
Molecular Profiling and the Interaction of Somatic Mutations with Transcriptomic Profiles in Non-Melanoma Skin Cancer (NMSC) in a Population Exposed to Arsenic

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Arsenic Exposure Measurement
2.3. Nucleic Acid Extraction
2.4. Somatic Mutation Assay
2.5. Gene Expression Assay
2.6. Statistical Method
3. Results
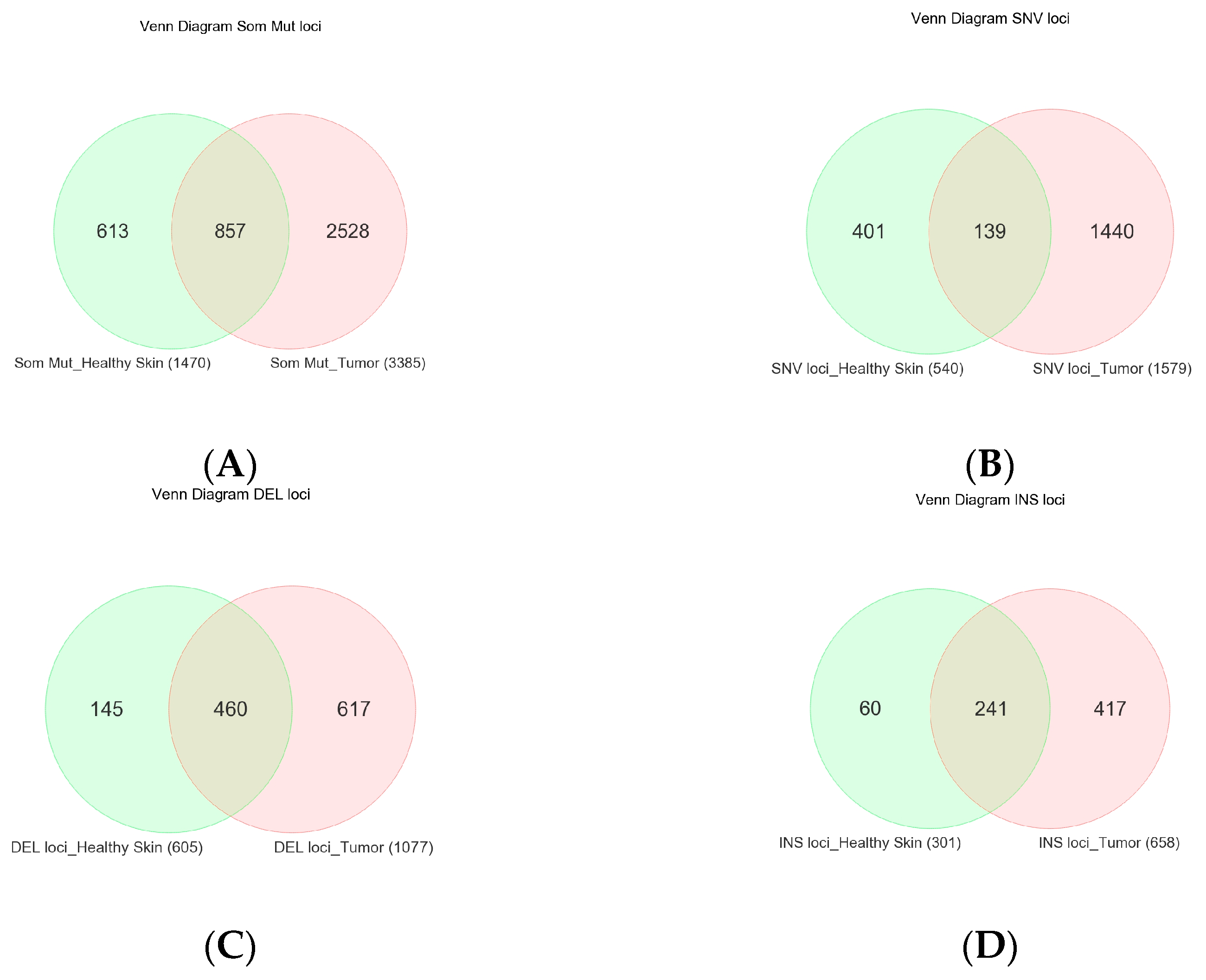
3.1. Somatic Mutation
3.2. NMSC-Associated Somatic SNVs
3.3. Somatic Mutation SNVs Common in NMSC and Healthy Skin Tissue
3.4. Somatic Mutation SNVs Detected Only in Healthy Skin Tissue
3.5. Association of Somatic Mutation and Differential Gene Expression in NMSC
3.5.1. Gene Level Analysis
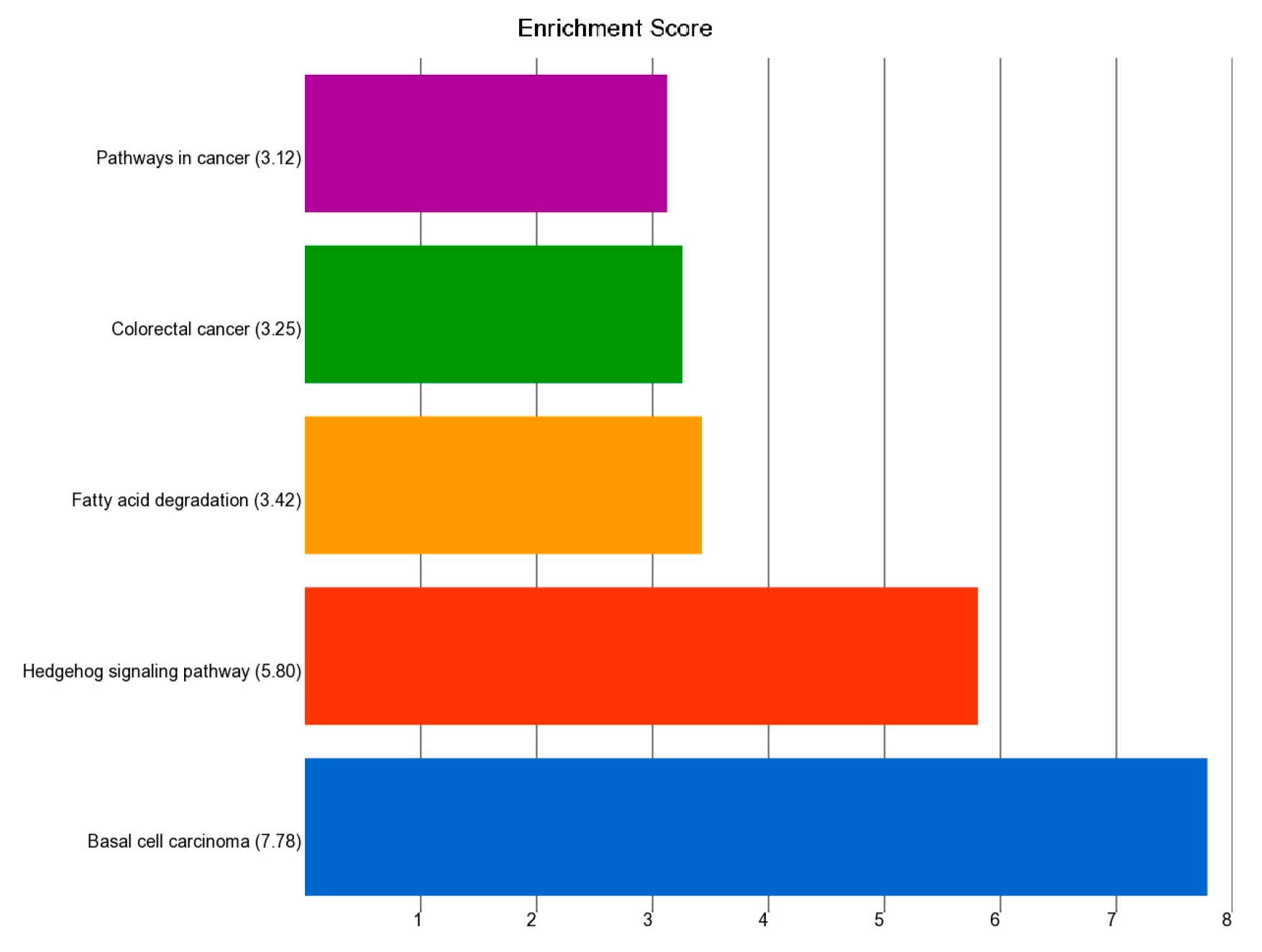
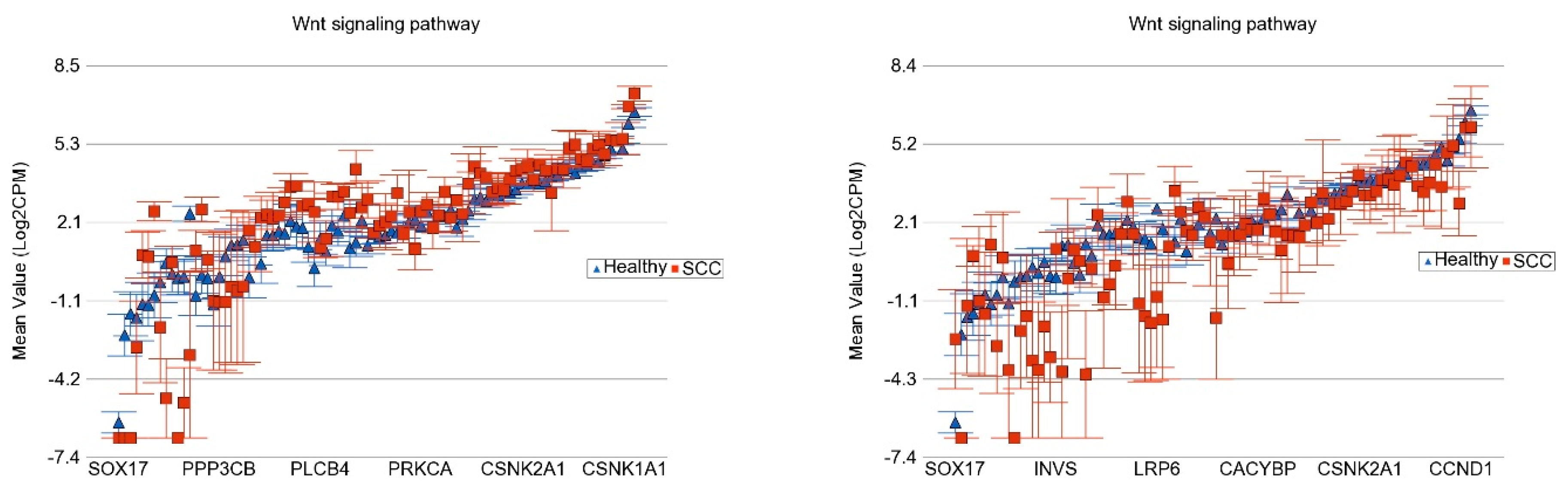
3.5.2. Pathway Level Analysis
- Association of the Somatic ns Mutation in the PTCH1 Gene and Dysregulated Pathways in BCC
- Association of the Somatic ns Mutation in the NOTCH1 Gene and Dysregulated Pathways in BCC
- Association of the Somatic ns Mutation in SYNE1 and PKHD1 Genes and Dysregulated Pathways in BCC
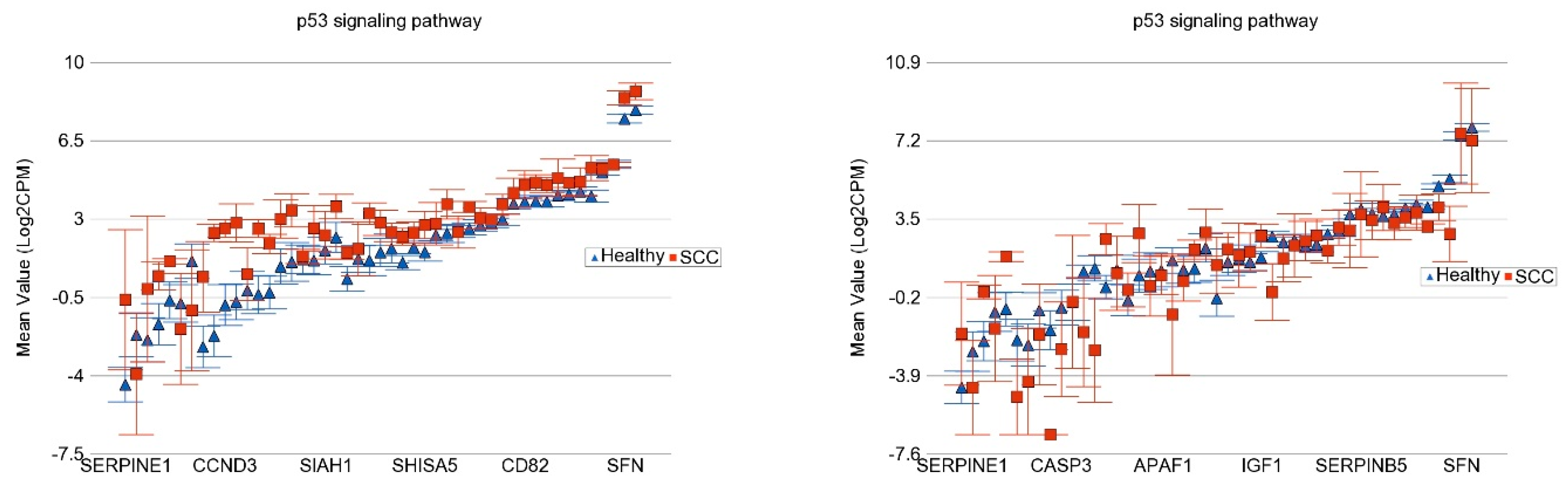
- Association of the Somatic ns Mutation in the TP53 Gene and Dysregulated Pathways in SCC
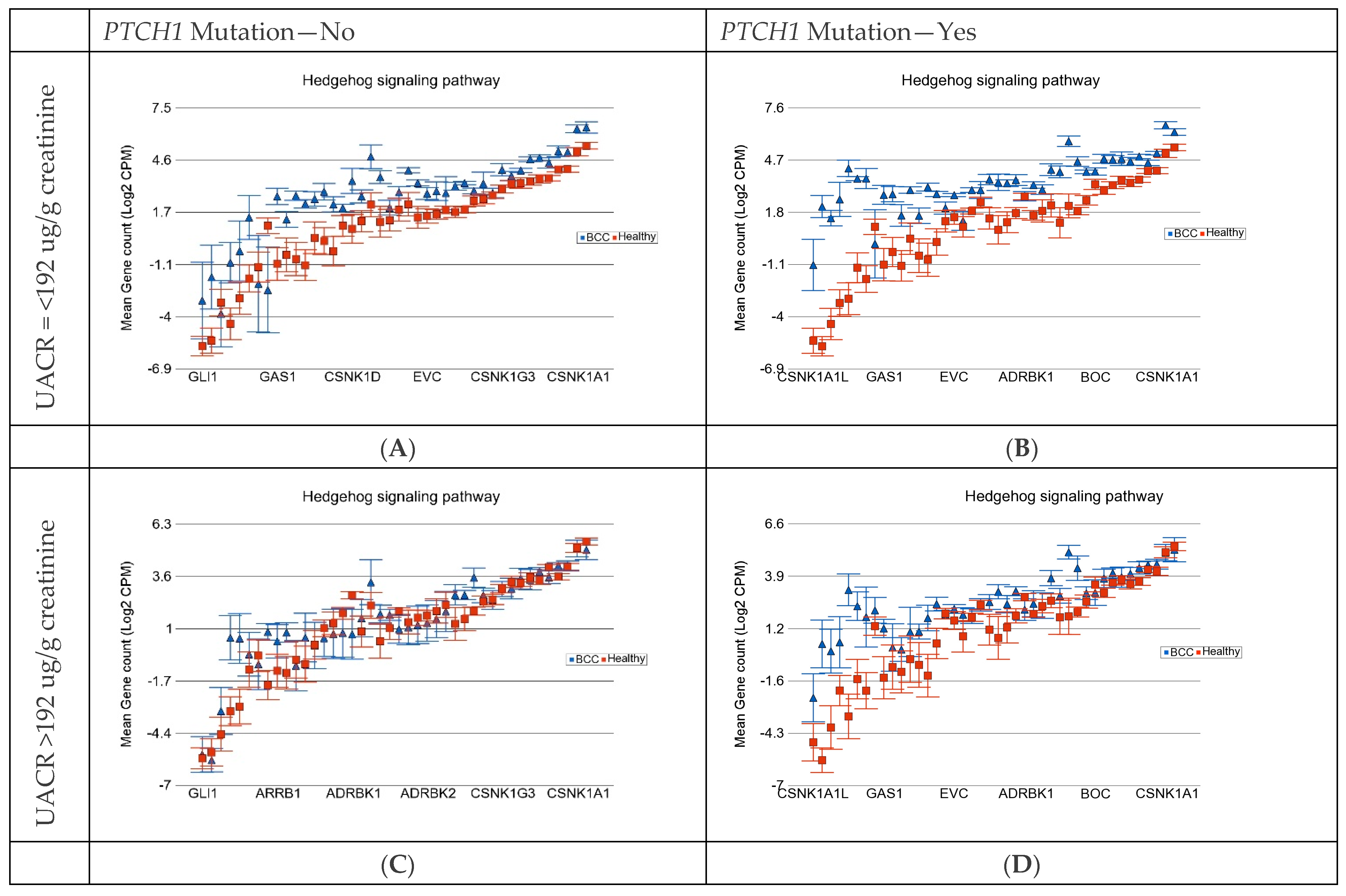
3.6. Gene–Environmental Interaction: Interaction of the Somatic Mutation and Degree of As Exposure on Gene Expression Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, W.; Zeng, W.; Jiang, A.; He, Z.; Shen, X.; Dong, X.; Feng, J.; Lu, H. Global, regional and national incidence, mortality and disability-adjusted life-years of skin cancers and trend analysis from 1990 to 2019: An analysis of the Global Burden of Disease Study 2019. Cancer Med. 2021, 10, 4905–4922. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goldenberg, G.; Karagiannis, T.; Palmer, J.B.; Lotya, J.; O’Neill, C.; Kisa, R.; Herrera, V.; Siegel, D.M. Incidence and prevalence of basal cell carcinoma (BCC) and locally advanced BCC (LABCC) in a large commercially insured population in the United States: A retrospective cohort study. J. Am. Acad. Dermatol. 2016, 75, 957–966.e2. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Armstrong, A.W. Nonmelanoma skin cancer. Dermatol. Clin. 2012, 30, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R.M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Fahradyan, A.; Howell, A.C.; Wolfswinkel, E.M.; Tsuha, M.; Sheth, P.; Wong, A.K. Updates on the Management of Non-Melanoma Skin Cancer (NMSC). Healthcare 2017, 5, 82. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Paolino, G.; Donati, M.; Didona, D.; Mercuri, S.R.; Cantisani, C. Histology of Non-Melanoma Skin Cancers: An Update. Biomedicines 2017, 5, 71. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jones, O.T.; Ranmuthu, C.K.I.; Hall, P.N.; Funston, G.; Walter, F.M. Recognising Skin Cancer in Primary Care. Adv. Ther. 2020, 37, 603–616. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Verkouteren, J.A.C.; Ramdas, K.H.R.; Wakkee, M.; Nijsten, T. Epidemiology of basal cell carcinoma: Scholarly review. Br. J. Dermatol. 2017, 177, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Staples, M.P.; Elwood, M.; Burton, R.C.; Williams, J.L.; Marks, R.; Giles, G.G. Non-melanoma skin cancer in Australia: The 2002 national survey and trends since 1985. Med. J. Aust. 2006, 184, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Abarca, J.F.; Casiccia, C.C. Skin cancer and ultraviolet-B radiation under the Antarctic ozone hole: Southern Chile, 1987–2000. Photodermatol. Photoimmunol. Photomed. 2002, 18, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Rawashdeh, M.A.; Matalka, I. Basal cell carcinoma of the maxillofacial region: Site distribution and incidence rates in Arab/Jordanians, 1991 to 2000. J. Oral Maxillofac. Surg. 2004, 62, 145–149. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Fowler, J.C.; Abnizova, I.; Sood, R.K.; Hall, M.W.J.; Szeverényi, I.; Tham, M.; Huang, J.; Young, S.M.; Hall, B.A.; et al. Somatic mutations in facial skin from countries of contrasting skin cancer risk. Nat. Genet. 2023, 55, 1440–1447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Flohil, S.C.; van der Leest, R.J.; Arends, L.R.; de Vries, E.; Nijsten, T. Risk of subsequent cutaneous malignancy in patients with prior keratinocyte carcinoma: A systematic review and meta-analysis. Eur. J. Cancer 2013, 49, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Van der Leest, R.J.; Flohil, S.C.; Arends, L.R.; de Vries, E.; Nijsten, T. Risk of subsequent cutaneous malignancy in patients with prior melanoma: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Housman, T.S.; Feldman, S.R.; Williford, P.M.; Fleischer, A.B., Jr.; Goldman, N.D.; Acostamadiedo, J.M.; Chen, G.J. Skin cancer is among the most costly of all cancers to treat for the Medicare population. J. Am. Acad. Dermatol. 2003, 48, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Guy, G.P., Jr.; Machlin, S.R.; Ekwueme, D.U.; Yabroff, K.R. Prevalence and costs of skin cancer treatment in the U.S., 2002–2006 and 2007–2011. Am. J. Prev. Med. 2015, 48, 183–187. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lai, V.; Cranwell, W.; Sinclair, R. Epidemiology of skin cancer in the mature patient. Clin. Dermatol. 2018, 36, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Savoia, P.; Veronese, F.; Camillo, L.; Tarantino, V.; Cremona, O.; Zavattaro, E. Multiple Basal Cell Carcinomas in Immunocompetent Patients. Cancers 2022, 14, 3211. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zambrano-Román, M.; Padilla-Gutiérrez, J.R.; Valle, Y.; Muñoz-Valle, J.F.; Valdés-Alvarado, E. Non-Melanoma Skin Cancer: A Genetic Update and Future Perspectives. Cancers 2022, 14, 2371. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hyeraci, M.; Papanikolau, E.S.; Grimaldi, M.; Ricci, F.; Pallotta, S.; Monetta, R.; Minafò, Y.A.; Di Lella, G.; Galdo, G.; Abeni, D.; et al. Systemic Photoprotection in Melanoma and Non-Melanoma Skin Cancer. Biomolecules 2023, 13, 1067. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nikolouzakis, T.K.; Falzone, L.; Lasithiotakis, K.; Krüger-Krasagakis, S.; Kalogeraki, A.; Sifaki, M.; Spandidos, D.A.; Chrysos, E.; Tsatsakis, A.; Tsiaoussis, J. Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer. J. Clin. Med. 2020, 9, 2868. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Azimi, A.; Fernandez-Peñas, P. Molecular Classifiers in Skin Cancers: Challenges and Promises. Cancers 2023, 15, 4463. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, H.R.; Yu, H.S.; Hu, H.; Monson, R.R. Arsenic in drinking water and skin cancers: Cell-type specificity (Taiwan, ROC). Cancer Causes Control 2001, 12, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.; Vahter, M.; Clemens, F.; Goessler, W.; Gurzau, E.; Hemminki, K.; Hough, R.; Koppova, K.; Kumar, R.; Rudnai, P.; et al. Inorganic arsenic and basal cell carcinoma in areas of Hungary, Romania, and Slovakia: A case-control study. Environ. Health Perspect. 2012, 120, 721–726. [Google Scholar] [PubMed] [PubMed Central]
- Maloney, M.E. Arsenic in Dermatology. Dermatol. Surg. 1996, 22, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.I.M.; Shabnam, N.; Ahsan, T.; Ahsan, S.M.A.; Kabir, M.S.; Khan, R.M.; Miah, M.A.; Uddin, M.K.; Liton, M.A.R. Cutaneous Malignancy due to Arsenicosis in Bangladesh: 12-Year Study in Tertiary Level Hospital. BioMed Res. Int. 2018, 2018, 4678362. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeng, Q.; Zhang, A. Assessing potential mechanisms of arsenic-induced skin lesions and cancers: Human and in vitro evidence. Environ. Pollut. 2020, 260, 113919. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Rachakonda, S.; Hielscher, T.; Calderazzo, S.; Rudnai, P.; Gurzau, E.; Koppova, K.; Fletcher, T.; Kumar, R. Telomere length, arsenic exposure and risk of basal cell carcinoma of skin. Carcinogenesis 2019, 40, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Speer, R.M.; Zhou, X.; Volk, L.B.; Liu, K.J.; Hudson, L.G. Arsenic and cancer: Evidence and mechanisms. Adv. Pharmacol. 2023, 96, 151–202. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kibriya, M.G.; Jasmine, F.; Munoz, A.; Islam, T.; Ahmed, A.; Tong, L.; Rakibuz-Zaman, M.; Shahriar, M.; Kamal, M.; Shea, C.R.; et al. Interaction of Arsenic Exposure and Transcriptomic Profile in Basal Cell Carcinoma. Cancers 2022, 14, 5598. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahsan, H.; Chen, Y.; Parvez, F.; Zablotska, L.; Argos, M.; Hussain, I.; Momotaj, H.; Levy, D.; Cheng, Z.; Slavkovich, V.; et al. Arsenic exposure from drinking water and risk of premalignant skin lesions in Bangladesh: Baseline results from the Health Effects of Arsenic Longitudinal Study. Am. J. Epidemiol. 2006, 163, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Jee, B.A.; Lim, H.; Kwon, S.M.; Jo, Y.; Park, M.C.; Lee, I.J.; Woo, H.G. Molecular classification of basal cell carcinoma of skin by gene expression profiling. Mol. Carcinog. 2015, 54, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.S.; Rayhan, D.J.; Hazany, S.; Kolodney, M.S. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J. Investig. Dermatol. 2014, 134, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutierrez Garcia-Rodrigo, C.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Di Bartolomeo, L.; Vaccaro, F.; Irrera, N.; Borgia, F.; Li Pomi, F.; Squadrito, F.; Vaccaro, M. Wnt Signaling Pathways: From Inflammation to Non-Melanoma Skin Cancers. Int. J. Mol. Sci. 2023, 24, 1575. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Liu, H.; Bian, Q. Identification of Potential Biomarkers Associated with Basal Cell Carcinoma. BioMed Res. Int. 2020, 2020, 2073690. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Argos, M.; Rahman, M.; Parvez, F.; Dignam, J.; Islam, T.; Quasem, I.; K. Hore, S.; T. Haider, A.; Hossain, Z.; I. Patwary, T.; et al. Baseline comorbidities in a skin cancer prevention trial in Bangladesh. Eur. J. Clin. Investig. 2013, 43, 579–588. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zilberg, C.; Lee, M.W.; Yu, B.; Ashford, B.; Kraitsek, S.; Ranson, M.; Shannon, K.; Cowley, M.; Iyer, N.G.; E Palme, C.; et al. Analysis of clinically relevant somatic mutations in high-risk head and neck cutaneous squamous cell carcinoma. Mod. Pathol. 2018, 31, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Asplund, A.; Gry Bjorklund, M.; Sundquist, C.; Stromberg, S.; Edlund, K.; Östman, A.; Nilsson, P.; Pontén, F.; Lundeberg, J. Expression profiling of microdissected cell populations selected from basal cells in normal epidermis and basal cell carcinoma. Br. J. Dermatol. 2008, 158, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Nindl, I.; Dang, C.; Forschner, T.; Kuban, R.J.; Meyer, T.; Sterry, W.; Stockfleth, E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol. Cancer 2006, 5, 30. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- O’Driscoll, L.; McMorrow, J.; Doolan, P.; McKiernan, E.; Mehta, J.P.; Ryan, E.; Gammell, P.; Joyce, H.; O’Donovan, N.; Walsh, N.; et al. Investigation of the molecular profile of basal cell carcinoma using whole genome microarrays. Mol. Cancer 2006, 5, 74. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yu, M.; Zloty, D.; Cowan, B.; Shapiro, J.; Haegert, A.; Bell, R.H.; Warshawski, L.; Carr, N.; McElwee, K.J. Superficial, nodular, and morpheiform basal-cell carcinomas exhibit distinct gene expression profiles. J. Investig. Dermatol. 2008, 128, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Heller, E.R.; Gor, A.; Wang, D.; Hu, Q.; Lucchese, A.; Kanduc, D.; Katdare, M.; Liu, S.; Sinha, A. Molecular signatures of basal cell carcinoma susceptibility and pathogenesis: A genomic approach. Int. J. Oncol. 2013, 42, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Dazard, J.E.; Gal, H.; Amariglio, N.; Rechavi, G.; Domany, E.; Givol, D. Genome-wide comparison of human keratinocyte and squamous cell carcinoma responses to UVB irradiation: Implications for skin and epithelial cancer. Oncogene 2003, 22, 2993–3006. [Google Scholar] [CrossRef] [PubMed]
- Dooley, T.P.; Reddy, S.P.; Wilborn, T.W.; Davis, R.L. Biomarkers of human cutaneous squamous cell carcinoma from tissues and cell lines identified by DNA microarrays and qRT-PCR. Biochem. Biophys. Res. Commun. 2003, 306, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Serewko, M.M.; Popa, C.; Dahler, A.L.; Smith, L.; Strutton, G.M.; Coman, W.; Dicker, A.J.; A Saunders, N. Alterations in gene expression and activity during squamous cell carcinoma development. Cancer Res. 2002, 62, 3759–3765. [Google Scholar] [PubMed]
- Nixon, D.E.; Mussmann, G.V.; Eckdahl, S.J.; Moyer, T.P. Total arsenic in urine: Palladium-persulfate vs nickel as a matrix modifier for graphite furnace atomic absorption spectrophotometry. Clin. Chem. 1991, 37, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Heinegård, D.; Tiderström, G. Determination of serum creatinine by a direct colorimetric method. Clin. Chim. Acta 1973, 43, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Jasmine, F.; Parvez, F.; Argos, M.; Roy, S.; Paul-Brutus, R.; Islam, T.; Ahmed, A.; Rakibuz-Zaman, M.; Shinkle, J.; et al. Association between genome-wide copy number variation and arsenic-induced skin lesions: A prospective study. Environ. Health 2017, 16, 75. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bisceglia, M.; Panniello, G.; Galliani, C.A.; Centola, M.; D’Errico, M.M.; Minenna, E.; Tucci, F.A.; Ben-Dor, D.J. Metastatic Basal Cell Carcinoma of the Skin: A Comprehensive Literature Review, Including Advances in Molecular Therapeutics. Adv. Anat. Pathol. 2020, 27, 331–353. [Google Scholar] [CrossRef] [PubMed]
- Gambini, D.; Passoni, E.; Nazzaro, G.; Beltramini, G.; Tomasello, G.; Ghidini, M.; Kuhn, E.; Garrone, O. Basal Cell Carcinoma and Hedgehog Pathway Inhibitors: Focus on Immune Response. Front. Med. 2022, 9, 893063. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martincorena, I.; Jones, P.H.; Campbell, P.J. Constrained positive selection on cancer mutations in normal skin. Proc. Natl. Acad. Sci. USA 2016, 113, E1128–E1129. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maturo, M.G.; Rachakonda, S.; Heidenreich, B.; Pellegrini, C.; Srinivas, N.; Requena, C.; Serra-Guillen, C.; Llombart, B.; Sanmartin, O.; Guillen, C.; et al. Coding and noncoding somatic mutations in candidate genes in basal cell carcinoma. Sci. Rep. 2020, 10, 8005. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nawrocka, P.M.; Galka-Marciniak, P.; Urbanek-Trzeciak, M.O.; M-Thirusenthilarasan, I.; Szostak, N.; Philips, A.; Susok, L.; Sand, M.; Kozlowski, P. Profile of Basal Cell Carcinoma Mutations and Copy Number Alterations-Focus on Gene-Associated Noncoding Variants. Front. Oncol. 2021, 11, 752579. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, Y.S.; Park, G.S.; Chung, Y.J.; Lee, J.H. Whole-exome sequencing of secondary tumors arising from nevus sebaceous revealed additional genomic alterations besides RAS mutations. J. Dermatol. 2023, 50, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Rawal, R.M.; Joshi, M.N.; Bhargava, P.; Shaikh, I.; Pandit, A.S.; Patel, R.P.; Patel, S.; Kothari, K.; Shah, M.; Saxena, A.; et al. Tobacco habituated and non-habituated subjects exhibit different mutational spectrums in head and neck squamous cell carcinoma. 3 Biotech 2015, 5, 685–696. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martinez, M.F.; Romano, M.V.; Martinez, A.P.; González, A.; Muchnik, C.; Stengel, F.M.; Mazzuoccolo, L.D.; Azurmendi, P.J. Nevoid Basal Cell Carcinoma Syndrome: PTCH1 Mutation Profile and Expression of Genes Involved in the Hedgehog Pathway in Argentinian Patients. Cells 2019, 8, 144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Somatic Variant Type | Variant Metrics | Median (BCC) | Median (SCC) | Median (Healthy Skin) |
|---|---|---|---|---|
| All Somatic Variants | Count | 10.00 | 44.00 | 15.00 |
| Coverage at the variant loci | 169.00 | 772.50 | 245.00 | |
| Frequency of the variant | 4.74 | 6.15 | 4.71 | |
| Q-score of the variant allele | 37.00 | 36.72 | 36.96 | |
| QUAL of the variant call | 200.00 | 200.00 | 200.00 | |
| SNVs | Count | 14.00 | 357.50 | 22.00 |
| Coverage at the variant loci | 114.00 | 974.50 | 154.00 | |
| Frequency of the variant | 8.33 | 43.89 | 17.78 | |
| Q-score of the variant allele | 37.00 | 36.86 | 37.00 | |
| QUAL of the variant call | 200.00 | 200.00 | 200.00 | |
| Insertion | Count | 9.00 | 37.50 | 15.00 |
| Coverage at the variant loci | 201.00 | 795.50 | 317.00 | |
| Frequency of the variant | 4.12 | 4.84 | 4.63 | |
| Q-score of the variant allele | 37.00 | 36.75 | 36.93 | |
| QUAL of the variant call | 200.00 | 200.00 | 200.00 | |
| Deletion | Count | 8.00 | 26.00 | 13.00 |
| Coverage at the variant loci | 189.00 | 616.00 | 297.00 | |
| Frequency of the variant | 4.17 | 4.07 | 3.91 | |
| Q-score of the variant allele | 37.00 | 36.32 | 36.69 | |
| QUAL of the variant call | 200.00 | 200.00 | 200.00 |
| BCC without PTCH1 Mutation | BCC with PTCH1 Mutation | ||||
|---|---|---|---|---|---|
| Gene Symbol | Fold Change | (95% CI) | Fold Change | (95% CI) | Interaction |
| p | |||||
| CHGA | 37.3 | (8.04–172.69) | 296.6 | (59.86–1469.36) | 0.0150 |
| MPPED1 | 35.6 | (8.13–156.0) | 51.3 | (10.99–239.68) | 0.6443 |
| CHCHD7 | 3.0 | (1.73–5.17) | 7.4 | (4.18–13.12) | 0.0035 |
| LGR5 | 20.5 | (4.81–87.16) | 84.4 | (18.63–382.35) | 0.0734 |
| ABI3BP | 6.3 | (2.39–16.80) | 27.3 | (9.88–75.45) | 0.0075 |
| IRS4 | 16.4 | (3.67–73.60) | 108.6 | (22.71–518.88) | 0.0229 |
| LEF1 | 9.6 | (3.02–30.61) | 20.4 | (6.1–68.31) | 0.2286 |
| RP11-368P15.3 | 15.4 | (3.83–62.07) | 36.6 | (8.55–156.32) | 0.2506 |
| DUSP10 | 12.0 | (3.05–47.20) | 41.5 | (9.96–173.2) | 0.0960 |
| PALM | 11.1 | (2.83–43.27) | 48.6 | (11.71–201.7) | 0.0480 |
| DIO2 | 3.1 | (1.57–6.05) | 8.4 | (4.14–16.91) | 0.0083 |
| VCAN | 5.4 | (1.94–15.17) | 26.2 | (8.95–76.53) | 0.0064 |
| RGCC | 25.0 | (5.30–117.63) | 30.5 | (6.05–153.7) | 0.8094 |
| LRRCC1 | 2.4 | (1.40–4.01) | 5.1 | (2.95–8.84) | 0.0090 |
| SOX4 | 2.4 | (1.39–3.97) | 5.0 | (2.88–8.58) | 0.0105 |
| SETBP1 | 2.0 | (1.26–3.05) | 4.9 | (3.1–7.82) | 0.0003 |
| DCP1B | 17.0 | (3.81–75.70) | 35.8 | (7.54–170.36) | 0.3530 |
| RP11-157G21.2 | 12.8 | (3.57–45.78) | 16.4 | (4.34–62.12) | 0.7144 |
| NNMT | 15.5 | (3.28–73.10) | 53.8 | (10.66–271.7) | 0.1390 |
| TGS1 | 2.8 | (1.61–4.86) | 3.6 | (2.04–6.44) | 0.3826 |
| SPON2 | 4.1 | (1.60–10.47) | 23.4 | (8.79–62.22) | 0.0012 |
| PROCR | 11.1 | (2.71–45.07) | 41.0 | (9.45–177.31) | 0.0875 |
| LPL | 12.4 | (2.97–51.52) | 35.6 | (8.04–157.49) | 0.1714 |
| BNC2 | 2.5 | (1.34–4.77) | 9.5 | (4.9–18.47) | 0.0003 |
| SYNJ1 | 7.1 | (2.21–22.57) | 21.2 | (6.3–71.02) | 0.0833 |
| TM4SF1 | 5.0 | (1.82–13.9) | 18.7 | (6.49–54.02) | 0.0196 |
| PTCH1 | 2.8 | (1.47–5.33) | 6.0 | (3.07–11.77) | 0.0311 |
| MMP11 | 11.4 | (2.45–53.18) | 77.3 | (15.54–384.35) | 0.0245 |
| MEX3A | 3.7 | (1.48–9.08) | 17.6 | (6.86–45.33) | 0.0023 |
| RP11-433O3.1 | 16.0 | (3.03–84.15) | 60.4 | (10.68–341.94) | 0.1400 |
| TUBA1A | 3.2 | (1.53–6.52) | 6.6 | (3.11–14.06) | 0.0622 |
| NXN | 2.8 | (1.51–5.11) | 4.3 | (2.29–8.13) | 0.1822 |
| TMCO3 | 10.3 | (2.53–42.01) | 30.9 | (7.14–133.59) | 0.1497 |
| PYCR2 | 14.3 | (2.87–71.24) | 51.0 | (9.55–272.2) | 0.1444 |
| ALCAM | 2.7 | (1.42–5.22) | 5.6 | (2.85–11.02) | 0.0433 |
| RP11-74M13.4 | −13.6 | (−56.71–3.24) | −20.3 | (-90.17–4.55) | 0.6003 |
| MARCKSL1 | 9.8 | (2.15–44.09) | 59.8 | (12.38–288.3) | 0.0294 |
| SCAMP5 | 8.2 | (2.07–32.72) | 38.7 | (9.18–163.16) | 0.0412 |
| VASH2 | 8.2 | (2.06–32.65) | 38.9 | (9.21–164.60) | 0.0403 |
| HDGFRP3 | 9.6 | (2.24–41.29) | 40.4 | (8.83–184.54) | 0.0715 |
| KRT17 | 4.7 | (1.74–12.72) | 12.2 | (4.31–34.25) | 0.0802 |
| DCLRE1A | 9.4 | (2.30–38.67) | 31.3 | (7.19–136.32) | 0.1176 |
| ATP5F1 | 4.7 | (1.81–12.24) | 9.7 | (3.56–26.24) | 0.1641 |
| RP11-366L20.2 | 11.2 | (2.50–50.45) | 36.1 | (7.53–172.85) | 0.1519 |
| BASP1 | 2.1 | (1.20–3.61) | 5.8 | (3.24–10.23) | 0.0012 |
| DSC2 | 3.2 | (1.4–7.29) | 10.7 | (4.5–25.24) | 0.0091 |
| IGF2BP2 | 2.2 | (1.21–3.94) | 6.3 | (3.39–11.55) | 0.0017 |
| FBN3 | 8.1 | (1.67–39.62) | 151.1 | (28.98–788.07) | 0.0013 |
| CSPG4 | 7.1 | (1.83–27.53) | 39.1 | (9.51–161.04) | 0.0228 |
| CCDC152 | 5.2 | (1.77–15.01) | 13.8 | (4.54–42.13) | 0.0902 |
| IGF2BP1 | −12.0 | (−48.57–2.94) | −17.7 | (−76.48–4.1) | 0.6008 |
| SOBP | 6.5 | (1.99–21.18) | 16.0 | (4.67–54.9) | 0.1587 |
| TMEM176B | 9.5 | (2.11–42.44) | 45.4 | (9.48–217.22) | 0.0565 |
| RP11-301G23.1 | 8.8 | (1.97–39.52) | 52.7 | (11.03–251.33) | 0.0307 |
| ACAA2 | 11.5 | (2.30–57.16) | 53.7 | (10.05–287.19) | 0.0781 |
| SHOX2 | 4.7 | (1.53–14.21) | 22.7 | (7.12–72.64) | 0.0108 |
| COA7 | 21.1 | (5.65–78.70) | 7.1 | (1.79–27.97) | 0.1268 |
| RUNX1T1 | 3.6 | (1.43–8.85) | 12.4 | (4.78–32.02) | 0.0139 |
| ITFG3 | 3.2 | (1.65–6.35) | 3.9 | (1.94–7.9) | 0.6011 |
| GRM5 | −9.3 | (−34.5–2.49) | −16.0 | (−62.86–4.05) | 0.4416 |
| PLAG1 | 9.6 | (2.26–41.08) | 29.8 | (6.57–135.5) | 0.1504 |
| SKIV2L | 12.8 | (3.14–52.23) | 15.2 | (3.5–65.88) | 0.8202 |
| HMGN1P38 | 5.5 | (2.1–14.27) | 6.5 | (2.41–17.76) | 0.7300 |
| CTB-167B5.2 | 9.9 | (2.25–43.22) | 31.5 | (6.75–147.23) | 0.1471 |
| VPS37D | 6.1 | (1.71–21.50) | 27.5 | (7.35–103.18) | 0.0302 |
| OSCP1 | 13.7 | (2.88–64.61) | 25.9 | (5.12–131.33) | 0.4421 |
| C1QTNF1 | 19.1 | (3.73–97.99) | 22.7 | (4.13–124.97) | 0.8438 |
| ACADL | −29.6 | (−148.16–5.92) | −13.3 | (−71.41–2.48) | 0.3556 |
| GJB6 | 5.5 | (1.93–15.78) | 9.7 | (3.24–29.02) | 0.3184 |
| PTCH2 | 9.5 | (1.8–49.61) | 101.3 | (17.97–570.53) | 0.0104 |
| RAB28 | 12.7 | (2.75–58.71) | 24.4 | (4.94–120.48) | 0.4273 |
| CASC14 | 12.1 | (2.4–60.91) | 39.6 | (7.33–213.81) | 0.1754 |
| LTBP1 | 2.9 | (1.25–6.59) | 12.1 | (5.09–28.84) | 0.0023 |
| ALYREF | 13.3 | (3.0–58.87) | 18.0 | (3.81–85.04) | 0.7035 |
| LITAF | 4.9 | (1.91–12.46) | 6.6 | (2.46–17.44) | 0.5564 |
| TMEM104 | 6.2 | (1.54–25.25) | 50.2 | (11.68–215.66) | 0.0078 |
| NUDCD1 | 12.4 | (2.95–52.09) | 15.2 | (3.39–67.72) | 0.7944 |
| SLCO2A1 | 13.7 | (2.77–67.39) | 26.5 | (5.02–140.11) | 0.4391 |
| SH3BP4 | 5.3 | (1.92–14.42) | 7.8 | (2.71–22.29) | 0.4691 |
| RP4-765C7.2 | 12.0 | (2.29–63.0) | 43.8 | (7.77–246.52) | 0.1502 |
| RP11-5N11.5 | 5.2 | (1.29–20.68) | 87.7 | (20.63–372.88) | 0.0004 |
| FMO2 | 6.4 | (1.95–20.71) | 12.3 | (3.59–42.36) | 0.2968 |
| AC005013.1 | 5.8 | (1.82–18.61) | 12.8 | (3.79–42.87) | 0.2120 |
| AC187652.1 | 11.9 | (2.99–47.46) | 11.8 | (2.78–49.71) | 0.9851 |
| GALNT15 | 9.6 | (2.41–38.38) | 15.6 | (3.67–65.88) | 0.5174 |
| EDN1 | 5.1 | (1.77–14.59) | 9.5 | (3.16–28.55) | 0.2714 |
| CREB5 | 2.3 | (1.13–4.65) | 8.7 | (4.15–18.04) | 0.0010 |
| RP11-73E6.2 | −8.1 | (−28.01–2.33) | −10.7 | (−39.29–2.93) | 0.6692 |
| TBX1 | 6.3 | (1.53–25.63) | 39.1 | (9.01–169.72) | 0.0190 |
| RAC3 | 13.1 | (3.15–54.56) | 11.9 | (2.69–52.64) | 0.8978 |
| RP3-512B11.3 | 9.3 | (2.06–41.58) | 27.4 | (5.71–131.35) | 0.1821 |
| C1QA | 11.8 | (2.11–66.46) | 52.4 | (8.66–316.86) | 0.1128 |
| COA3 | 13.0 | (2.8–60.32) | 18.3 | (3.69–90.76) | 0.6775 |
| RP11-384C21.9 | −7.9 | (−31.63–1.98) | −20.2 | (−85.61–4.76) | 0.2112 |
| TGFB2 | 8.3 | (2.22–31.06) | 13.6 | (3.42–53.58) | 0.4900 |
| STMN1 | 2.6 | (1.19–5.42) | 8.0 | (3.61–17.48) | 0.0072 |
| GLI2 | 7.5 | (1.86–30.38) | 22.6 | (5.26–96.75) | 0.1466 |
| UBAC2 | 7.2 | (2.09–24.74) | 11.3 | (3.11–40.92) | 0.4971 |
| SOX18 | 10.9 | (1.99–59.42) | 48.9 | (8.33–286.95) | 0.1039 |
| IFT88 | 5.4 | (1.86–15.56) | 8.0 | (2.65–24.26) | 0.4840 |
| RP11-496I9.1 | 11.6 | (2.19–60.82) | 35.5 | (6.27–201.05) | 0.2107 |
| RN7SL776P | −13.7 | (−57.39–3.25) | −10.8 | (−48.48–2.42) | 0.7635 |
| FSCN1 | 7.0 | (1.96–25.13) | 13.4 | (3.542–50.60) | 0.3471 |
| SLC7A2 | 3.8 | (1.37–10.49) | 12.3 | (4.24–35.44) | 0.0361 |
| RBP1 | 10.9 | (2.52–46.79) | 15.4 | (3.36–70.83) | 0.6533 |
| TMEM217 | −10.5 | (−45.1–2.45) | −15.7 | (−71.82–3.44) | 0.6061 |
| LRIG3 | 4.7 | (1.6–13.51) | 10.1 | (3.31–30.56) | 0.1816 |
| PELI2 | 4.1 | (1.44–11.52) | 11.5 | (3.89–34.03) | 0.0676 |
| TRIL | 8.3 | (1.98–34.59) | 20.2 | (4.55–89.76) | 0.2476 |
| ARPC1B | 3.8 | (1.52–9.69) | 7.3 | (2.78–19.23) | 0.1974 |
| CAV1 | 2.2 | (1.13–4.06) | 5.7 | (2.94–11.13) | 0.0062 |
| GPX8 | 7.5 | (1.92–29.08) | 17.1 | (4.15–70.41) | 0.2594 |
| INPP4A | 5.2 | (1.75–15.31) | 9.0 | (2.92–28.01) | 0.3391 |
| ADAM23 | 8.3 | (1.84–36.98) | 27.6 | (5.79–131.96) | 0.1376 |
| PNMA1 | 9.1 | (1.93–43.07) | 29.3 | (5.8–148.32) | 0.1646 |
| MGAT5 | 3.7 | (1.50–8.99) | 6.6 | (2.6–16.79) | 0.2251 |
| IRX1 | 5.4 | (1.22–23.96) | 74.3 | (15.73–351.2) | 0.0020 |
| UST | 8.4 | (1.92–36.55) | 22.6 | (4.86–104.94) | 0.2130 |
| Coordinate | Reference Allele | Variant Allele | Exact Match | Coding Region Change | Amino Acid Change |
|---|---|---|---|---|---|
| chr9:98211572 | T | A | clinvar_hg19 | NM_000264.3:c.3583A>T; NM_001083602.1:c.3385A>T; NM_001083603.1:c.3580A>T; NM_001083604.1:c.3130A>T; NM_001083605.1:c.3130A>T; NM_001083606.1:c.3130A>T; NM_001083607.1:c.3130A>T | NP_000255.2:p.Thr1195Ser; NP_001077071.1:p.Thr1129Ser; NP_001077072.1:p.Thr1194Ser; NP_001077073.1:p.Thr1044Ser; NP_001077074.1:p.Thr1044Ser; NP_001077075.1:p.Thr1044Ser; NP_001077076.1:p.Thr1044Ser |
| chr9:98239132 | G | T | clinvar_hg19 | NM_000264.3:c.1511C>A; NM_001083602.1:c.1313C>A; NM_001083603.1:c.1508C>A; NM_001083604.1:c.1058C>A; NM_001083605.1:c.1058C>A; NM_001083606.1:c.1058C>A; NM_001083607.1:c.1058C>A | NP_000255.2:p.Pro504Gln; NP_001077071.1:p.Pro438Gln; NP_001077072.1:p.Pro503Gln; NP_001077073.1:p.Pro353Gln; NP_001077074.1:p.Pro353Gln; NP_001077075.1:p.Pro353Gln; NP_001077076.1:p.Pro353Gln |
| chr9:98242779 | C | A | NM_000264.3:c.838G>T; NM_001083602.1:c.640G>T; NM_001083603.1:c.835G>T; NM_001083604.1:c.385G>T; NM_001083605.1:c.385G>T; NM_001083606.1:c.385G>T; NM_001083607.1:c.385G>T | NP_000255.2:p.Glu280*; NP_001077071.1:p.Glu214*; NP_001077072.1:p.Glu279*; NP_001077073.1:p.Glu129*; NP_001077074.1:p.Glu129*; NP_001077075.1:p.Glu129*; NP_001077076.1:p.Glu129* | |
| chr9:98242797 | G | A | NM_000264.3:c.820C>T; NM_001083602.1:c.622C>T; NM_001083603.1:c.817C>T; NM_001083604.1:c.367C>T; NM_001083605.1:c.367C>T; NM_001083606.1:c.367C>T; NM_001083607.1:c.367C>T | NP_000255.2:p.Gln274*; NP_001077071.1:p.Gln208*; NP_001077072.1:p.Gln273*; NP_001077073.1:p.Gln123*; NP_001077074.1:p.Gln123*; NP_001077075.1:p.Gln123*; NP_001077076.1:p.Gln123* | |
| chr9:98248001 | G | A | NM_000264.3:c.550C>T; NM_001083602.1:c.352C>T; NM_001083603.1:c.547C>T; NM_001083604.1:c.97C>T; NM_001083605.1:c.97C>T; NM_001083606.1:c.97C>T; NM_001083607.1:c.97C>T | NP_000255.2:p.Gln184*; NP_001077071.1:p.Gln118*; NP_001077072.1:p.Gln183*; NP_001077073.1:p.Gln33*; NP_001077074.1:p.Gln33*; NP_001077075.1:p.Gln33*; NP_001077076.1:p.Gln33* | |
| chr9:98244299 | A | T | NM_000264.3:c.678T>A; NM_001083602.1:c.480T>A; NM_001083603.1:c.675T>A; NM_001083604.1:c.225T>A; NM_001083605.1:c.225T>A; NM_001083606.1:c.225T>A; NM_001083607.1:c.225T>A | NP_000255.2:p.Cys226*; NP_001077071.1:p.Cys160*; NP_001077072.1:p.Cys225*; NP_001077073.1:p.Cys75*; NP_001077074.1:p.Cys75*; NP_001077075.1:p.Cys75*; NP_001077076.1:p.Cys75* | |
| chr9:98268818 | T | A | NM_000264.3:c.265A>T; NM_001083602.1:c.67A>T; NM_001083603.1:c.262A>T; NM_001083604.1:c.-189A>T; NM_001083605.1:c.-189A>T; NM_001083606.1:c.-189A>T; NM_001083607.1:c.-189A>T | NP_000255.2:p.Lys89*; NP_001077071.1:p.Lys23*; NP_001077072.1:p.Lys88* | |
| chr9:98241336 | C | T | clinvar_hg19 | NM_000264.3:c.1161G>A; NM_001083602.1:c.963G>A; NM_001083603.1:c.1158G>A; NM_001083604.1:c.708G>A; NM_001083605.1:c.708G>A; NM_001083606.1:c.708G>A; NM_001083607.1:c.708G>A | NP_000255.2:p.Trp387*; NP_001077071.1:p.Trp321*; NP_001077072.1:p.Trp386*; NP_001077073.1:p.Trp236*; NP_001077074.1:p.Trp236*; NP_001077075.1:p.Trp236*; NP_001077076.1:p.Trp236* |
| chr9:98268719 | C | A | NM_000264.3:c.364G>T; NM_001083602.1:c.166G>T; NM_001083603.1:c.361G>T; NM_001083604.1:c.-90G>T; NM_001083605.1:c.-90G>T; NM_001083606.1:c.-90G>T; NM_001083607.1:c.-90G>T | NP_000255.2:p.Glu122*; NP_001077071.1:p.Glu56*; NP_001077072.1:p.Glu121* | |
| chr9:98215785 | C | A | NM_000264.3:c.3424G>T; NM_001083602.1:c.3226G>T; NM_001083603.1:c.3421G>T; NM_001083604.1:c.2971G>T; NM_001083605.1:c.2971G>T; NM_001083606.1:c.2971G>T; NM_001083607.1:c.2971G>T | NP_000255.2:p.Gly1142*; NP_001077071.1:p.Gly1076*; NP_001077072.1:p.Gly1141*; NP_001077073.1:p.Gly991*; NP_001077074.1:p.Gly991*; NP_001077075.1:p.Gly991*; NP_001077076.1:p.Gly991* | |
| chr9:98218658 | C | T | NM_000264.3:c.3206G>A; NM_001083602.1:c.3008G>A; NM_001083603.1:c.3203G>A; NM_001083604.1:c.2753G>A; NM_001083605.1:c.2753G>A; NM_001083606.1:c.2753G>A; NM_001083607.1:c.2753G>A | NP_000255.2:p.Gly1069Asp; NP_001077071.1:p.Gly1003Asp; NP_001077072.1:p.Gly1068Asp; NP_001077073.1:p.Gly918Asp; NP_001077074.1:p.Gly918Asp; NP_001077075.1:p.Gly918Asp; NP_001077076.1:p.Gly918Asp | |
| chr9:98239830 | T | A | NM_000264.3:c.1502A>T; NM_001083602.1:c.1304A>T; NM_001083603.1:c.1499A>T; NM_001083604.1:c.1049A>T; NM_001083605.1:c.1049A>T; NM_001083606.1:c.1049A>T; NM_001083607.1:c.1049A>T | NP_000255.2:p.Gln501Leu; NP_001077071.1:p.Gln435Leu; NP_001077072.1:p.Gln500Leu; NP_001077073.1:p.Gln350Leu; NP_001077074.1:p.Gln350Leu; NP_001077075.1:p.Gln350Leu; NP_001077076.1:p.Gln350Leu |
| Basal Cell Carcinoma | Hedgehog Signaling Pathway | Antigen Processing and Presentation | ||||||
|---|---|---|---|---|---|---|---|---|
| UACR | PTCH1 Mut −ve | PTCH1 Mut +ve | PTCH1 Mut −ve | PTCH1 Mut +ve | PTCH1 Mut −ve | PTCH1 Mut +ve | ||
| ≤192 µg/g | FC | 4.05 | 6.92 | 2.93 | 5.58 | 4.77 | 6.25 | |
| 95% CI | (3.03–5.4) | (5.30–9.04) | (2.23–3.85) | (4.34–7.17) | (3.72–6.09) | (4.98–7.83) | ||
| ≥192 µg/g | FC | 1.33 | 2.98 | 1.37 | 3.01 | 1.08 | 2.05 | |
| 95% CI | (1.08–1.65) | (2.35–3.7) | (1.12–1.67) | (2.40–3.76) | (−1.098–1.30) | (1.67–2.50) | ||
| Interaction p | 1.68 × 10−31 | 3.17 × 10−24 | 2.75 × 10−52 | |||||
| IL17 Signaling Pathway | Antigen Processing and Presentation | p53 Signaling Pathway | ||||||
|---|---|---|---|---|---|---|---|---|
| UACR | NOTCH1 Mut −ve | NOTCH1 Mut +ve | NOTCH1 Mut −ve | NOTCH1 Mut +ve | NOTCH1 Mut −ve | NOTCH1 Mut +ve | ||
| ≤192 µg/g | FC | 5.13 | 6.45 | 5.44 | 6.09 | 4.96 | 5.77 | |
| 95% CI | (4.18–6.31) | (4.93–8.43) | (4.39–6.71) | (4.62–8.02) | (4.04–6.08) | (4.42–7.53) | ||
| ≥192 µg/g | FC | 1.12 | 2.29 | 1.17 | 2.58 | 1.26 | 2.66 | |
| 95% CI | (−1.04–1.32) | (1.80–2.91) | (−1.01–1.38) | (2.01–3.30) | (1.07–1.48) | (2.09–3.37) | ||
| Interaction p | 6.27 × 10−60 | 2.45 × 10−54 | 2.49 × 10−48 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasmine, F.; Argos, M.; Khamkevych, Y.; Islam, T.; Rakibuz-Zaman, M.; Shahriar, M.; Shea, C.R.; Ahsan, H.; Kibriya, M.G. Molecular Profiling and the Interaction of Somatic Mutations with Transcriptomic Profiles in Non-Melanoma Skin Cancer (NMSC) in a Population Exposed to Arsenic. Cells 2024, 13, 1056. https://doi.org/10.3390/cells13121056
Jasmine F, Argos M, Khamkevych Y, Islam T, Rakibuz-Zaman M, Shahriar M, Shea CR, Ahsan H, Kibriya MG. Molecular Profiling and the Interaction of Somatic Mutations with Transcriptomic Profiles in Non-Melanoma Skin Cancer (NMSC) in a Population Exposed to Arsenic. Cells. 2024; 13(12):1056. https://doi.org/10.3390/cells13121056
Chicago/Turabian StyleJasmine, Farzana, Maria Argos, Yuliia Khamkevych, Tariqul Islam, Muhammad Rakibuz-Zaman, Mohammad Shahriar, Christopher R. Shea, Habibul Ahsan, and Muhammad G. Kibriya. 2024. "Molecular Profiling and the Interaction of Somatic Mutations with Transcriptomic Profiles in Non-Melanoma Skin Cancer (NMSC) in a Population Exposed to Arsenic" Cells 13, no. 12: 1056. https://doi.org/10.3390/cells13121056
APA StyleJasmine, F., Argos, M., Khamkevych, Y., Islam, T., Rakibuz-Zaman, M., Shahriar, M., Shea, C. R., Ahsan, H., & Kibriya, M. G. (2024). Molecular Profiling and the Interaction of Somatic Mutations with Transcriptomic Profiles in Non-Melanoma Skin Cancer (NMSC) in a Population Exposed to Arsenic. Cells, 13(12), 1056. https://doi.org/10.3390/cells13121056