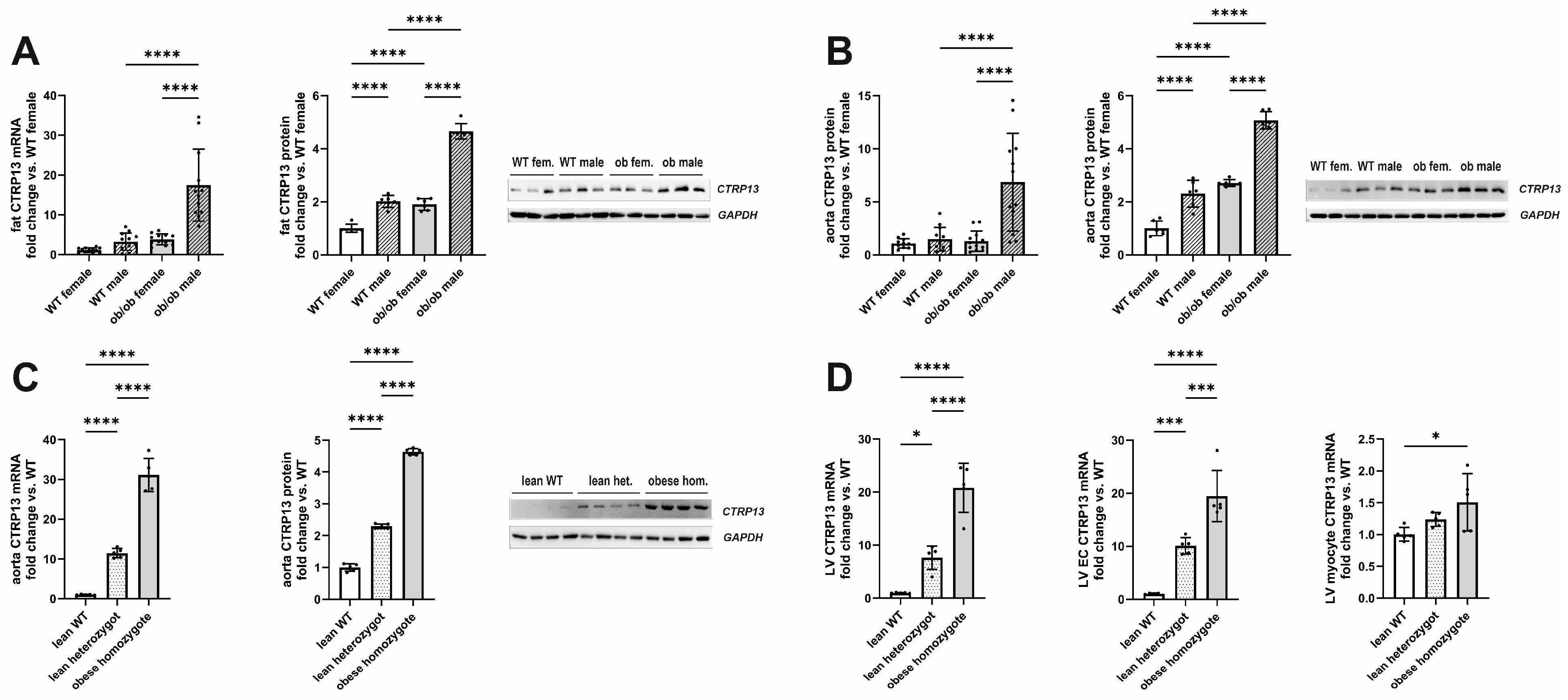
3.1. Impact of Obesity on CTRP13 Expression
3.1.1. Impact of Obesity on CTRP13 Expression in Mice and Rats
3.1.2. Impact of Obesity on CTRP13 in Patient Samples
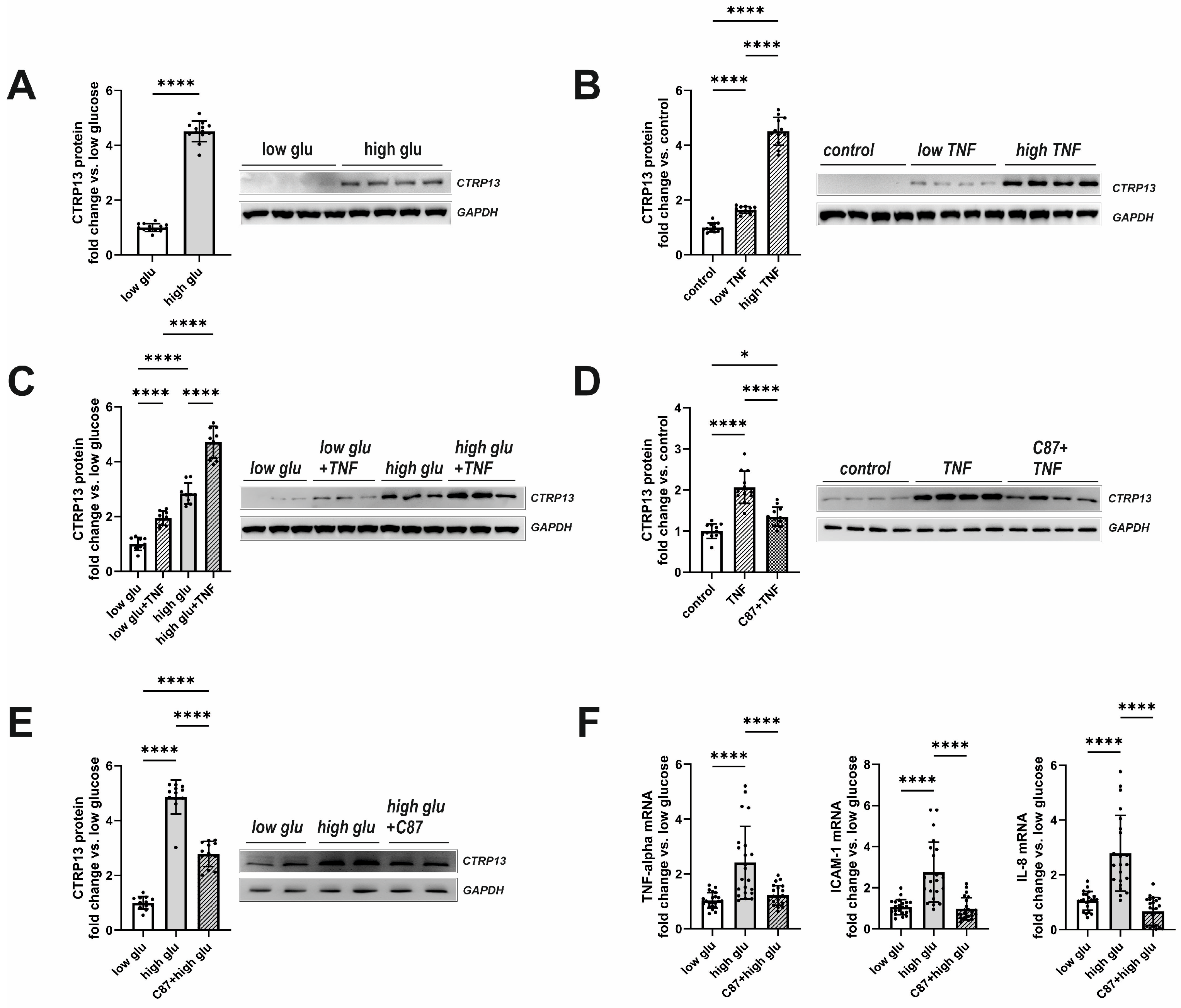
3.2. Mechanisms Involved in Obesity-Associated CTRP13 Induction in Ecs
3.3. Functional Impact of CTRP13 on Endothelial Cells
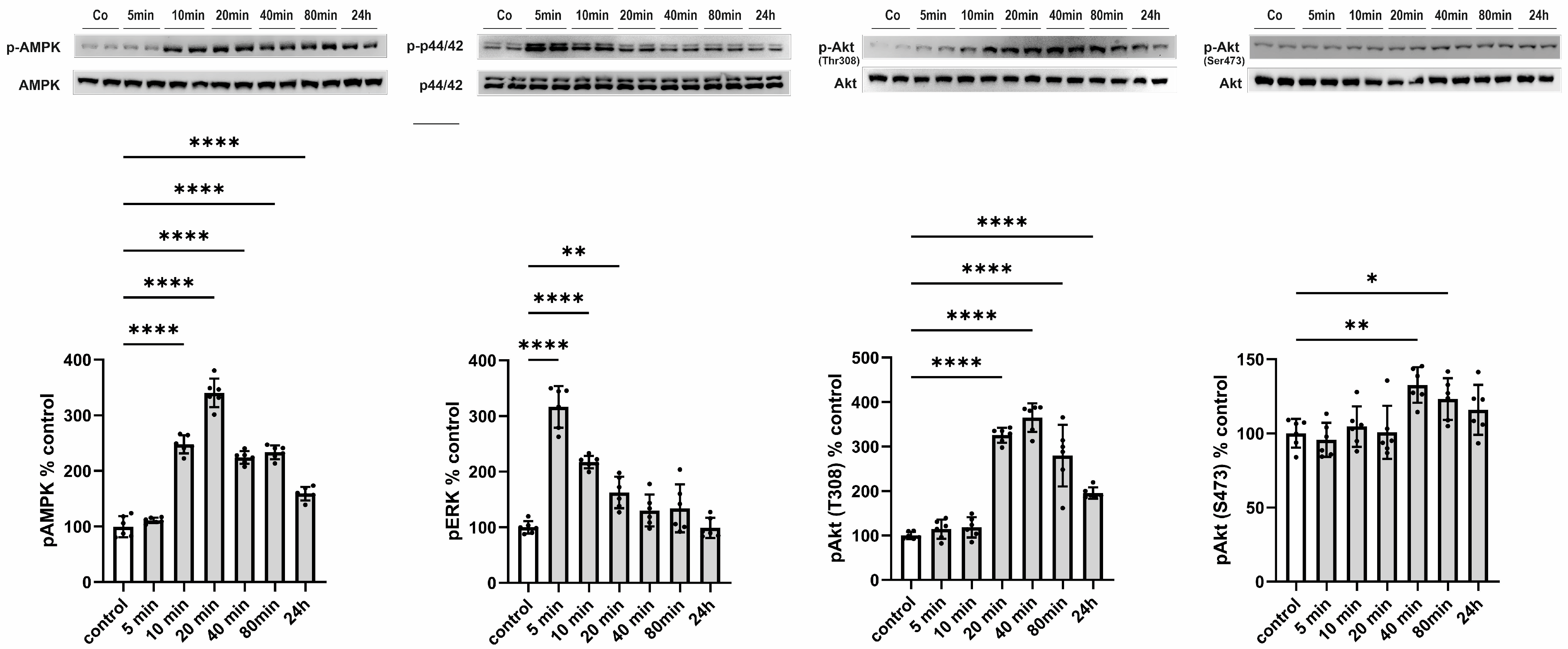
3.3.1. Signal Transduction Induced by CTRP13
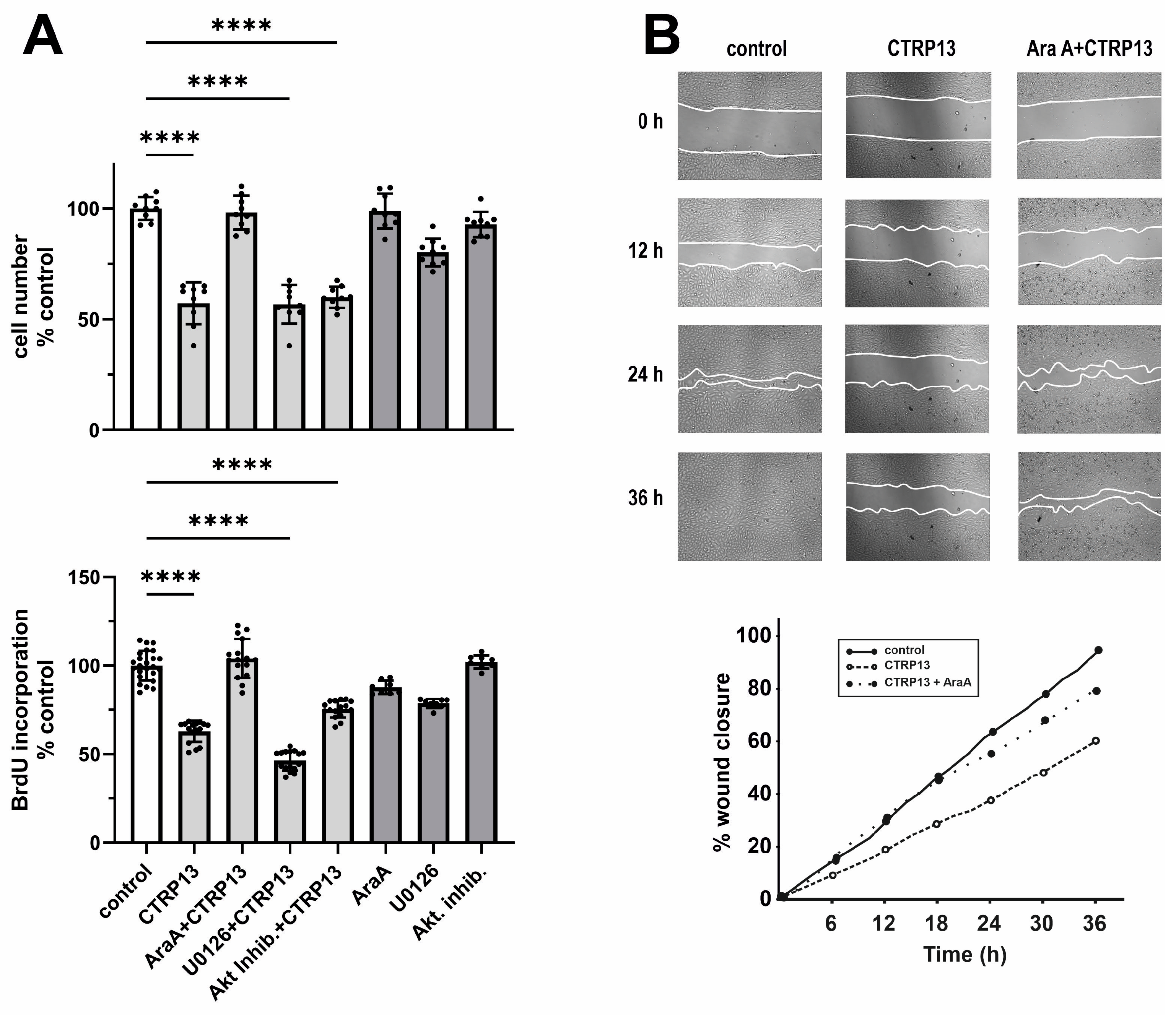
3.3.2. EC Proliferation and Migration
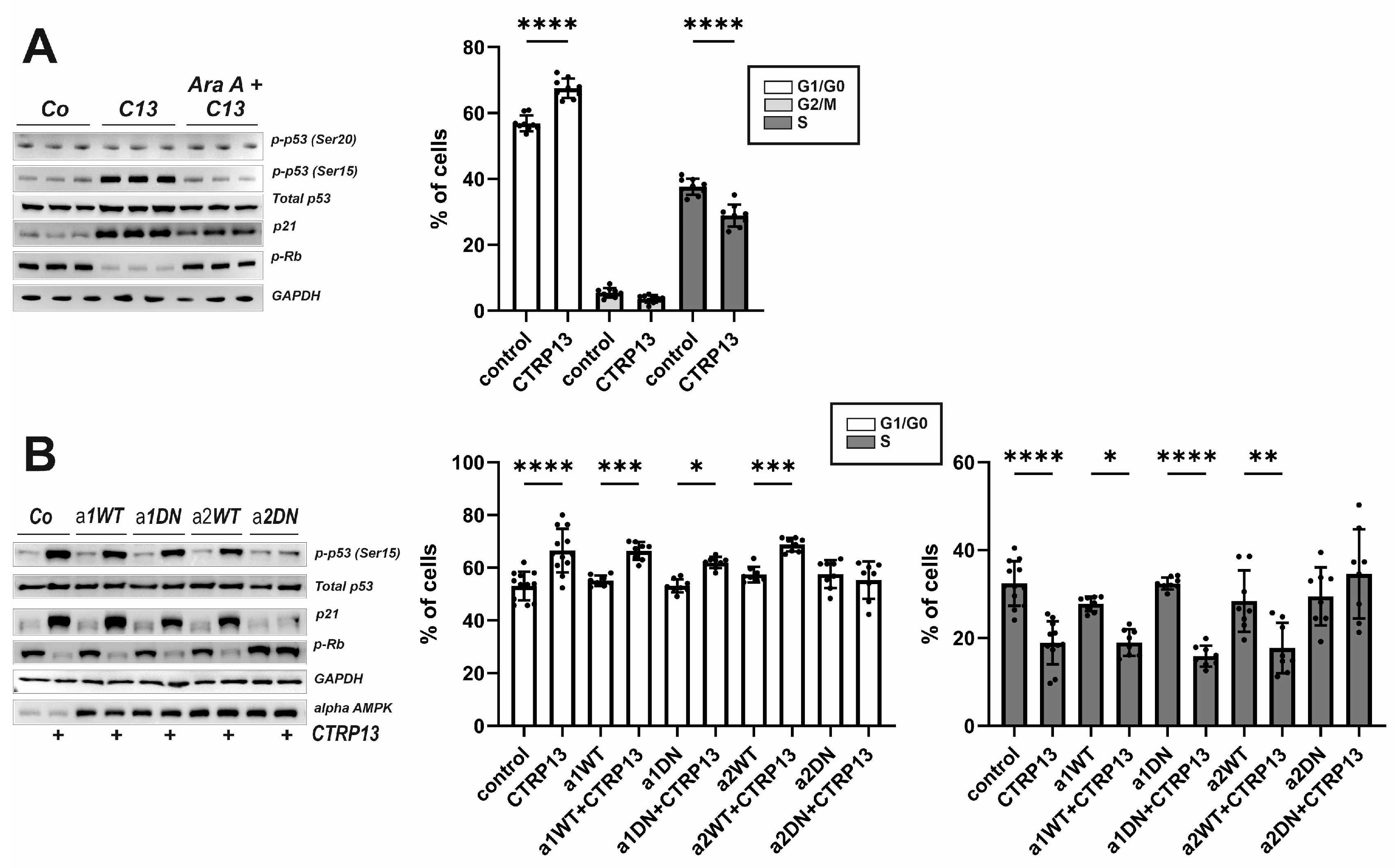
3.3.3. EC Cell Cycle Progression and Role of AMPK Isoforms
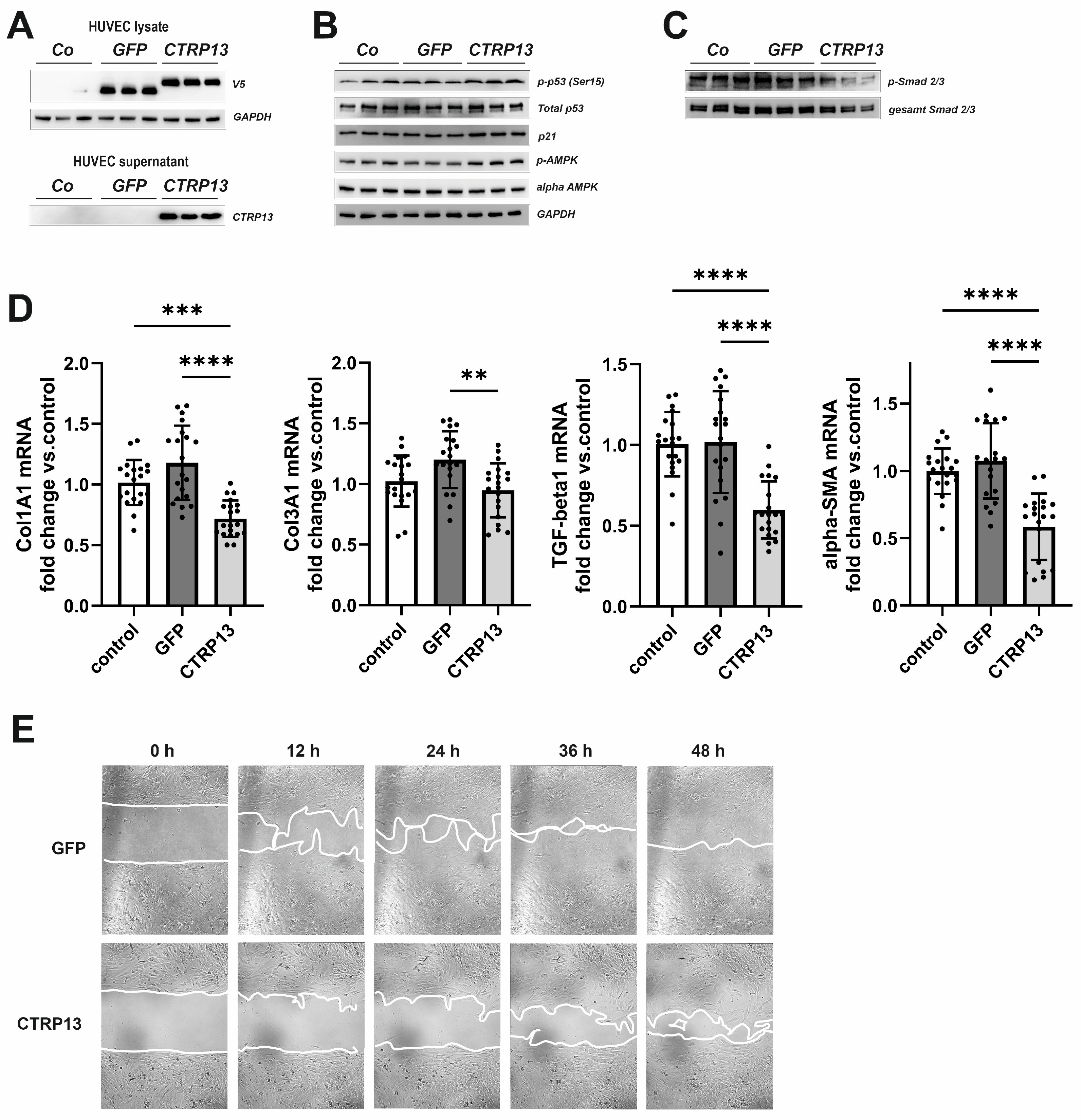
3.4. Functional Impact of Endothelial CTRP13 on Human Fibroblasts
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Models Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Qiu, S.; Yang, G.; Wu, Q. Adiponectin and metabolic cardiovascular diseases: Therapeutic opportunities and challenges. Genes Dis. 2023, 10, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kihara, S.; Arita, Y.; Maeda, K.; Kuriyama, H.; Okamoto, Y.; Hotta, K.; Nishida, M.; Takahashi, M.; Nakamura, T.; et al. Novel modulator for endothelial adhesion molecules: Adipocyte-derived plasma protein adiponectin. Circulation 1999, 100, 2473–2476. [Google Scholar] [CrossRef] [PubMed]
- Maahs, D.M.; Ogden, L.G.; Kinney, G.L.; Wadwa, P.; Snell-Bergeon, J.K.; Dabelea, D.; Hokanson, J.E.; Ehrlich, J.; Eckel, R.H.; Rewers, M. Low plasma adiponectin levels predict progression of coronary artery calcification. Circulation 2005, 111, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Pischon, T.; Girman, C.J.; Hotamisligil, G.S.; Rifai, N.; Hu, F.B.; Rimm, E.B. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004, 291, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Davey Smith, G.; Ebrahim, S.; Thompson, C.; Sattar, N. Plasma adiponectin levels are associated with insulin resistance, but do not predict future risk of coronary heart disease in women. J. Clin. Endocrinol. Metab. 2005, 90, 5677–5683. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, S.; Pedersen, S.H.; Mogelvang, R.; Bjerre, M.; Frystyk, J.; Flyvbjerg, A.; Galatius, S.; Jensen, J.S. Usefulness of adiponectin as a predictor of all cause mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Am. J. Cardiol. 2012, 109, 492–496. [Google Scholar] [CrossRef]
- Sattar, N.; Wannamethee, G.; Sarwar, N.; Tchernova, J.; Cherry, L.; Wallace, A.M.; Danesh, J.; Whincup, P.H. Adiponectin and coronary heart disease: A prospective study and meta-analysis. Circulation 2006, 114, 623–629. [Google Scholar] [CrossRef]
- Schanbacher, C.; Hermanns, H.M.; Lorenz, K.; Wajant, H.; Lang, I. Complement 1q/Tumor Necrosis Factor-Related Proteins (CTRPs): Structure, Receptors and Signaling. Biomedicines 2023, 11, 559. [Google Scholar] [CrossRef]
- Li, L.; Aslam, M.; Siegler, B.H.; Niemann, B.; Rohrbach, S. Comparative Analysis of CTRP-Mediated Effects on Cardiomyocyte Glucose Metabolism: Cross Talk between AMPK and Akt Signaling Pathway. Cells 2021, 10, 905. [Google Scholar] [CrossRef]
- Niemann, B.; Li, L.; Siegler, D.; Siegler, B.H.; Knapp, F.; Hanna, J.; Aslam, M.; Kracht, M.; Schulz, R.; Rohrbach, S. CTRP9 Mediates Protective Effects in Cardiomyocytes via AMPK- and Adiponectin Receptor-Mediated Induction of Anti-Oxidant Response. Cells 2020, 9, 1229. [Google Scholar] [CrossRef]
- Byerly, M.S.; Swanson, R.; Wei, Z.; Seldin, M.M.; McCulloh, P.S.; Wong, G.W. A central role for C1q/TNF-related protein 13 (CTRP13) in modulating food intake and body weight. PLoS ONE 2013, 8, e62862. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Sarver, D.C.; Saqib, M.; Zhou, M.; Aja, S.; Seldin, M.M.; Wong, G.W. CTRP13 ablation improves systemic glucose and lipid metabolism. Mol. Metab. 2023, 78, 101824. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Fan, W.; Sun, L. A Review of the Relationship Between CTRP Family and Coronary Artery Disease. Curr. Atheroscler. Rep. 2020, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, W.; Liang, M.; Huang, D.; Huang, K. CTRP13 inhibits atherosclerosis via autophagy-lysosome-dependent degradation of CD36. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chao, Y.; Xu, W.; Liang, M.; Deng, S.; Zhang, D.; Huang, K. CTRP13 Preserves Endothelial Function by Targeting GTP Cyclohydrolase 1 in Diabetes. Diabetes 2020, 69, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Chao, Y.; Zhang, F.; Wang, C. CTRP13 attenuates vascular calcification by regulating Runx2. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 9627–9637. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chao, Y.; Liang, M.; Huang, K.; Wang, C. CTRP13 Mitigates Abdominal Aortic Aneurysm Formation via NAMPT1. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 324–337. [Google Scholar] [CrossRef]
- Gunduz, D.; Klewer, M.; Bauer, P.; Tanislav, C.; Sedding, D.; Rohrbach, S.; Schulz, R.; Aslam, M. Compound C inhibits in vitro angiogenesis and ameliorates thrombin-induced endothelial barrier failure. Eur. J. Pharmacol. 2015, 768, 165–172. [Google Scholar] [CrossRef]
- Turban, S.; Stretton, C.; Drouin, O.; Green, C.J.; Watson, M.L.; Gray, A.; Ross, F.; Lantier, L.; Viollet, B.; Hardie, D.G.; et al. Defining the contribution of AMP-activated protein kinase (AMPK) and protein kinase C (PKC) in regulation of glucose uptake by metformin in skeletal muscle cells. J. Biol. Chem. 2012, 287, 20088–20099. [Google Scholar] [CrossRef]
- Wei, Z.; Peterson, J.M.; Wong, G.W. Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): Activation of AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J. Biol. Chem. 2011, 286, 15652–15665. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K.; Liaqat, A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J. Cell. Biochem. 2018, 119, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Ma, L.; Gong, H.; Zhu, H.; Ji, Q.; Su, P.; Liu, P.; Cao, S.; Yao, J.; Jiang, L.; Han, M.; et al. A novel small-molecule tumor necrosis factor alpha inhibitor attenuates inflammation in a hepatitis mouse model. J. Biol. Chem. 2014, 289, 12457–12466. [Google Scholar] [CrossRef] [PubMed]
- Herbert, S.P.; Stainier, D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. New Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef]
- Chen, H.; Montagnani, M.; Funahashi, T.; Shimomura, I.; Quon, M.J. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J. Biol. Chem. 2003, 278, 45021–45026. [Google Scholar] [CrossRef]
- Ouedraogo, R.; Gong, Y.; Berzins, B.; Wu, X.; Mahadev, K.; Hough, K.; Chan, L.; Goldstein, B.J.; Scalia, R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J. Clin. Investig. 2007, 117, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Szmitko, P.E.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Chan, L.; Al-Omran, M.; Teoh, H.; et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H656–H663. [Google Scholar] [CrossRef]
- Han, S.H.; Sakuma, I.; Shin, E.K.; Koh, K.K. Antiatherosclerotic and anti-insulin resistance effects of adiponectin: Basic and clinical studies. Prog. Cardiovasc. Dis. 2009, 52, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Shanaki, M.; Fadaei, R.; Moradi, N.; Emamgholipour, S.; Poustchi, H. The Circulating CTRP13 in Type 2 Diabetes and Non-Alcoholic Fatty Liver Patients. PLoS ONE 2016, 11, e0168082. [Google Scholar] [CrossRef] [PubMed]
- Afrookhteh, A.; Emamgholipour, S.; Alipoor, B.; Moradi, N.; Meshkani, R.; Nasli-Esfahani, E.; Rahimipour, A.; Shanaki, M. The Circulating Levels of Complement-C1q/TNF-Related Protein 13 (CTRP13) in Patients with Type 2 Diabetes and its Association with Insulin Resistance. Clin. Lab. 2017, 63, 327–333. [Google Scholar] [CrossRef]
- Erbas, I.M.; Paketci, A.; Turan, S.; Sisman, A.R.; Demir, K.; Bober, E.; Abaci, A. Low Complement C1q/TNF-related Protein-13 Levels are Associated with Childhood Obesity But not Binge Eating Disorder. J. Clin. Res. Pediatr. Endocrinol. 2022, 14, 179–187. [Google Scholar] [CrossRef]
- Shanaki, M.; Moradi, N.; Fadaei, R.; Zandieh, Z.; Shabani, P.; Vatannejad, A. Lower circulating levels of CTRP12 and CTRP13 in polycystic ovarian syndrome: Irrespective of obesity. PLoS ONE 2018, 13, e0208059. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y. Adiponectin: A key player in obesity related disorders. Curr. Pharm. Des. 2010, 16, 1896–1901. [Google Scholar] [CrossRef]
- Iwabu, M.; Okada-Iwabu, M.; Yamauchi, T.; Kadowaki, T. Adiponectin/AdipoR Research and Its Implications for Lifestyle-Related Diseases. Front. Cardiovasc. Med. 2019, 6, 116. [Google Scholar] [CrossRef]
- Hajri, T.; Tao, H.; Wattacheril, J.; Marks-Shulman, P.; Abumrad, N.N. Regulation of adiponectin production by insulin: Interactions with tumor necrosis factor-alpha and interleukin-6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E350–E360. [Google Scholar] [CrossRef]
- Ruan, H.; Miles, P.D.; Ladd, C.M.; Ross, K.; Golub, T.R.; Olefsky, J.M.; Lodish, H.F. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: Implications for insulin resistance. Diabetes 2002, 51, 3176–3188. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Dobrescu, C.; Bagby, G.J. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 1992, 130, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Niu, X.; Tian, L.; Liu, J.; Niu, R.; Quan, J.; Yu, J.; Lin, W.; Qian, Z.; Zeng, P. CTRP13 attenuates the expression of LN and CAV-1 Induced by high glucose via CaMKKbeta/AMPK pathway in rLSECs. Aging 2020, 12, 11485–11499. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Yuan, Y.; Yi, W.; Lau, W.B.; Wang, Y.; Wang, X.; Sun, Y.; Lopez, B.L.; Christopher, T.A.; Peterson, J.M.; et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2616–2623. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Choi, J.H.; Ryu, H.S.; Pak, Y.K.; Park, K.S.; Lee, H.K.; Lee, W. C1q tumor necrosis factor alpha-related protein isoform 5 is increased in mitochondrial DNA-depleted myocytes and activates AMP-activated protein kinase. J. Biol. Chem. 2009, 284, 27780–27789. [Google Scholar] [CrossRef] [PubMed]
- Bolliger, M.F.; Martinelli, D.C.; Sudhof, T.C. The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 2534–2539. [Google Scholar] [CrossRef]
- Kaur, B.; Brat, D.J.; Calkins, C.C.; Van Meir, E.G. Brain angiogenesis inhibitor 1 is differentially expressed in normal brain and glioblastoma independently of p53 expression. Am. J. Pathol. 2003, 162, 19–27. [Google Scholar] [CrossRef]
- Hamoud, N.; Tran, V.; Aimi, T.; Kakegawa, W.; Lahaie, S.; Thibault, M.P.; Pelletier, A.; Wong, G.W.; Kim, I.S.; Kania, A.; et al. Spatiotemporal regulation of the GPCR activity of BAI3 by C1qL4 and Stabilin-2 controls myoblast fusion. Nat. Commun. 2018, 9, 4470. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Snead, D.; Mitchell, D. An investigation into the potential role of brain angiogenesis inhibitor protein 3 (BAI3) in the tumorigenesis of small-cell carcinoma: A review of the surrounding literature. J. Recept. Signal Transduct. Res. 2017, 37, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Tan, A.; Yang, R.; Xue, Y.; Zhang, M.; Chen, L.; Xiao, L.; Yang, X.; Yu, Y. C1ql1/Ctrp14 and C1ql4/Ctrp11 promote angiogenesis of endothelial cells through activation of ERK1/2 signal pathway. Mol. Cell. Biochem. 2017, 424, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Hug, C.; Wang, J.; Ahmad, N.S.; Bogan, J.S.; Tsao, T.S.; Lodish, H.F. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl. Acad. Sci. USA 2004, 101, 10308–10313. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Dong, L.Q. Adiponectin signaling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Munoz, M.; Contreras, C.; Prieto, D. AMPK, metabolism, and vascular function. FEBS J. 2021, 288, 3746–3771. [Google Scholar] [CrossRef]
- Bu, L.L.; Yuan, H.H.; Xie, L.L.; Guo, M.H.; Liao, D.F.; Zheng, X.L. New Dawn for Atherosclerosis: Vascular Endothelial Cell Senescence and Death. Int. J. Mol. Sci. 2023, 24, 15160. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, G.; Chen, N.; Zhang, W.; Gao, Q.; Li, T.; Yuan, N.; Jin, H. CTRP13 alleviates palmitic acid-induced inflammation, oxidative stress, apoptosis and endothelial cell dysfunction in HUVECs. Tissue Cell 2023, 86, 102232. [Google Scholar] [CrossRef]
- Baretella, O.; Xu, A.; Vanhoutte, P.M. No Protective Effect of Constitutive Activation of AMPK in Endothelial Cells on Vascular Function in Aged Obese Mice but Augmented alpha1-Adrenergic Contractions in Renal Arteries Reversible by Weight Loss. J. Vasc. Res. 2018, 55, 189–202. [Google Scholar] [CrossRef]
- Liang, Y.; Huang, B.; Song, E.; Bai, B.; Wang, Y. Constitutive activation of AMPK alpha1 in vascular endothelium promotes high-fat diet-induced fatty liver injury: Role of COX-2 induction. Br. J. Pharmacol. 2014, 171, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Zippel, N.; Loot, A.E.; Stingl, H.; Randriamboavonjy, V.; Fleming, I.; Fisslthaler, B. Endothelial AMP-Activated Kinase alpha1 Phosphorylates eNOS on Thr495 and Decreases Endothelial NO Formation. Int. J. Mol. Sci. 2018, 19, 2753. [Google Scholar] [CrossRef]
- Peyton, K.J.; Liu, X.M.; Yu, Y.; Yates, B.; Durante, W. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J. Pharmacol. Exp. Ther. 2012, 342, 827–834. [Google Scholar] [CrossRef]
- Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Protective actions of globular and full-length adiponectin on human endothelial cells: Novel insights into adiponectin-induced angiogenesis. J. Vasc. Res. 2012, 49, 534–543. [Google Scholar] [CrossRef]
- Lu, Y.; Gao, X.; Wang, R.; Sun, J.; Guo, B.; Wei, R.; Jia, Y. Adiponectin inhibits proliferation of vascular endothelial cells induced by Ox-LDL by promoting dephosphorylation of Caveolin-1 and depolymerization of eNOS and up-regulating release of NO. Int. Immunopharmacol. 2019, 73, 424–434. [Google Scholar] [CrossRef]
- Igata, M.; Motoshima, H.; Tsuruzoe, K.; Kojima, K.; Matsumura, T.; Kondo, T.; Taguchi, T.; Nakamaru, K.; Yano, M.; Kukidome, D.; et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ. Res. 2005, 97, 837–844. [Google Scholar] [CrossRef]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Wild-Type (n = 12) | ob/ob (n = 12) |
|---|---|---|
| body weight (g) | 31.5 ± 2.1 | 56.5 ± 4.8 *** |
| glucose (mmol/L) | 7.1 ± 0.5 | 12.8 ± 0.9 * |
| insulin (µg/L) | 1.1 ± 0.4 | 16.4 ± 0.5 *** |
| TNF-alpha (pg/mL) | 20.5 ± 1.8 | 84.9 ± 2.1 *** |
| Parameter | Fa/Fa (n = 5) | Fa/fa (n = 5) | fa/fa (n = 5) |
|---|---|---|---|
| body weight (g) | 331.6 ± 8.5 | 358.3 ± 10.2 | 465.5 ± 12.6 ** |
| glucose (mmol/L) | 5.5 ± 0.4 | 6.9 ± 0.6 * | 16.3 ± 2.1 ** |
| insulin (µg/L) | 0.4 ± 0.3 | 1.7 ± 0.8 * | 4.9 ± 0.6 ** |
| TNF-alpha (pg/mL) | 3.8 ± 0.4 | 9.7 ± 0.5 ** | 17.9 ± 1.5 *** |
| Parameter | Lean (n = 15) | Obese (n = 20) |
|---|---|---|
| BMI (kg/m2) | 24.2 ± 0.4 | 33.5 ± 0.7 *** |
| age | 69.0 ± 3.4 | 62.8 ± 3.2 |
| male gender (n) | 11 | 10 |
| ejection fraction (%) | 63.1 ± 2.2 | 59.9 ± 2.4 |
| number of grafts (n) | 3.6 ± 0.2 | 4.0 ± 0.3 |
| cross-clamping time (min) | 61.3 ± 4.6 | 59.7 ± 4.9 |
| cardiopulm. Bypass (min) | 102.9 ± 6.2 | 97.8 ± 7.7 |
| preoperative CRP (mg/L) | 5.0 ± 1.7 | 6.6 ± 2.0 |
| preoperative leucocytes (109/L) | 7.4 ± 0.4 | 8.3 ± 0.4 |
| preoperative HbA1c (%) | 5.8 ± 0.1 | 6.0 ± 0.1 |
| fasting glucose (mmol/L) | 5.3 ± 0.9 | 7.5 ± 1.2 * |
| Beta-blocker (n) | 10 | 10 |
| ACE inhibitor (n) | 8 | 7 |
| ARB (n) | 5 | 6 |
| Anticoagulation (n) | 15 | 15 |
| Statin (n) | 12 | 15 |
| Diuretic (n) | 6 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, M.; Li, L.; Nürnberger, S.; Niemann, B.; Rohrbach, S. CTRP13-Mediated Effects on Endothelial Cell Function and Their Potential Role in Obesity. Cells 2024, 13, 1291. https://doi.org/10.3390/cells13151291
Aslam M, Li L, Nürnberger S, Niemann B, Rohrbach S. CTRP13-Mediated Effects on Endothelial Cell Function and Their Potential Role in Obesity. Cells. 2024; 13(15):1291. https://doi.org/10.3390/cells13151291
Chicago/Turabian StyleAslam, Muhammad, Ling Li, Sina Nürnberger, Bernd Niemann, and Susanne Rohrbach. 2024. "CTRP13-Mediated Effects on Endothelial Cell Function and Their Potential Role in Obesity" Cells 13, no. 15: 1291. https://doi.org/10.3390/cells13151291
APA StyleAslam, M., Li, L., Nürnberger, S., Niemann, B., & Rohrbach, S. (2024). CTRP13-Mediated Effects on Endothelial Cell Function and Their Potential Role in Obesity. Cells, 13(15), 1291. https://doi.org/10.3390/cells13151291