
High-Density Lipoprotein Modifications: Causes and Functional Consequences in Type 2 Diabetes Mellitus



Abstract

1. Introduction
2. HDL Structure and Function
2.1. HDL Structure
2.2. HDL Function
2.2.1. Reverse Cholesterol Transport
2.2.2. Cellular Interactions
2.2.3. Detoxification of Hazardous Molecules
3. Generation of HDL Modifications in T2DM
3.1. Post-Translational Modifications
3.1.1. Glycation
3.1.2. Oxidation
3.1.3. Carbamylation
3.2. Other Modifications in Lipid and Protein
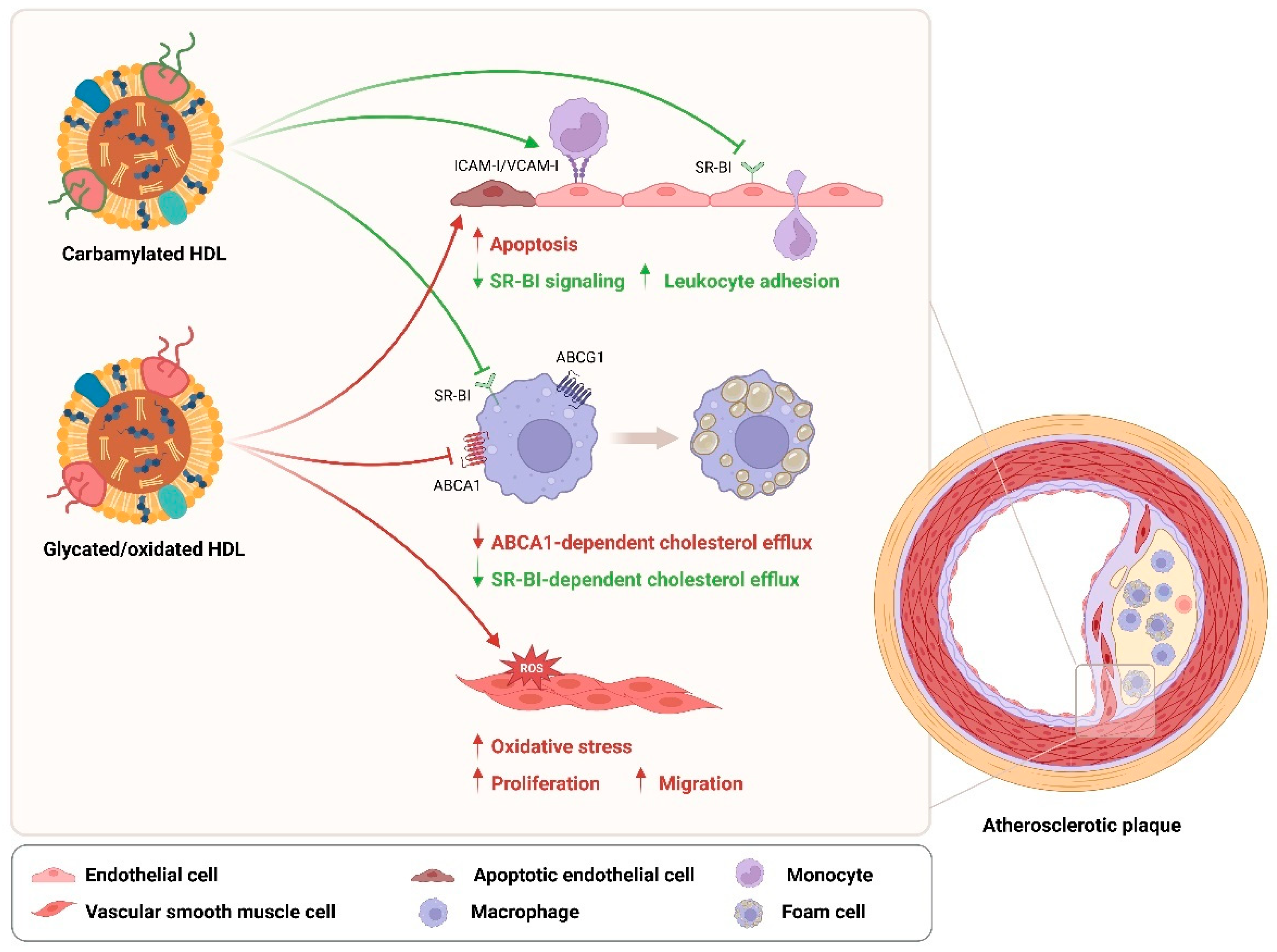
4. Effects of HDL Modifications on T2DM
5. Effects of HDL Modifications on T2DM Cardiovascular Complications
6. Conclusions and Perspective
Funding
Acknowledgments
Conflicts of Interest
References
- Ahmad, E.; Lim, S.; Lamptey, R.; Webb, D.R.; Davies, M.J. Type 2 diabetes. Lancet 2022, 400, 1803–1820. [Google Scholar] [CrossRef] [PubMed]
- Morrish, N.J.; Wang, S.L.; Stevens, L.K.; Fuller, J.H.; Keen, H. Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia 2001, 44 (Suppl. 2), S14–S21. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.D.; Vani, A.K.; Aleman, J.O.; Weintraub, H.S.; Berger, J.S.; Schwartzbard, A.Z. The Changing Landscape of Diabetes Therapy for Cardiovascular Risk Reduction: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1856–1869. [Google Scholar] [CrossRef]
- Martagon, A.J.; Zubirán, R.; González-Arellanes, R.; Praget-Bracamontes, S.; Rivera-Alcántara, J.A.; Aguilar-Salinas, C.A. HDL abnormalities in type 2 diabetes: Clinical implications. Atherosclerosis 2023, 394, 117213. [Google Scholar] [CrossRef]
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev. Cardiovasc. Ther. 2012, 10, 353–361. [Google Scholar] [CrossRef]
- Assmann, G.; Schulte, H.; von Eckardstein, A.; Huang, Y. High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis 1996, 124, S11–S20. [Google Scholar] [CrossRef]
- Miller, G.; Miller, N. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet 1975, 305, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.; Pignone, M.; Mulrow, C. Framingham-based tools to calculate the global risk of coronary heart disease: A systematic review of tools for clinicians. J. Gen. Intern. Med. 2003, 18, 1039–1052. [Google Scholar] [CrossRef]
- Assmann, G.; Gotto, A.M., Jr. HDL cholesterol and protective factors in atherosclerosis. Circulation 2004, 109, III-8–III-14. [Google Scholar] [CrossRef]
- Rye, K.-A.; Barter, P.J. Cardioprotective functions of HDLs1. J. Lipid Res. 2014, 55, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Xepapadaki, E.; Nikdima, I.; Sagiadinou, E.C.; Zvintzou, E.; Kypreos, K.E. HDL and type 2 diabetes: The chicken or the egg? Diabetologia 2021, 64, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; De La Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Investigators, A.-H. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Group, H.-T.C. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar]
- Xiang, D.; Zhang, Q.; Wang, Y.-T. Effectiveness of niacin supplementation for patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Medicine 2020, 99, e21235. [Google Scholar] [CrossRef]
- Barter, P.J.; Rye, K.-A.; Tardif, J.-C.; Waters, D.D.; Boekholdt, S.M.; Breazna, A.; Kastelein, J.J. Effect of torcetrapib on glucose, insulin, and hemoglobin A1c in subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial. Circulation 2011, 124, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.; Jankowski, V.; Bender, G.; Zewinger, S.; Rye, K.A.; van der Vorst, E.P.C. Interaction between high-density lipoproteins and inflammation: Function matters more than concentration! Adv. Drug Deliv. Rev. 2020, 159, 94–119. [Google Scholar] [CrossRef] [PubMed]
- Lui, D.T.W.; Cheung, C.L.; Lee, A.C.H.; Wong, Y.; Shiu, S.W.M.; Tan, K.C.B. Carbamylated HDL and Mortality Outcomes in Type 2 Diabetes. Diabetes Care 2021, 44, 804–809. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Márquez, A.B.; Nazir, S.; van der Vorst, E.P. High-density lipoprotein modifications: A pathological consequence or cause of disease progression? Biomedicines 2020, 8, 549. [Google Scholar] [CrossRef] [PubMed]
- Bonilha, I.; Zimetti, F.; Zanotti, I.; Papotti, B.; Sposito, A.C. Dysfunctional high-density lipoproteins in type 2 diabetes mellitus: Molecular mechanisms and therapeutic implications. J. Clin. Med. 2021, 10, 2233. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [CrossRef] [PubMed]
- von Eckardstein, A.; Nordestgaard, B.G.; Remaley, A.T.; Catapano, A.L. High-density lipoprotein revisited: Biological functions and clinical relevance. Eur. Heart J. 2023, 44, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Westerterp, M.; Von Eckardstein, A.; Remaley, A.; Rye, K.-A. HDL in the 21st century: A multifunctional roadmap for future HDL research. Circulation 2021, 143, 2293–2309. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Chapman, M.J.; Fazio, S.; Hussain, M.M.; Kontush, A.; Krauss, R.M.; Otvos, J.D.; Remaley, A.T.; Schaefer, E.J. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011, 57, 392–410. [Google Scholar] [CrossRef]
- Cheung, M.C.; Albers, J.J. Distribution of high density lipoprotein particles with different apoprotein composition: Particles with AI and A-II and particles with AI but no A-II. J. Lipid Res. 1982, 23, 747–753. [Google Scholar] [CrossRef]
- Delalla, O.F.; Elliott, H.A.; Gofman, J.W. Ultracentrifugal studies of high density serum lipoproteins in clinically healthy adults. Am. J. Physiol.-Leg. Content 1954, 179, 333–337. [Google Scholar] [CrossRef]
- Barter, P.; Ha, Y.; Calvert, G. Studies of esterified cholesterol in sub-fractions of plasma high density lipoproteins. Atherosclerosis 1981, 38, 165–175. [Google Scholar] [CrossRef]
- Nichols, A.V.; Krauss, R.M.; Musliner, T.A. [24] Nondenaturing polyacrylamide gradient gel electrophoresis. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1986; Volume 128, pp. 417–431. [Google Scholar]
- Williams, P.T.; Krauss, R.M.; Vranizan, K.M.; Stefanick, M.L.; Wood, P.; Lindgren, F. Associations of lipoproteins and apolipoproteins with gradient gel electrophoresis estimates of high density lipoprotein subfractions in men and women. Arterioscler. Thromb. A J. Vasc. Biol. 1992, 12, 332–340. [Google Scholar] [CrossRef]
- Asztalos, B.F.; Schaefer, E.J. HDL in atherosclerosis: Actor or bystander? Atheroscler. Suppl. 2003, 4, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zannis, V.I.; Cole, F.S.; Jackson, C.L.; Kurnit, D.M.; Karathanasis, S.K. Distribution of apolipoprotein A-I, C-II, C-III, and E mRNA in fetal human tissues. Time-dependent induction of apolipoprotein E mRNA by cultures of human monocyte-macrophages. Biochemistry 1985, 24, 4450–4455. [Google Scholar] [CrossRef]
- Brunham, L.R.; Kruit, J.K.; Iqbal, J.; Fievet, C.; Timmins, J.M.; Pape, T.D.; Coburn, B.A.; Bissada, N.; Staels, B.; Groen, A.K. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Investig. 2006, 116, 1052–1062. [Google Scholar] [CrossRef]
- Timmins, J.M.; Lee, J.-Y.; Boudyguina, E.; Kluckman, K.D.; Brunham, L.R.; Mulya, A.; Gebre, A.K.; Coutinho, J.M.; Colvin, P.L.; Smith, T.L. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J. Clin. Investig. 2005, 115, 1333–1342. [Google Scholar] [CrossRef]
- Kingwell, B.A.; Chapman, M.J.; Kontush, A.; Miller, N.E. HDL-targeted therapies: Progress, failures and future. Nat. Rev. Drug Discov. 2014, 13, 445–464. [Google Scholar] [CrossRef]
- Asztalos, B.F.; Schaefer, E.J. High-density lipoprotein subpopulations in pathologic conditions. Am. J. Cardiol. 2003, 91, 12–17. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Barrera, G.C.; Nakamura, K.; Baldán, Á.; Tarr, P.; Fishbein, M.C.; Frank, J.; Francone, O.L.; Edwards, P.A. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005, 1, 121–131. [Google Scholar] [CrossRef]
- Anastasius, M.; Luquain-Costaz, C.; Kockx, M.; Jessup, W.; Kritharides, L. A critical appraisal of the measurement of serum ‘cholesterol efflux capacity’ and its use as surrogate marker of risk of cardiovascular disease. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 1257–1273. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A. HDL and reverse remnant-cholesterol transport (RRT): Relevance to cardiovascular disease. Trends Mol. Med. 2020, 26, 1086–1100. [Google Scholar] [CrossRef]
- Wang, M.; Briggs, M.R. HDL: The metabolism, function, and therapeutic importance. Chem. Rev. 2004, 104, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Jiang, X.C.; Lagrost, L.; Tall, A.R. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J. Lipid Res. 2009, 50, S201–S206. [Google Scholar] [CrossRef]
- Clay, M.A.; Newnham, H.H.; Forte, T.M.; Barter, P.I. Cholesteryl ester transfer protein and hepatic lipase activity promote shedding of apo A-I from HDL and subsequent formation of discoidal HDL. Biochim. Biophys. Acta 1992, 1124, 52–58. [Google Scholar] [CrossRef]
- Jahangiri, A.; Rader, D.; Marchadier, D.; Curtiss, L.; Bonnet, D.; Rye, K.A. Evidence that endothelial lipase remodels high density lipoproteins without mediating the dissociation of apolipoprotein AI. J. Lipid Res. 2005, 46, 896–903. [Google Scholar] [CrossRef]
- Martinez, L.O.; Jacquet, S.; Esteve, J.-P.; Rolland, C.; Cabezón, E.; Champagne, E.; Pineau, T.; Georgeaud, V.; Walker, J.E.; Tercé, F.J.N. Ectopic β-chain of ATP synthase is an apolipoprotein AI receptor in hepatic HDL endocytosis. Nature 2003, 421, 75–79. [Google Scholar] [CrossRef]
- Furtado, J.D.; Yamamoto, R.; Melchior, J.T.; Andraski, A.B.; Gamez-Guerrero, M.; Mulcahy, P.; He, Z.; Cai, T.; Davidson, W.S.; Sacks, F.M. Distinct proteomic signatures in 16 HDL (high-density lipoprotein) subspecies. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2827–2842. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef]
- Catapano, A.L.; Pirillo, A.; Bonacina, F.; Norata, G.D. HDL in innate and adaptive immunity. Cardiovasc. Res. 2014, 103, 372–383. [Google Scholar] [CrossRef]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.; Lapergue, B.; Burillo, E.; Michel, J.; Levoye, A.; Martin-Ventura, J. HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef]
- von Eckardstein, A.; Widmann, C. High-density lipoprotein, beta cells, and diabetes. Cardiovasc. Res. 2014, 103, 384–394. [Google Scholar] [CrossRef]
- Van Linthout, S.; Foryst-Ludwig, A.; Spillmann, F.; Peng, J.; Feng, Y.; Meloni, M.; Van Craeyveld, E.; Kintscher, U.; Schultheiss, H.-P.; De Geest, B. Impact of HDL on adipose tissue metabolism and adiponectin expression. Atherosclerosis 2010, 210, 438–444. [Google Scholar] [CrossRef]
- Manandhar, B.; Cochran, B.J.; Rye, K.A. Role of high-density lipoproteins in cholesterol homeostasis and glycemic control. J. Am. Heart Assoc. 2020, 9, e013531. [Google Scholar] [CrossRef]
- Nofer, J.-R. Signal transduction by HDL: Agonists, receptors, and signaling cascades. High Density Lipoproteins Biol. Underst. Clin. Exploit. 2015, 224, 229–256. [Google Scholar]
- Didichenko, S.A.; Navdaev, A.V.; Cukier, A.M.; Gille, A.; Schuetz, P.; Spycher, M.O.; Thérond, P.; Chapman, M.J.; Kontush, A.; Wright, S.D. Enhanced HDL functionality in small HDL species produced upon remodeling of HDL by reconstituted HDL, CSL112: Effects on cholesterol efflux, anti-inflammatory and antioxidative activity. Circ. Res. 2016, 119, 751–763. [Google Scholar] [CrossRef]
- Dorighello, G.G.; Assis, L.H.; Rentz, T.; Morari, J.; Santana, M.F.; Passarelli, M.; Ridgway, N.D.; Vercesi, A.E.; Oliveira, H.C. Novel role of CETP in macrophages: Reduction of mitochondrial oxidants production and modulation of cell immune-metabolic profile. Antioxidants 2022, 11, 1734. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; Labzin, L.I.; Kono, H.; Seki, R.; Schmidt, S.V.; Beyer, M.; Xu, D.; Zimmer, S.; Lahrmann, C.; Schildberg, F.A. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 2014, 15, 152–160. [Google Scholar] [CrossRef]
- Fotakis, P.; Kothari, V.; Thomas, D.G.; Westerterp, M.; Molusky, M.M.; Altin, E.; Abramowicz, S.; Wang, N.; He, Y.; Heinecke, J.W. Anti-inflammatory effects of HDL (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e253–e272. [Google Scholar] [CrossRef]
- van Der Vorst, E.P.; Theodorou, K.; Wu, Y.; Hoeksema, M.A.; Goossens, P.; Bursill, C.A.; Aliyev, T.; Huitema, L.F.; Tas, S.W.; Wolfs, I.M. High-density lipoproteins exert pro-inflammatory effects on macrophages via passive cholesterol depletion and PKC-NF-κB/STAT1-IRF1 signaling. Cell Metab. 2017, 25, 197–207. [Google Scholar] [CrossRef] [PubMed]
- van der Vorst, E.P.; Theodorou, K.; Biessen, E.A.; Donners, M.M. Disease-or storage-associated structural modifications are unlikely to explain HDL pro-inflammatory effects on macrophages. Cell Metab. 2017, 26, 4–5. [Google Scholar] [CrossRef]
- Ito, A.; Hong, C.; Oka, K.; Salazar, J.V.; Diehl, C.; Witztum, J.L.; Diaz, M.; Castrillo, A.; Bensinger, S.J.; Chan, L. Cholesterol accumulation in CD11c+ immune cells is a causal and targetable factor in autoimmune disease. Immunity 2016, 45, 1311–1326. [Google Scholar] [CrossRef]
- Umemoto, T.; Han, C.Y.; Mitra, P.; Averill, M.M.; Tang, C.; Goodspeed, L.; Omer, M.; Subramanian, S.; Wang, S.; Den Hartigh, L.J. Apolipoprotein AI and high-density lipoprotein have anti-inflammatory effects on adipocytes via cholesterol transporters: ATP-binding cassette A-1, ATP-binding cassette G-1, and scavenger receptor B-1. Circ. Res. 2013, 112, 1345–1354. [Google Scholar] [CrossRef]
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 2001, 7, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Xepapadaki, E.; Zvintzou, E.; Kalogeropoulou, C.; Filou, S.; Kypreos, K.E. Τhe antioxidant function of HDL in atherosclerosis. Angiology 2020, 71, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Frej, C.; Holmér, A.; Guo, L.J.; Tran, S.; Dahlbäck, B. High-density lipoprotein–associated apolipoprotein M limits endothelial inflammation by delivering sphingosine-1-phosphate to the sphingosine-1-phosphate receptor 1. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 118–129. [Google Scholar] [CrossRef]
- Han, R.; Lai, R.; Ding, Q.; Wang, Z.; Luo, X.; Zhang, Y.; Cui, G.; He, J.; Liu, W.; Chen, Y. Apolipoprotein AI stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia 2007, 50, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Meilhac, O.; Tanaka, S.; Couret, D. High-density lipoproteins are bug scavengers. Biomolecules 2020, 10, 598. [Google Scholar] [CrossRef]
- Cazita, P.M.; Barbeiro, D.F.; Moretti, A.I.; Quintão, E.C.; Soriano, F.G. Human cholesteryl ester transfer protein expression enhances the mouse survival rate in an experimental systemic inflammation model: A novel role for CETP. Shock 2008, 30, 590–595. [Google Scholar] [CrossRef]
- Camont, L.; Chapman, M.J.; Kontush, A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 2011, 17, 594–603. [Google Scholar] [CrossRef]
- Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol. Cell. Biochem. 2018, 440, 167–187. [Google Scholar] [CrossRef]
- Ulrich, P.; Cerami, A. Protein glycation, diabetes, and aging. Recent Prog. Horm. Res. 2001, 56, 1–21. [Google Scholar] [CrossRef]
- Vlassara, H.; Palace, M.R. Diabetes and advanced glycation endproducts. J. Intern. Med. 2002, 251, 87–101. [Google Scholar] [CrossRef]
- Pan, B.; Ren, H.; Ma, Y.; Liu, D.; Yu, B.; Ji, L.; Pan, L.; Li, J.; Yang, L.; Lv, X.; et al. High-density lipoprotein of patients with type 2 diabetes mellitus elevates the capability of promoting migration and invasion of breast cancer cells. Int. J. Cancer 2012, 131, 70–82. [Google Scholar] [CrossRef]
- Godfrey, L.; Yamada-Fowler, N.; Smith, J.; Thornalley, P.J.; Rabbani, N. Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr. Diabetes 2014, 4, e134. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Klein, R.L.; Semler, A.J.; Januszewski, A.S. Lipoprotein glycation in diabetes mellitus. In Lipoproteins in Diabetes Mellitus; Springer: Berlin/Heidelberg, Germany, 2023; pp. 275–318. [Google Scholar]
- Domingo-Espín, J.; Nilsson, O.; Bernfur, K.; Del Giudice, R.; Lagerstedt, J. Site-specific glycations of apolipoprotein AI lead to differentiated functional effects on lipid-binding and on glucose metabolism. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 2822–2834. [Google Scholar] [CrossRef]
- Liu, D.; Ji, L.; Zhao, M.; Wang, Y.; Guo, Y.; Li, L.; Zhang, D.; Xu, L.; Pan, B.; Su, J.; et al. Lysine glycation of apolipoprotein A-I impairs its anti-inflammatory function in type 2 diabetes mellitus. J. Mol. Cell Cardiol. 2018, 122, 47–57. [Google Scholar] [CrossRef]
- Nobecourt, E.; Davies, M.J.; Brown, B.E.; Curtiss, L.K.; Bonnet, D.J.; Charlton, F.; Januszewski, A.S.; Jenkins, A.J.; Barter, P.J.; Rye, K.A. The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin:cholesterol acyltransferase. Diabetologia 2007, 50, 643–653. [Google Scholar] [CrossRef]
- Fournier, N.; Myara, I.; Atger, V.; Moatti, N. Reactivity of lecithin-cholesterol acyl transferase (LCAT) towards glycated high-density lipoproteins (HDL). Clin. Chim. Acta 1995, 234, 47–61. [Google Scholar] [CrossRef]
- Ferretti, G.; Bacchetti, T.; Marchionni, C.; Caldarelli, L.; Curatola, G. Effect of glycation of high density lipoproteins on their physicochemical properties and on paraoxonase activity. Acta Diabetol. 2001, 38, 163–169. [Google Scholar] [CrossRef]
- Passarelli, M.; Catanozi, S.; Nakandakare, E.; Rocha, J.; Morton, R.; Shimabukuro, A.; Quintão, E.C. Plasma lipoproteins from patients with poorly controlled diabetes mellitus and “in vitro” glycation of lipoproteins enhance the transfer rate of cholesteryl ester from HDL to apo-B-containing lipoproteins. Diabetologia 1997, 40, 1085–1093. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Why is HDL functionally deficient in type 2 diabetes? Curr. Diabetes Rep. 2008, 8, 51–59. [Google Scholar] [CrossRef]
- Shao, B.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Myeloperoxidase: An oxidative pathway for generating dysfunctional high-density lipoprotein. Chem. Res. Toxicol. 2010, 23, 447–454. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Rocha, M.; Falcon, R.; de Pablo, C.; Alvarez, A.; Jover, A.; Hernandez-Mijares, A.; Victor, V.M. Is myeloperoxidase a key component in the ROS-induced vascular damage related to nephropathy in type 2 diabetes? Antioxid. Redox Signal. 2013, 19, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.T.; Dasari, P.S.; Tryggestad, J.B.; Aston, C.E.; Teague, A.M.; Short, K.R. Oxidized HDL and LDL in adolescents with type 2 diabetes compared to normal weight and obese peers. J. Diabetes Complicat. 2015, 29, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Settle, M.; Brubaker, G.; Schmitt, D.; Hazen, S.L.; Smith, J.D.; Kinter, M. Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J. Biol. Chem. 2005, 280, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Xie, S.; Li, J.; Tian, R.; Peng, Y.Y. Myeloperoxidase-mediated oxidation targets serum apolipoprotein A-I in diabetic patients and represents a potential mechanism leading to impaired anti-apoptotic activity of high density lipoprotein. Clin. Chim. Acta 2015, 441, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.; Doerries, C.; Heinemann, M. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wang, Y.; Yao, X.; Liu, S. High levels of oxidized fatty acids in HDL impair the antioxidant function of HDL in patients with diabetes. Front. Endocrinol. 2022, 13, 993193. [Google Scholar] [CrossRef]
- Chang, C.M.; Hsieh, C.J.; Huang, J.C.; Huang, I.C. Acute and chronic fluctuations in blood glucose levels can increase oxidative stress in type 2 diabetes mellitus. Acta Diabetol. 2012, 49, S171–S177. [Google Scholar] [CrossRef]
- Denimal, D.; Monier, S.; Bouillet, B.; Vergès, B.; Duvillard, L. High-density lipoprotein alterations in type 2 diabetes and obesity. Metabolites 2023, 13, 253. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Sakane, N.; Ueda, M.; Mashiba, S.; Hayase, Y.; Tsuzaki, K.; Yamada, T.; Remaley, A.T. Oxidized high-density lipoprotein is associated with increased plasma glucose in non-diabetic dyslipidemic subjects. Clin. Chim. Acta 2012, 414, 125–129. [Google Scholar] [CrossRef]
- Sartore, G.; Seraglia, R.; Burlina, S.; Bolis, A.; Marin, R.; Manzato, E.; Ragazzi, E.; Traldi, P.; Lapolla, A. High-density lipoprotein oxidation in type 2 diabetic patients and young patients with premature myocardial infarction. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 418–425. [Google Scholar] [CrossRef]
- Jaisson, S.; Pietrement, C.; Gillery, P. Protein carbamylation: Chemistry, pathophysiological involvement, and biomarkers. Adv. Clin. Chem. 2018, 84, 1–38. [Google Scholar]
- Delanghe, S.; Delanghe, J.R.; Speeckaert, R.; Van Biesen, W.; Speeckaert, M.M. Mechanisms and consequences of carbamoylation. Nat. Rev. Nephrol. 2017, 13, 580–593. [Google Scholar] [CrossRef]
- Chen, Z.; Ding, S.; Wang, Y.P.; Chen, L.; Mao, J.Y.; Yang, Y.; Sun, J.T.; Yang, K. Association of carbamylated high-density lipoprotein with coronary artery disease in type 2 diabetes mellitus: Carbamylated high-density lipoprotein of patients promotes monocyte adhesion. J. Transl. Med. 2020, 18, 460. [Google Scholar] [CrossRef]
- Battle, S.; Gogonea, V.; Willard, B.; Wang, Z.; Fu, X.; Huang, Y.; Graham, L.M.; Cameron, S.J.; DiDonato, J.A.; Crabb, J.; et al. The pattern of apolipoprotein AI lysine carbamylation reflects its lipidation state and the chemical environment within human atherosclerotic aorta. J. Biol. Chem. 2022, 298, 101832. [Google Scholar] [CrossRef]
- Holzer, M.; Zangger, K.; El-Gamal, D.; Binder, V.; Curcic, S.; Konya, V.; Schuligoi, R.; Heinemann, A.; Marsche, G. Myeloperoxidase-Derived Chlorinating Species Induce Protein Carbamylation Through Decomposition of Thiocyanate and Urea: Novel Pathways Generating Dysfunctional High-Density Lipoprotein. Antioxid. Redox Signal. 2012, 17, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.; Otvos, J.D.; Rosenson, R.S.; Pradhan, A.; Buring, J.E.; Ridker, P.M. Lipoprotein particle size and concentration by nuclear magnetic resonance and incident type 2 diabetes in women. Diabetes 2010, 59, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Bakogianni, M.C.; Kalofoutis, C.A.; Skenderi, K.; Kalofoutis, A.T. Clinical evaluation of plasma high-density lipoprotein subfractions (HDL2, HDL3) in non-insulin-dependent diabetics with coronary artery disease. J. Diabetes Its Complicat. 2001, 15, 265–269. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.; Perton, F.G.; Dallinga-Thie, G.M.; van Roon, A.M.; Wolffenbuttel, B.H.; van Tol, A.; Dullaart, R.P. Plasma cholesteryl ester transfer is a determinant of intima-media thickness in type 2 diabetic and nondiabetic subjects: Role of CETP and triglycerides. Diabetes 2005, 54, 3554–3559. [Google Scholar] [CrossRef]
- Von Eckardstein, A.; Nofer, J.-R.; Assmann, G. High density lipoproteins and arteriosclerosis: Role of cholesterol efflux and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Watanabe, T.; Sakaue, T.; Lewis, G.F. Mechanisms of HDL lowering in insulin resistant, hypertriglyceridemic states: The combined effect of HDL triglyceride enrichment and elevated hepatic lipase activity. Clin. Biochem. 2003, 36, 421–429. [Google Scholar] [CrossRef]
- Brahimaj, A.; Ligthart, S.; Ikram, M.A.; Hofman, A.; Franco, O.H.; Sijbrands, E.J.; Kavousi, M.; Dehghan, A. Serum levels of apolipoproteins and incident type 2 diabetes: A prospective cohort study. Diabetes Care 2017, 40, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.-H.; Gao, J.-L.; Pu, C.; Feng, G.; Wang, L.-Z.; Huang, L.-Z.; Zhang, Y. A single-nucleotide polymorphism C-724/del in the proter region of the apolipoprotein M gene is associated with type 2 diabetes mellitus. Lipids Health Dis. 2016, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Brinck, J.W.; Thomas, A.; Lauer, E.; Jornayvaz, F.R.; Brulhart-Meynet, M.-C.; Prost, J.-C.; Pataky, Z.; Löfgren, P.; Hoffstedt, J.; Eriksson, M. Diabetes mellitus is associated with reduced high-density lipoprotein sphingosine-1-phosphate content and impaired high-density lipoprotein cardiac cell protection. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Shiu, S.; Wong, Y.; Wong, W.; Tam, S. Plasma apolipoprotein E concentration is an important determinant of phospholipid transfer protein activity in type 2 diabetes mellitus. Diabetes/Metab. Res. Rev. 2006, 22, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Dahlback, B. A novel human apolipoprotein (apoM). J. Biol. Chem. 1999, 274, 31286–31290. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Shigenaga, J.K.; Chui, L.G.; Moser, A.; Khovidhunkit, W.; Grunfeld, C. Infection and inflammation decrease apolipoprotein M expression. Atherosclerosis 2008, 199, 19–26. [Google Scholar] [CrossRef]
- Bach-Ngohou, K.; Ouguerram, K.; Nazih, H.; Maugere, P.; Ripolles-Piquer, B.; Zair, Y.; Frenais, R.; Krempf, M.; Bard, J. Apolipoprotein E kinetics: Influence of insulin resistance and type 2 diabetes. Int. J. Obes. 2002, 26, 1451–1458. [Google Scholar] [CrossRef]
- Poh, R.; Muniandy, S. Paraoxonase 1 activity as a predictor of cardiovascular disease in type 2 diabetes. Southeast Asian J. Trop. Med. Public Health 2010, 41, 1231–1246. [Google Scholar]
- Kotur-Stevuljević, J.; Vekić, J.; Stefanović, A.; Zeljković, A.; Ninić, A.; Ivanišević, J.; Miljković, M.; Sopić, M.; Munjas, J.; Mihajlović, M. Paraoxonase 1 and atherosclerosis-related diseases. Biofactors 2020, 46, 193–205. [Google Scholar] [CrossRef]
- Griffiths, K.; Pazderska, A.; Ahmed, M.; McGowan, A.; Maxwell, A.P.; McEneny, J.; Gibney, J.; McKay, G.J. Type 2 diabetes in young females results in increased serum amyloid a and changes to features of high density lipoproteins in both HDL 2 and HDL 3. J. Diabetes Res. 2017, 2017, 1314864. [Google Scholar] [CrossRef]
- Xepapadaki, E.; Maulucci, G.; Constantinou, C.; Karavia, E.A.; Zvintzou, E.; Daniel, B.; Sasson, S.; Kypreos, K.E. Impact of apolipoprotein A1-or lecithin: Cholesterol acyltransferase-deficiency on white adipose tissue metabolic activity and glucose homeostasis in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation Reduces the Stability of ApoAI and Increases HDL Dysfunction in Diet-Controlled Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef]
- Brites, F.D.; Cavallero, E.; de Geitere, C.; Nicolaiew, N.; Jacotot, B.; Rosseneu, M.; Fruchart, J.C.; Wikinski, R.L.; Castro, G.R. Abnormal capacity to induce cholesterol efflux and a new LpA-I pre-beta particle in type 2 diabetic patients. Clin. Chim. Acta 1999, 279, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Syvänne, M.; Castro, G.; Dengremont, C.; De Geitere, C.; Jauhiainen, M.; Ehnholm, C.; Michelagnoli, S.; Franceschini, G.; Kahri, J.; Taskinen, M.-R. Cholesterol efflux from Fu5AH hepatoma cells induced by plasma of subjects with or without coronary artery disease and non-insulin-dependent diabetes: Importance of LpA-I: A-II particles and phospholipid transfer protein. Atherosclerosis 1996, 127, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Kruit, J.K.; Brunham, L.R.; Verchere, C.B.; Hayden, M.R. HDL and LDL cholesterol significantly influence β-cell function in type 2 diabetes mellitus. Curr. Opin. Lipidol. 2010, 21, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Head, W.S.; Gunawardana, S.C.; Hasty, A.H.; Piston, D.W. Direct effect of cholesterol on insulin secretion: A novel mechanism for pancreatic beta-cell dysfunction. Diabetes 2007, 56, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, N.; Nagai, R.; Miyazaki, A.; Ikemoto, M.; Arai, H.; Horiuchi, S.; Nakayama, H. Scavenger receptor class B type I-mediated reverse cholesterol transport is inhibited by advanced glycation end products. J. Biol. Chem. 2001, 276, 13348–13355. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, M.; Tang, C.; McDonald, T.O.; O’Brien, K.D.; Gerrity, R.G.; Heinecke, J.W.; Oram, J.F. Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes 2005, 54, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Kruit, J.; Kremer, P.; Dai, L.; Tang, R.; Ruddle, P.; de Haan, W.; Brunham, L.; Verchere, C.; Hayden, M. Cholesterol efflux via ATP-binding cassette transporter A1 (ABCA1) and cholesterol uptake via the LDL receptor influences cholesterol-induced impairment of beta cell function in mice. Diabetologia 2010, 53, 1110–1119. [Google Scholar] [CrossRef]
- Sanchez-Aguilera, P.; Diaz-Vegas, A.; Campos, C.; Quinteros-Waltemath, O.; Cerda-Kohler, H.; Barrientos, G.; Contreras-Ferrat, A.; Llanos, P. Role of ABCA1 on membrane cholesterol content, insulin-dependent Akt phosphorylation and glucose uptake in adult skeletal muscle fibers from mice. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1469–1477. [Google Scholar] [CrossRef]
- Low, H.; Hoang, A.; Forbes, J.; Thomas, M.; Lyons, J.G.; Nestel, P.; Bach, L.A.; Sviridov, D. Advanced glycation end-products (AGEs) and functionality of reverse cholesterol transport in patients with type 2 diabetes and in mouse models. Diabetologia 2012, 55, 2513–2521. [Google Scholar] [CrossRef]
- Bouillet, B.; Gautier, T.; Blache, D.; Pais de Barros, J.P.; Duvillard, L.; Petit, J.M.; Lagrost, L.; Verges, B. Glycation of apolipoprotein C1 impairs its CETP inhibitory property: Pathophysiological relevance in patients with type 1 and type 2 diabetes. Diabetes Care 2014, 37, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Shokri, Y.; Variji, A.; Nosrati, M.; Khonakdar-Tarsi, A.; Kianmehr, A.; Kashi, Z.; Bahar, A.; Bagheri, A.; Mahrooz, A. Importance of paraoxonase 1 (PON1) as an antioxidant and antiatherogenic enzyme in the cardiovascular complications of type 2 diabetes: Genotypic and phenotypic evaluation. Diabetes Res. Clin. Pract. 2020, 161, 108067. [Google Scholar] [CrossRef] [PubMed]
- Bacchetti, T.; Masciangelo, S.; Armeni, T.; Bicchiega, V.; Ferretti, G. Glycation of human high density lipoprotein by methylglyoxal: Effect on HDL-paraoxonase activity. Metabolism 2014, 63, 307–311. [Google Scholar] [CrossRef]
- Koren-Gluzer, M.; Aviram, M.; Hayek, T. Paraoxonase1 (PON1) reduces insulin resistance in mice fed a high-fat diet, and promotes GLUT4 overexpression in myocytes, via the IRS-1/Akt pathway. Atherosclerosis 2013, 229, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Koren-Gluzer, M.; Aviram, M.; Meilin, E.; Hayek, T. The antioxidant HDL-associated paraoxonase-1 (PON1) attenuates diabetes development and stimulates beta-cell insulin release. Atherosclerosis 2011, 219, 510–518. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, H.K.; Choi, Y.J.; Eum, W.S.; Park, J.; Choi, S.Y.; Kwon, H.Y. PEP-1-paraoxonase 1 fusion protein prevents cytokine-induced cell destruction and impaired insulin secretion in rat insulinoma cells. BMB Rep. 2018, 51, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Tamama, K.; Tomura, H.; Sato, K.; Malchinkhuu, E.; Damirin, A.; Kimura, T.; Kuwabara, A.; Murakami, M.; Okajima, F. High-density lipoprotein inhibits migration of vascular smooth muscle cells through its sphingosine 1-phosphate component. Atherosclerosis 2005, 178, 19–23. [Google Scholar] [CrossRef]
- Van Der Vorst, E.P.; Vanags, L.Z.; Dunn, L.L.; Prosser, H.C.; Rye, K.A.; Bursill, C.A. High-density lipoproteins suppress chemokine expression and proliferation in human vascular smooth muscle cells. FASEB J. 2013, 27, 1413–1425. [Google Scholar] [CrossRef]
- Shen, Y.; Ding, F.H.; Sun, J.T.; Pu, L.J.; Zhang, R.Y.; Zhang, Q.; Chen, Q.J.; Shen, W.F.; Lu, L. Association of elevated apoA-I glycation and reduced HDL-associated paraoxonase1, 3 activity, and their interaction with angiographic severity of coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2015, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Pennathur, S.; Heinecke, J.W. Myeloperoxidase targets apolipoprotein AI, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J. Biol. Chem. 2012, 287, 6375–6386. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Denimal, D.; Monier, S.; Simoneau, I.; Duvillard, L.; Vergès, B.; Bouillet, B. HDL functionality in type 1 diabetes: Enhancement of cholesterol efflux capacity in relationship with decreased HDL carbamylation after improvement of glycemic control. Cardiovasc. Diabetol. 2022, 21, 154. [Google Scholar] [CrossRef]
- Hadfield, K.A.; Pattison, D.I.; Brown, B.E.; Hou, L.; Rye, K.-A.; Davies, M.J.; Hawkins, C.L. Myeloperoxidase-derived oxidants modify apolipoprotein AI and generate dysfunctional high-density lipoproteins: Comparison of hypothiocyanous acid (HOSCN) with hypochlorous acid (HOCl). Biochem. J. 2013, 449, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Rifici, V.A.; Khachadurian, A.K. Oxidation of high density lipoproteins: Characterization and effects on cholesterol efflux from J774 macrophages. Biochim. Et Biophys. Acta (BBA)-Lipids Lipid Metab. 1996, 1299, 87–94. [Google Scholar] [CrossRef]
- Wu, Z.; Jankowski, V.; Jankowski, J.J.M.A.o.M. Irreversible post-translational modifications–Emerging cardiovascular risk factors. Mol Asp. Med. 2022, 86, 101010. [Google Scholar] [CrossRef]
- Okuda, L.S.; Castilho, G.; Rocco, D.D.F.M.; Nakandakare, E.R.; Catanozi, S.; Passarelli, M. Advanced glycated albumin impairs HDL anti-inflammatory activity and primes macrophages for inflammatory response that reduces reverse cholesterol transport. Biochim. Et Biophys. Acta-Mol. Cell Biol. Lipids 2012, 1821, 1485–1492. [Google Scholar] [CrossRef]
- Shao, B.; Tang, C.; Heinecke, J.W.; Oram, J.F. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J. Lipid Res. 2010, 51, 1849–1858. [Google Scholar] [CrossRef]
- Sun, J.T.; Yang, K.; Lu, L.; Zhu, Z.B.; Zhu, J.Z.; Ni, J.W.; Han, H.; Chen, N.; Zhang, R.Y. Increased carbamylation level of HDL in end-stage renal disease: Carbamylated-HDL attenuated endothelial cell function. Am. J. Physiol. Ren. Physiol. 2016, 310, F511–F517. [Google Scholar] [CrossRef]
- Matsunaga, T.; Iguchi, K.; Nakajima, T.; Koyama, I.; Miyazaki, T.; Inoue, I.; Kawai, S.; Katayama, S.; Hirano, K.; Hokari, S.; et al. Glycated high-density lipoprotein induces apoptosis of endothelial cells via a mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2001, 287, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Nakajima, T.; Miyazaki, T.; Koyama, I.; Hokari, S.; Inoue, I.; Kawai, S.; Shimomura, H.; Katayama, S.; Hara, A.; et al. Glycated high-density lipoprotein regulates reactive oxygen species and reactive nitrogen species in endothelial cells. Metabolism 2003, 52, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, L.; Jiang, R.; Zheng, L.; Liu, D. Oxidized high-density lipoprotein induces the proliferation and migration of vascular smooth muscle cells by promoting the production of ROS. J. Atheroscler. Thromb. 2014, 21, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Qian, M.-M.; Liu, P.-L.; Zhang, L.; Wang, Y.; Liu, D.-H. Glycation of high-density lipoprotein triggers oxidative stress and promotes the proliferation and migration of vascular smooth muscle cells. J. Geriatr. Cardiol. JGC 2017, 14, 473. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Measurement | Subfraction |
|---|---|---|
| Ultracentrifugation | Density | HDL2 (1.063–1.125 g/mL) HDL3 (1.125–1.21 g/mL) |
| Non-denaturing polyacrylamide gradient gel electrophoresis | Size | HDL2b (9.7–12.0 nm) HDL2a (8.8–9.7 nm) HDL3a (8.2–8.8 nm) HDL3b (7.8–8.2 nm) HDL3c (7.2–7.8 nm) |
| Nuclear magnetic resonance | Size | Large HDL (8.8–13.0 nm) Medium HDL (8.2–8.8 nm) Small HDL (7.3–8.2 nm) |
| Agarose gel electrophoresis | Charge and shape | α-HDL (spherical) preβ-HDL (discoidal) |
| 2-dimensional electrophoresis | Charge and size | preβ-HDL (preβ1 and preβ2) α-HDL (α1, α2, α3 and α4) preα-HDL (preα1, preα2, preα3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; van der Vorst, E.P.C. High-Density Lipoprotein Modifications: Causes and Functional Consequences in Type 2 Diabetes Mellitus. Cells 2024, 13, 1113. https://doi.org/10.3390/cells13131113
Zhang X, van der Vorst EPC. High-Density Lipoprotein Modifications: Causes and Functional Consequences in Type 2 Diabetes Mellitus. Cells. 2024; 13(13):1113. https://doi.org/10.3390/cells13131113
Chicago/Turabian StyleZhang, Xiaodi, and Emiel P. C. van der Vorst. 2024. "High-Density Lipoprotein Modifications: Causes and Functional Consequences in Type 2 Diabetes Mellitus" Cells 13, no. 13: 1113. https://doi.org/10.3390/cells13131113
APA StyleZhang, X., & van der Vorst, E. P. C. (2024). High-Density Lipoprotein Modifications: Causes and Functional Consequences in Type 2 Diabetes Mellitus. Cells, 13(13), 1113. https://doi.org/10.3390/cells13131113