
Targeted Therapy of Multiple Sclerosis: A Case for Antigen-Specific Tregs


Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. CNS Is Not Immune-Privileged
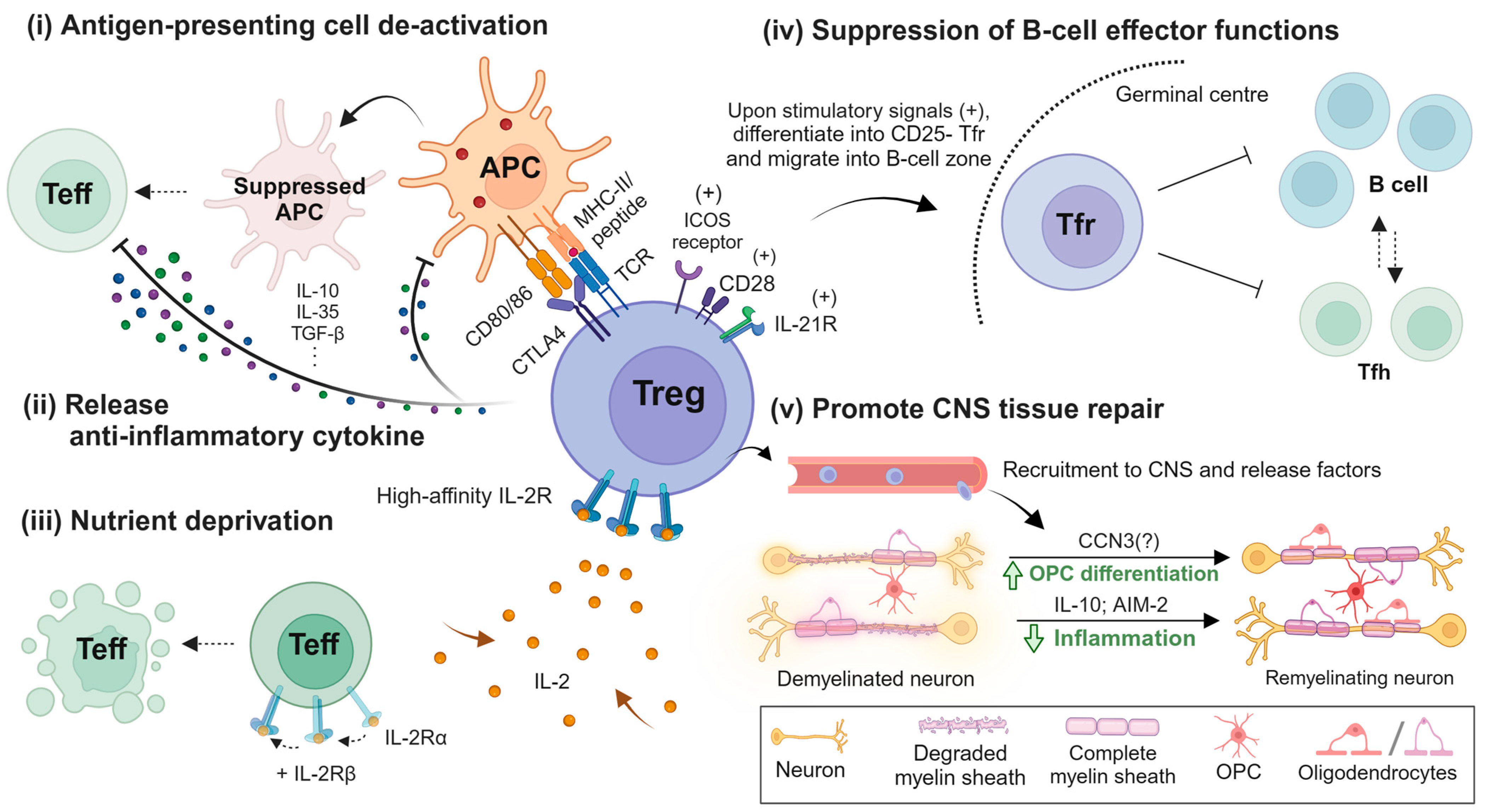
3. Treg Biology and Its Role in MS
4. In Vivo Boosting of Treg Cells for MS Treatment
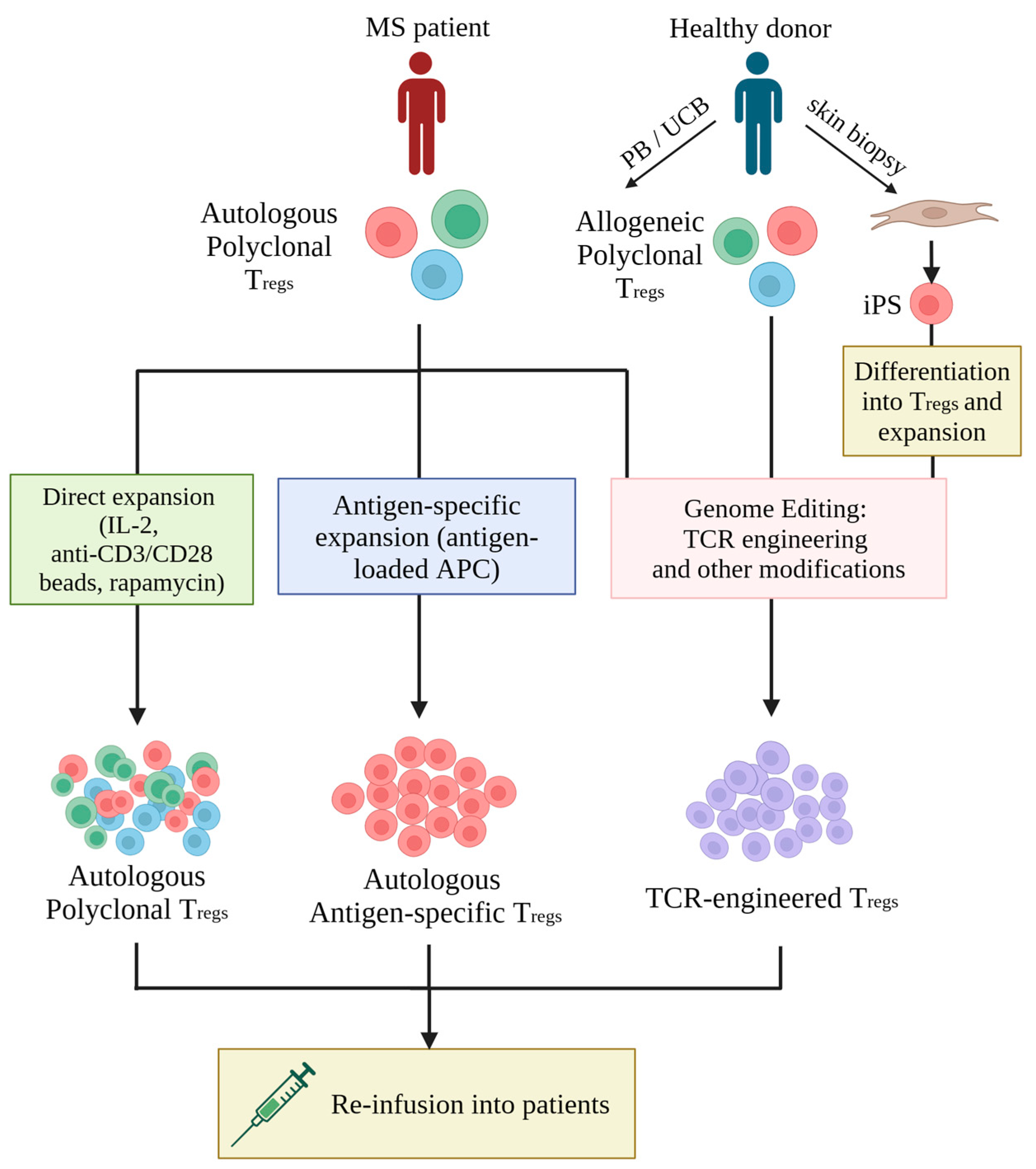
5. Optimizing Adoptive Treg Cell Therapy for MS
6. The Risks and Challenges of Antigen-Specific Treg Cell Therapy
7. Risks of TCR Gene Therapy
8. Improving the Safety of Engineered Treg Therapy
9. Conclusions
10. Future Directions
Funding
Conflicts of Interest
References
- Lorscheider, J.; Buzzard, K.; Jokubaitis, V.; Spelman, T.; Havrdova, E.; Horakova, D.; Trojano, M.; Izquierdo, G.; Girard, M.; Duquette, P.; et al. Defining secondary progressive multiple sclerosis. Brain J. Neurol. 2016, 139, 2395–2405. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Bar-Or, A. Emerging therapies to target CNS pathophysiology in multiple sclerosis. Nat. Rev. Neurol. 2022, 18, 466–475. [Google Scholar] [CrossRef]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 1. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Antel, J.; Kuhlmann, T. Inflammation in multiple sclerosis: Consequences for remyelination and disease progression. Nat. Rev. Neurol. 2023, 19, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood–brain barriers. Trends Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Merkler, D.; Vincenti, I.; Masson, F.; Liblau, S.R. Tissue-resident CD8 T cells in central nervous system inflammatory diseases: Present at the crime scene and …guilty. Curr. Opin. Immunol. 2022, 77, 102211. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, I.; Page, N.; Steinbach, K.; Yermanos, A.; Lemeille, S.; Nunez, N.; Kreutzfeldt, M.; Klimek, B.; Di Liberto, G.; Egervari, K.; et al. Tissue-resident memory CD8+ T cells cooperate with CD4+ T cells to drive compartmentalized immunopathology in the CNS. Sci. Transl. Med. 2022, 14, eabl6058. [Google Scholar] [CrossRef] [PubMed]
- Frieser, D.; Pignata, A.; Khajavi, L.; Shlesinger, D.; Gonzalez-Fierro, C.; Nguyen, X.-H.; Yermanos, A.; Merkler, D.; Höftberger, R.; Desestret, V.; et al. Tissue-resident CD8+ T cells drive compartmentalized and chronic autoimmune damage against CNS neurons. Sci. Transl. Med. 2022, 14, eabl6157. [Google Scholar] [CrossRef]
- Clark, L.B.; Appleby, M.W.; Brunkow, M.E.; Wilkinson, J.E.; Ziegler, S.F.; Ramsdell, F. Cellular and molecular characterization of the scurfy mouse mutant. J. Immunol. 1999, 162, 2546–2554. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.S.; Boesteanu, A.; Reed, A.J.; Petrone, A.L.; Holenbeck, A.E.; Lerman, M.A.; Naji, A.; Caton, A.J. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001, 2, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2019, 20, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25−Naive T Cells to CD4+CD25+Regulatory T Cells by TGF-β Induction of Transcription FactorFoxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Toker, A.; Engelbert, D.; Garg, G.; Polansky, J.K.; Floess, S.; Miyao, T.; Baron, U.; Düber, S.; Geffers, R.; Giehr, P.; et al. Active Demethylation of the Foxp3 Locus Leads to the Generation of Stable Regulatory T Cells within the Thymus. J. Immunol. 2013, 190, 3180–3188. [Google Scholar] [CrossRef] [PubMed]
- Dhamne, C.; Chung, Y.; Alousi, A.M.; Cooper, L.J.N.; Tran, D.Q. Peripheral and Thymic Foxp3+ Regulatory T Cells in Search of Origin, Distinction, and Function. Front. Immunol. 2013, 4, 253. [Google Scholar] [CrossRef] [PubMed]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.-D.; Bopp, T.; Schmitt, E.; et al. Epigenetic Control of the foxp3 Locus in Regulatory T Cells. PLoS Biol. 2007, 5, e38. [Google Scholar] [CrossRef]
- Baron, U.; Floess, S.; Wieczorek, G.; Baumann, K.; Grützkau, A.; Dong, J.; Thiel, A.; Boeld, T.J.; Hoffmann, P.; Edinger, M.; et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur. J. Immunol. 2007, 37, 2378–2389. [Google Scholar] [CrossRef]
- Duffy, S.S.; Keating, B.A.; Moalem-Taylor, G. Adoptive Transfer of Regulatory T Cells as a Promising Immunotherapy for the Treatment of Multiple Sclerosis. Front. Neurosci. 2019, 13, 1107. [Google Scholar] [CrossRef]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9, 585. [Google Scholar] [CrossRef]
- Arpaia, N.; Green Jesse, A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting Piper, M.; Rudensky Alexander, Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.H.; Tripathi, R.B.; Foerster, S.; McKenzie, I.; Kougioumtzidou, E.; Grist, M.; Richardson, W.D.; Franklin, R.J.M. Pre-Existing Mature Oligodendrocytes Do Not Contribute to Remyelination following Toxin-Induced Spinal Cord Demyelination. Am. J. Pathol. 2016, 186, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Naughton, M.; Moffat, J.; Eleftheriadis, G.; Gallardo, N.d.l.V.; Young, A.; Falconer, J.; Hawkins, K.; Pearson, B.; Perbal, B.; Hogan, A.; et al. CCN3 is dynamically regulated by treatment and disease state in multiple sclerosis. J. Neuroinflammation 2020, 17, 349. [Google Scholar] [CrossRef] [PubMed]
- De la Vega Gallardo, N.; Penalva, R.; Dittmer, M.; Naughton, M.J.; Falconer, J.L.; Moffat, J.; Guzman, A.; Hombrebueno, J.R.; Lin, Z.; Perbal, B.; et al. Dynamic CCN3 expression in the murine CNS does not confer essential roles in myelination or remyelination. Proc. Natl. Acad. Sci. USA 2020, 117, 18018–18028. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-C.; Guo, Z.; Guo, H.; Chen, L.; Zhang, G.; Liang, K.; Xie, L.; Tan, X.; Gibson, S.A.; Rampanelli, E.; et al. AIM2 in regulatory T cells restrains autoimmune diseases. Nature 2021, 591, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Sage, P.T.; Ron-Harel, N.; Juneja, V.R.; Sen, D.R.; Maleri, S.; Sungnak, W.; Kuchroo, V.K.; Haining, W.N.; Chevrier, N.; Haigis, M.; et al. Suppression by TFR cells leads to durable and selective inhibition of B cell effector functions. Nat. Immunol. 2016, 17, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.S.; Keating, B.A.; Perera, C.J.; Moalem-Taylor, G. The role of regulatory T cells in nervous system pathologies. J. Neurosci. Res. 2017, 96, 951–968. [Google Scholar] [CrossRef]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of Functional Suppression by CD4+CD25+Regulatory T Cells in Patients with Multiple Sclerosis. J. Exp. Med. 2004, 199, 971–979. [Google Scholar] [CrossRef]
- Haas, J.; Fritzsching, B.; Trübswetter, P.; Korporal, M.; Milkova, L.; Fritz, B.; Vobis, D.; Krammer, P.H.; Suri-Payer, E.; Wildemann, B. Prevalence of Newly Generated Naive Regulatory T Cells (Treg) Is Critical for Treg Suppressive Function and Determines Treg Dysfunction in Multiple Sclerosis. J. Immunol. 2007, 179, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Villar, M.; Baecher-Allan, C.M.; Hafler, D.A. Identification of T helper type 1–like, Foxp3+ regulatory T cells in human autoimmune disease. Nat. Med. 2011, 17, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Zhang, A.-H.; Yoon, J.; Culp, W.E.; Lees, J.R.; Wucherpfennig, K.W.; Scott, D.W. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J. Autoimmun. 2018, 92, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.A.; Malpass, K.H.; Anderton, S.M. Curing CNS autoimmune disease with myelin-reactive Foxp3+Treg. Eur. J. Immunol. 2009, 39, 1108–1117. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]
- Rhodes, K.R.; Tzeng, S.Y.; Iglesias, M.; Lee, D.; Storm, K.; Neshat, S.Y.; VanDyke, D.; Lowmaster, S.M.; Spangler, J.B.; Raimondi, G.; et al. Bioengineered particles expand myelin-specific regulatory T cells and reverse autoreactivity in a mouse model of multiple sclerosis. Sci. Adv. 2023, 9, eadd8693. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [PubMed]
- Motwani, K.; Peters, L.D.; Vliegen, W.H.; El-sayed, A.G.; Seay, H.R.; Lopez, M.C.; Baker, H.V.; Posgai, A.L.; Brusko, M.A.; Perry, D.J.; et al. Human Regulatory T Cells from Umbilical Cord Blood Display Increased Repertoire Diversity and Lineage Stability Relative to Adult Peripheral Blood. Front. Immunol. 2020, 11, 611. [Google Scholar] [CrossRef] [PubMed]
- Seay, H.R.; Putnam, A.L.; Cserny, J.; Posgai, A.L.; Rosenau, E.H.; Wingard, J.R.; Girard, K.F.; Kraus, M.; Lares, A.P.; Brown, H.L.; et al. Expansion of Human Tregs from Cryopreserved Umbilical Cord Blood for GMP-Compliant Autologous Adoptive Cell Transfer Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 178–191. [Google Scholar] [CrossRef]
- Haque, M.; Song, J.; Fino, K.; Sandhu, P.; Song, X.; Lei, F.; Zheng, S.; Ni, B.; Fang, D.; Song, J. Stem cell-derived tissue-associated regulatory T cells ameliorate the development of autoimmunity. Sci. Rep. 2016, 6, 20588. [Google Scholar] [CrossRef]
- Haque, R.; Lei, F.; Xiong, X.; Bian, Y.; Zhao, B.; Wu, Y.; Song, J. Programming of Regulatory T Cells from Pluripotent Stem Cells and Prevention of Autoimmunity. J. Immunol. 2012, 189, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Bronge, M.; Högelin, K.A.; Thomas, O.G.; Ruhrmann, S.; Carvalho-Queiroz, C.; Nilsson, O.B.; Kaiser, A.; Zeitelhofer, M.; Holmgren, E.; Linnerbauer, M.; et al. Identification of four novel T cell autoantigens and personal autoreactive profiles in multiple sclerosis. Sci. Adv. 2022, 8, eabn1823. [Google Scholar] [CrossRef] [PubMed]
- McGovern, J.L.; Holler, A.; Thomas, S.; Stauss, H.J. Forced Fox-P3 expression can improve the safety and antigen-specific function of engineered regulatory T cells. J. Autoimmun. 2022, 132, 102888. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Henriksen, K.J.; Bi, M.; Finger, E.B.; Szot, G.; Ye, J.; Masteller, E.L.; McDevitt, H.; Bonyhadi, M.; Bluestone, J.A. In Vitro–expanded Antigen-specific Regulatory T Cells Suppress Autoimmune Diabetes. J. Exp. Med. 2004, 199, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Tarbell, K.V.; Yamazaki, S.; Olson, K.; Toy, P.; Steinman, R.M. CD25+ CD4+ T Cells, Expanded with Dendritic Cells Presenting a Single Autoantigenic Peptide, Suppress Autoimmune Diabetes. J. Exp. Med. 2004, 199, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Borojevic, R.; Streutker, C.; Snider, D.; Liang, H.; Croitoru, K. Expression of Dual TCR on DO11.10 T Cells Allows for Ovalbumin-Induced Oral Tolerance to Prevent T Cell-Mediated Colitis Directed against Unrelated Enteric Bacterial Antigens. J. Immunol. 2004, 172, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Fujio, K.; Okamoto, A.; Araki, Y.; Shoda, H.; Tahara, H.; Tsuno, N.H.; Takahashi, K.; Kitamura, T.; Yamamoto, K. Gene Therapy of Arthritis with TCR Isolated from the Inflamed Paw. J. Immunol. 2006, 177, 8140–8147. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.-S.; Tanriver, Y.; Jiang, S.; Xue, S.-A.; Ratnasothy, K.; Chen, D.; Stauss, H.J.; Bucy, R.P.; Lombardi, G.; Lechler, R. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J. Clin. Investig. 2008, 118, 3619–3628. [Google Scholar] [CrossRef] [PubMed]
- Serra P & Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 2019, 37, 238–251. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-shelf” allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Miyao, T.; Floess, S.; Setoguchi, R.; Luche, H.; Fehling, H.; Waldmann, H.; Huehn, J.; Hori, S. Plasticity of Foxp3+ T Cells Reflects Promiscuous Foxp3 Expression in Conventional T Cells but Not Reprogramming of Regulatory T Cells. Immunity 2012, 36, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.-L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A Serious Adverse Event after Successful Gene Therapy for X-Linked Severe Combined Immunodeficiency. New Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Bendle, G.M.; Linnemann, C.; I Hooijkaas, A.; Bies, L.; A de Witte, M.; Jorritsma, A.; Kaiser, A.D.M.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010, 16, 565–570. [Google Scholar] [CrossRef]
- Cohen, C.J.; Li, Y.; El-Gamil, M.; Robbins, P.D.; Rosenberg, S.A.; Morgan, R.A. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors with a Second Disulfide Bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef] [PubMed]
- Bethune, M.T.; Gee, M.H.; Bunse, M.; Lee, M.S.; Gschweng, E.H.; Pagadala, M.S.; Zhou, J.; Cheng, D.; Heath, J.R.; Kohn, D.B.; et al. Domain-swapped T cell receptors improve the safety of TCR gene therapy. eLife 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Uckert, W. Minimal Amino Acid Exchange in Human TCR Constant Regions Fosters Improved Function of TCR Gene-Modified T Cells. J. Immunol. 2010, 184, 6223–6231. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Mohammed, F.; Reijmers, R.M.; Woolston, A.; Stauss, T.; Kennedy, A.; Stirling, D.; Holler, A.; Green, L.; Jones, D.; et al. Framework engineering to produce dominant T cell receptors with enhanced antigen-specific function. Nat. Commun. 2019, 10, 4451. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.V.; Vignali, D.A.A. Once a Treg, always a Treg? Immunol. Rev. 2014, 259, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Bucktrout Samantha, L.; Martinez-Llordella, M.; Zhou, X.; Anthony, B.; Rosenthal, W.; Luche, H.; Fehling Hans, J.; Bluestone Jeffrey, A. Self-antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity 2013, 39, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Honaker, Y.; Hubbard, N.; Xiang, Y.; Fisher, L.; Hagin, D.; Sommer, K.; Song, Y.; Yang, S.J.; Lopez, C.; Tappen, T.; et al. Gene editing to induce FOXP3 expression in human CD4 + T cells leads to a stable regulatory phenotype and function. Sci. Transl. Med. 2020, 12, eaay6422. [Google Scholar] [CrossRef]
- Wright, G.A.; Notley, C.A.; Xue, S.-A.; Bendle, G.M.; Holler, A.; Schumacher, T.N.; Ehrenstein, M.R.; Stauss, H.J. Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 19078–19083. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, Y.; Stauss, H.J. Targeted Therapy of Multiple Sclerosis: A Case for Antigen-Specific Tregs. Cells 2024, 13, 797. https://doi.org/10.3390/cells13100797
Zhong Y, Stauss HJ. Targeted Therapy of Multiple Sclerosis: A Case for Antigen-Specific Tregs. Cells. 2024; 13(10):797. https://doi.org/10.3390/cells13100797
Chicago/Turabian StyleZhong, Yiya, and Hans J. Stauss. 2024. "Targeted Therapy of Multiple Sclerosis: A Case for Antigen-Specific Tregs" Cells 13, no. 10: 797. https://doi.org/10.3390/cells13100797
APA StyleZhong, Y., & Stauss, H. J. (2024). Targeted Therapy of Multiple Sclerosis: A Case for Antigen-Specific Tregs. Cells, 13(10), 797. https://doi.org/10.3390/cells13100797