
A Potent PDK4 Inhibitor for Treatment of Heart Failure with Reduced Ejection Fraction

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
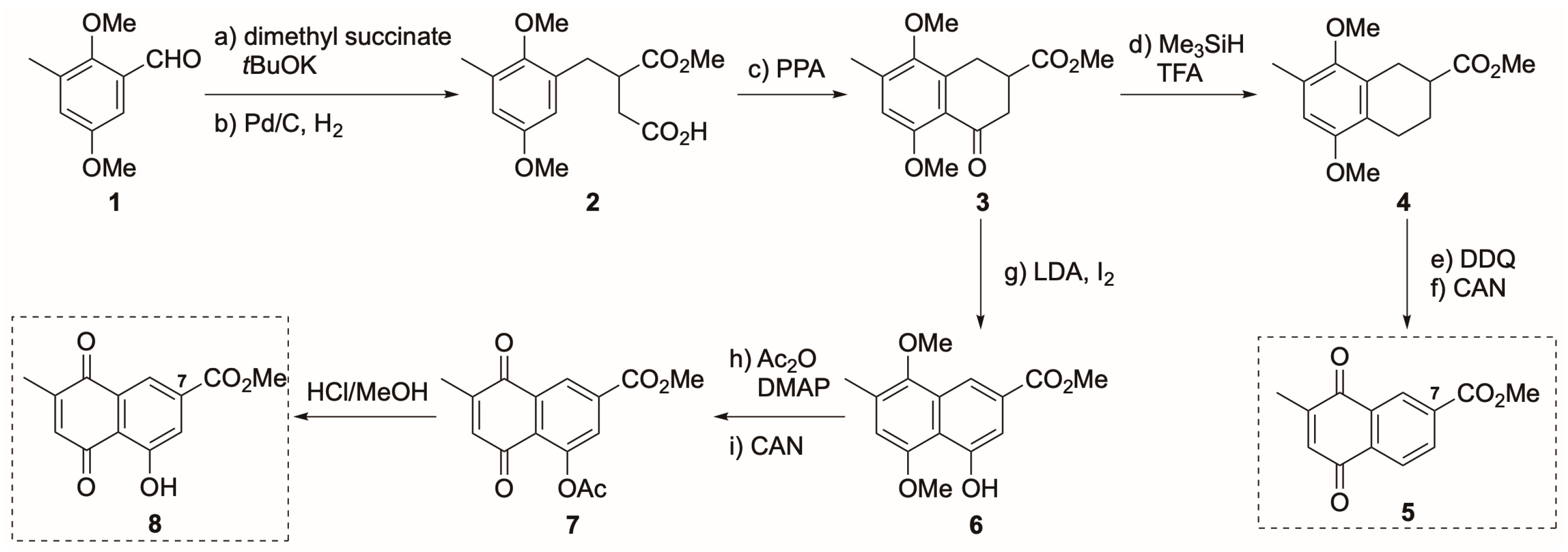
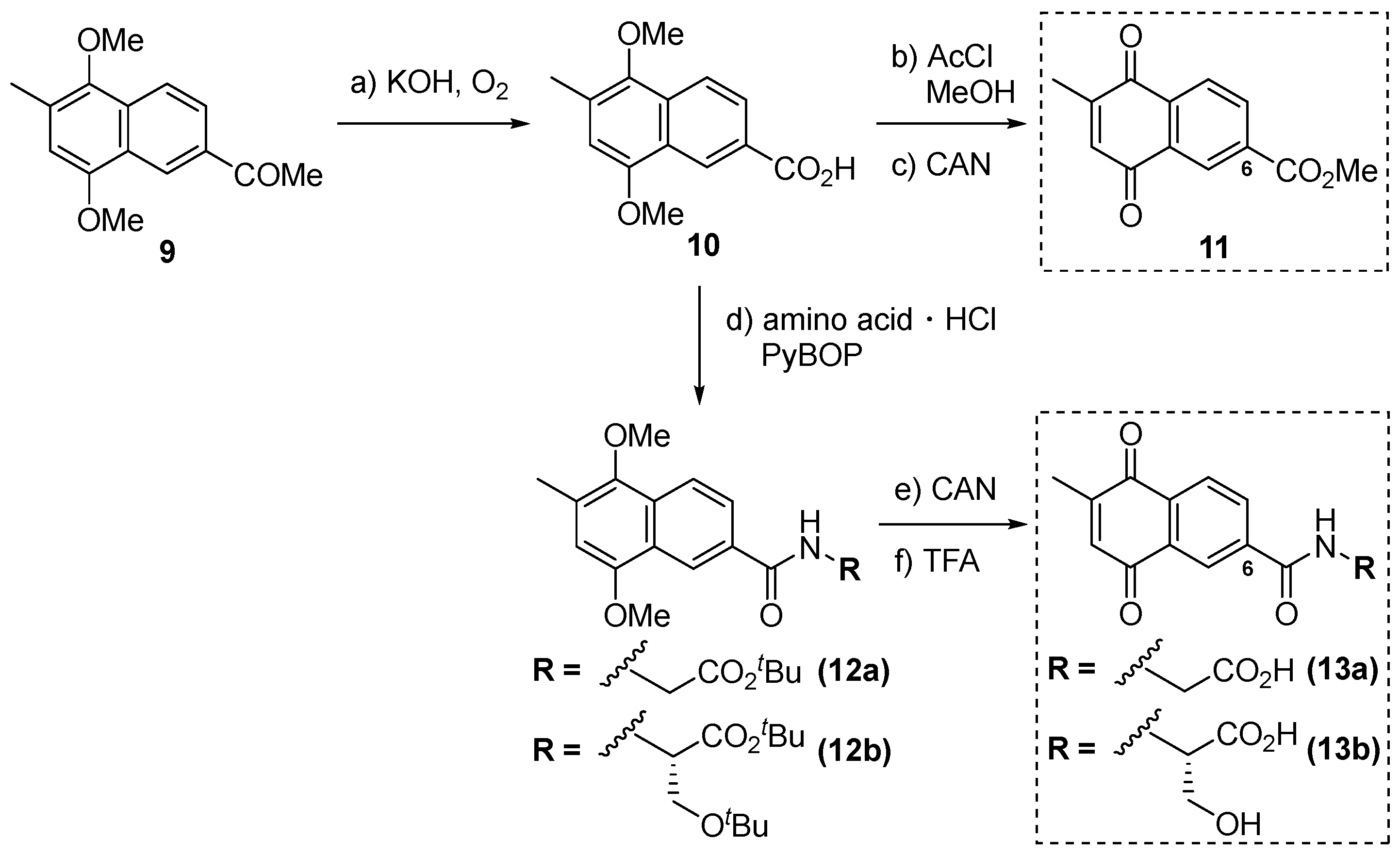
2.1. Synthesis of Novel PDK4 Inhibitors
2.2. Assays for PDK4 Inhibition Capacity and Toxicity
2.2.1. Description of PDK4
2.2.2. Preparation of Solution of Derivatives
2.2.3. Off-Chip Mobility Shift Assay (MSA) (Carna Biosciences, Inc., Kobe, Japan) (Table 1)
2.2.4. Calculation
2.2.5. Cytotoxicity Assay against Human Fibroblast Cells
2.3. Animals
2.4. Transverse Aortic Constriction (TAC)
2.5. Echocardiography
2.6. Injection
2.7. Sample Collection
2.8. Picrosirius Red Staining
2.9. Immunoblot Analysis
2.10. PDH Activity
2.11. Statistical Analysis
3. Results
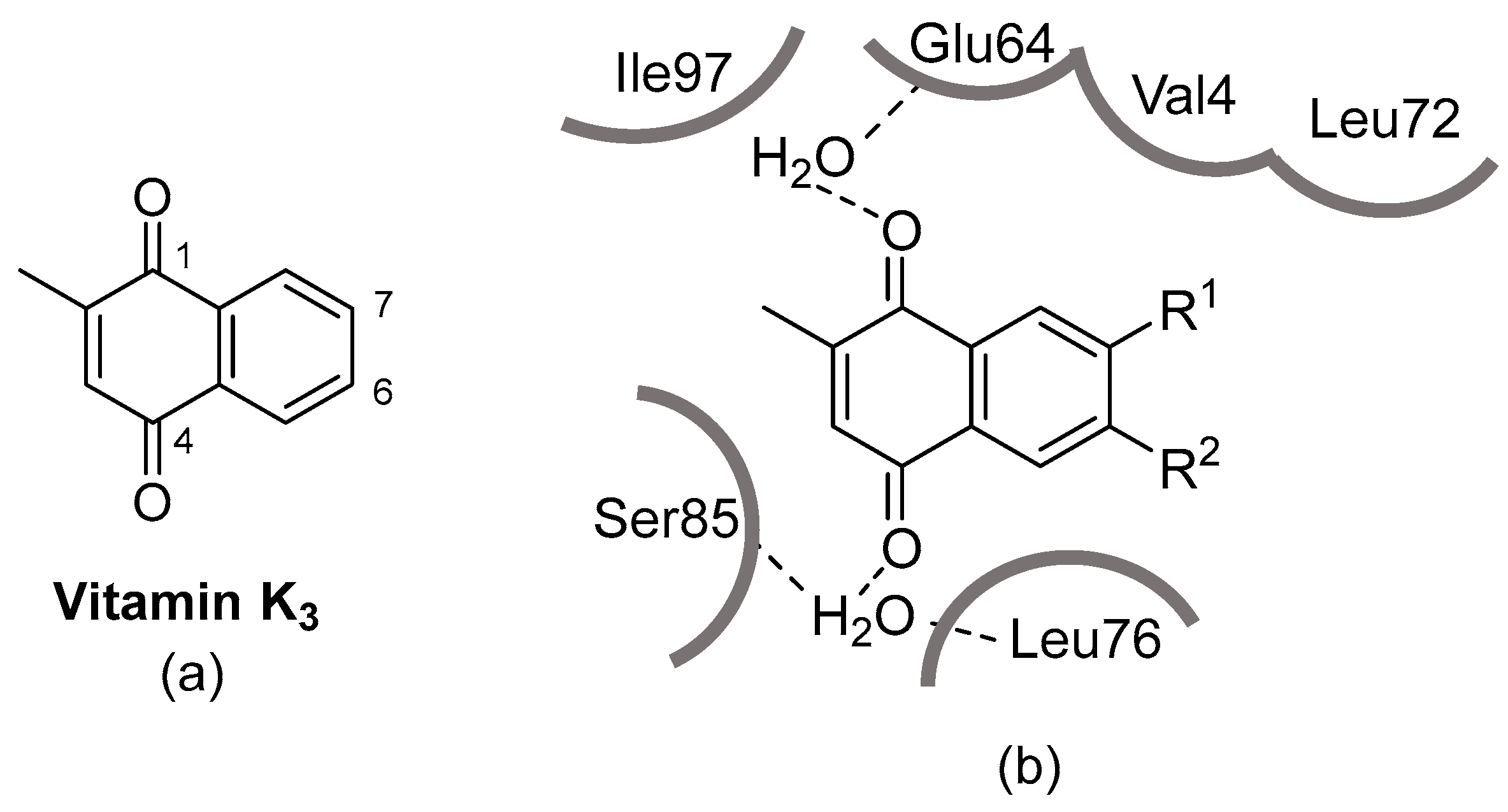
3.1. Synthesis of Derivatives of Vitamin K3 for PDK4 Inhibitors
3.2. PDK4 Inhibitory Activity, Toxicity, and Cardioprotection of PDK4 Inhibitor Candidates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PDK4 Inhibitory Activity (%) | Toxicity (IC50 (µg/mL)) |
|---|---|---|
| Vitamin K3 | 65.9 | 18 |
| 5 | 34.6 | 8 |
| 8 | 81.0 | 1 |
| 11 | 65.4 | 7 |
| 13a | 66.8 | >110 |
| 13b | 63.2 | >99 |
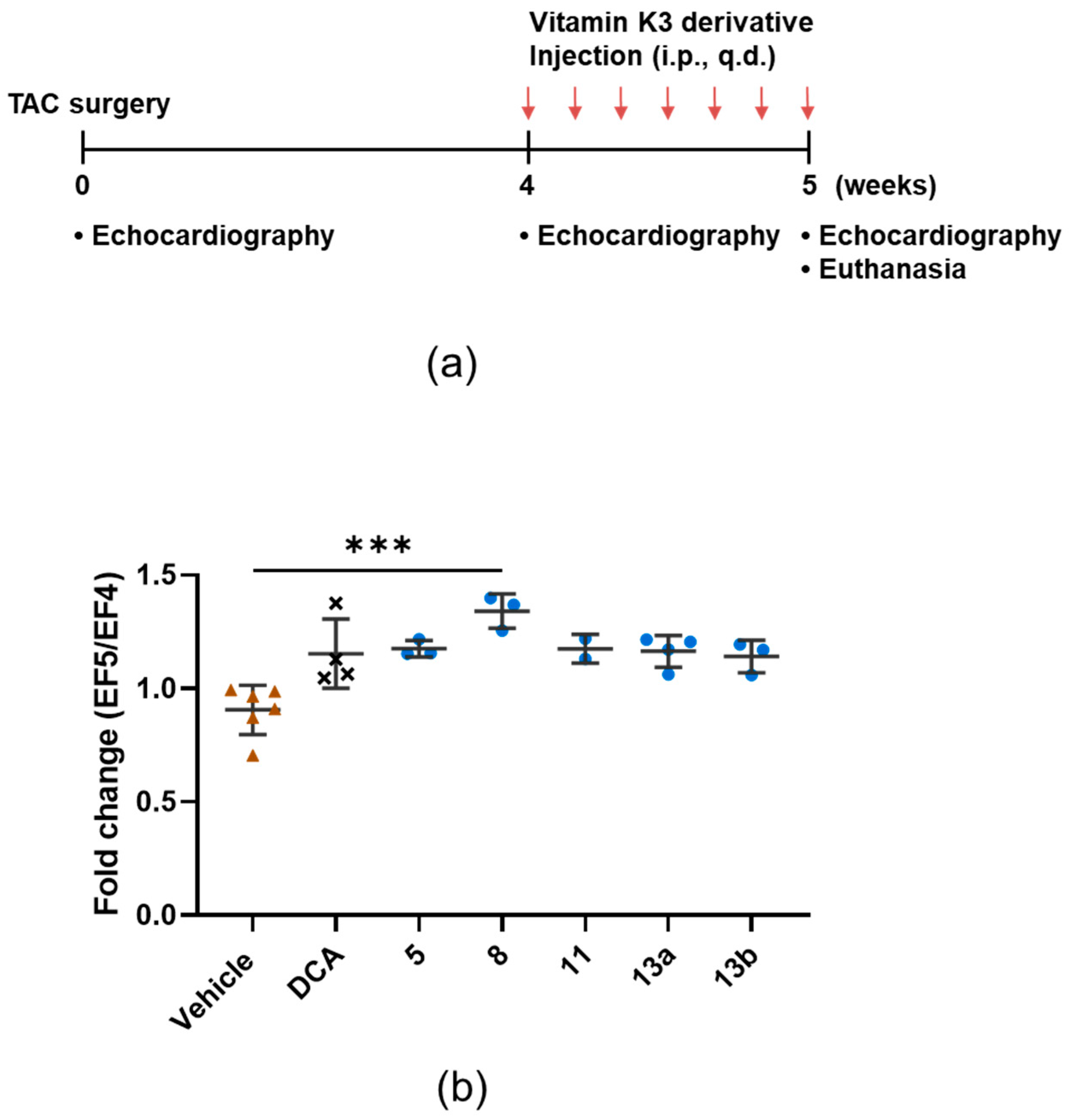
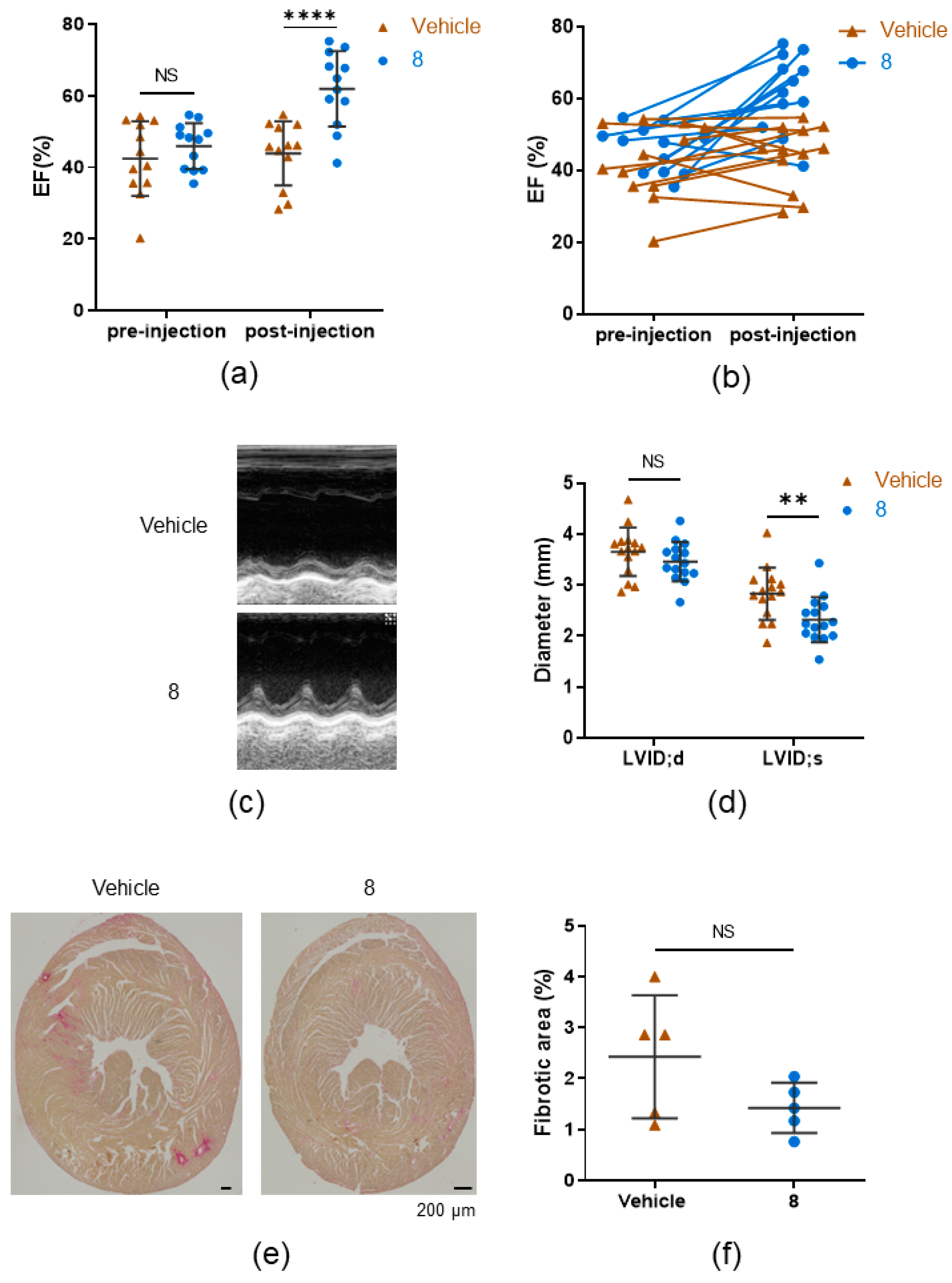
3.3. 8 Improves the Ejection Fraction
3.4. Compound 8 Treats Heart Failure by Activating Pyruvate Dehydrogenase
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.S.; Tu, J.V.; Lee, D.S.; Austin, P.C.; Fang, J.; Haouzi, A.; Gong, Y.; Liu, P.P. Outcome of heart failure with preserved ejection fraction in a population-based study. N. Engl. J. Med. 2006, 355, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef] [PubMed]
- Pellicori, P.; Khan, M.J.I.; Graham, F.J.; Cleland, J.G.F. New perspectives and future directions in the treatment of heart failure. Heart Fail Rev. 2020, 25, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Chen, J.; Alimonti, A.; Cavalli, A. How phosphorylation influences E1 subunit pyruvate dehydrogenase: A computational study. Sci. Rep. 2018, 8, 14683. [Google Scholar] [CrossRef]
- Wang, X.; Shen, X.; Yan, Y.; Li, H. Pyruvate dehydrogenase kinases (PDKs): An overview toward clinical applications. Biosci. Rep. 2021, 41, BSR20204402. [Google Scholar] [CrossRef]
- Kiilerich, K.; Adser, H.; Jakobsen, A.; Pedersen, P.; Hardie, D.; Wojtaszewski, J.; Pilegaard, H. PGC-1 increases PDH content but does not change acute PDH regulation in mouse skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1350–R1359. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Zhabyeyev, P.; Gandhi, M.; Mori, J.; Basu, R.; Kassiri, Z.; Clanachan, A.; Lopaschuk, G.D.; Oudit, G.Y. Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc. Res. 2013, 97, 676–685. [Google Scholar] [CrossRef]
- Moon, S.S.; Lee, J.E.; Lee, Y.S.; Kim, S.W.; Jeoung, N.H.; Lee, I.K.; Kim, J.G. Association of pyruvate dehydrogenase kinase 4 gene polymorphisms with type 2 diabetes and metabolic syndrome. Diabetes Res. Clin. Pract. 2012, 95, 230–236. [Google Scholar] [CrossRef]
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Agonist-Induced Hypertrophy and Diastolic Dysfunction Are Associated With Selective Reduction in Glucose Oxidation. Circ. Heart Fail. 2012, 5, 493–503. [Google Scholar] [CrossRef]
- Yamane, K.; Indalao, I.L.; Chida, J.; Yamamoto, Y.; Hanawa, M.; Kido, H. Diisopropylamine Dichloroacetate, a Novel Pyruvate Dehydrogenase Kinase 4 Inhibitor, as a Potential Therapeutic Agent for Metabolic Disorders and Multiorgan Failure in Severe Influenza. PLoS ONE 2014, 9, e98032. [Google Scholar] [CrossRef]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail 2010, 3, 420–430. [Google Scholar] [CrossRef]
- Felitsyn, N.; Stacpoole, P.W.; Notterpek, L. Dichloroacetate causes reversible demyelination in vitro: Potential mechanism for its neuropathic effect. J. Neurochem. 2007, 100, 429–436. [Google Scholar] [CrossRef]
- Yao, M.; Yao, D.; Yamaguchi, M.; Chida, J.; Yao, D.; Kido, H. Bezafibrate upregulates carnitine palmitoyltransferase II expression and promotes mitochondrial energy crisis dissipation in fibroblasts of patients with influenza-associated encephalopathy. Mol. Genet. Metab. 2011, 104, 265–272. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Adams, M.A.; Jia, Z. Structural and Biochemical Evidence for an Enzymatic Quinone Redox Cycle in Escherichia coli: Identification of a novel quinol monooxygenase * [boxs]. J. Biol. Chem. 2005, 280, 8358–8363. [Google Scholar] [CrossRef]
- Maloney, D.J.; Hecht, S.M. A Stereocontrolled Synthesis of δ-trans-Tocotrienoloic Acid. Org. Lett. 2005, 7, 4297–4300. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L.; Seely, G.; Liddell, P.; Barrett, D.; Harding, L.O.; Ma, X.C.; Lee, S.-J.; Gao, F. A carotenoid-porphyrin-diquinone tetrad: Synthesis, electrochemistry and photoinitiated electron transfer. Tetrahedron 1989, 45, 4867–4891. [Google Scholar] [CrossRef]
- Nyland, R.L., II; Luo, M.; Kelley, M.R.; Borch, R.F. Design and Synthesis of Novel Quinone Inhibitors Targeted to the Redox Function of Apurinic/Apyrimidinic Endonuclease 1/Redox Enhancing Factor-1 (Ape1/Ref-1). J. Med. Chem. 2010, 53, 1200–1210. [Google Scholar] [CrossRef]
- Cui, X.-R.; Saito, R.; Kubo, T.; Kon, D.; Hirano, Y.; Saito, S. Preparations of Anthraquinone and Naphthoquinone Derivatives and Their Cytotoxic Effects. Chem. Pharm. Bull. 2011, 59, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Nakano, H.; Yamaji, K.; Yamamoto, T.; Kido, H.; Chida, J.; Yamane, K. Pdk4 Inhibitor and Use Thereof. U.S. Patent US20150335610A1, 3 July 2014. [Google Scholar]
- Methatham, T.; Nagai, R.; Aizawa, K. A New Hypothetical Concept in Metabolic Understanding of Cardiac Fibrosis: Glycolysis Combined with TGF-β and KLF5 Signaling. Int. J. Mol. Sci. 2022, 23, 4302. [Google Scholar] [PubMed]
- Methatham, T.; Tomida, S.; Kimura, N.; Imai, Y.; Aizawa, K. Inhibition of the canonical Wnt signaling pathway by a β-catenin/CBP inhibitor prevents heart failure by ameliorating cardiac hypertrophy and fibrosis. Sci. Rep. 2021, 11, 14886. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Jasinska-Piadlo, A.; Campbell, P. Management of patients with heart failure and preserved ejection fraction. Heart 2023, 109, 874–883. [Google Scholar] [CrossRef]
- Morciano, G.; Vitto, V.A.M.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial Bioenergetics and Dynamism in the Failing Heart. Life 2021, 11, 436. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y. Branched chain amino acid metabolic reprogramming in heart failure. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 2270–2275. [Google Scholar] [CrossRef]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.-Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Uddin, G.M.; Zhang, L.; Shah, S.; Fukushima, A.; Wagg, C.S.; Gopal, K.; Al Batran, R.; Pherwani, S.; Ho, K.L.; Boisvenue, J.; et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc. Diabetol. 2019, 18, 86. [Google Scholar] [CrossRef] [PubMed]
- Bersin, R.M.; Stacpoole, P.W. Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am. Heart J. 1997, 134 Pt 1, 841–855. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Mehta, H.S.; Barker, C.M.; Van Houten, J.; Mollenkopf, S.; Gunnarsson, C.; Ryan, M.; Cork, D.P. Mortality and guideline-directed medical therapy in real-world heart failure patients with reduced ejection fraction. Clin. Cardiol. 2021, 44, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.L.; Dhekne, V.V.; Rao, A.S. Synthesis of 6-acetyl-1,4-dimethoxy-2-methylnaphthalene. Indian J. Chem. B 1983, 22, 23–26. [Google Scholar]

| Kinase | Platform | Substrate | ATP (μM) | Metal | Positive Control | |||
|---|---|---|---|---|---|---|---|---|
| Name | (nM) | Km | Assay | Name | (mM) | |||
| PDK4 | MSA | PDHKtide | 1000 | 19 | 25 | Mg + K | 5 + 25 | CHCl2CO2H |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aizawa, K.; Ikeda, A.; Tomida, S.; Hino, K.; Sugita, Y.; Hirose, T.; Sunazuka, T.; Kido, H.; Yokoyama, S.; Nagai, R. A Potent PDK4 Inhibitor for Treatment of Heart Failure with Reduced Ejection Fraction. Cells 2024, 13, 87. https://doi.org/10.3390/cells13010087
Aizawa K, Ikeda A, Tomida S, Hino K, Sugita Y, Hirose T, Sunazuka T, Kido H, Yokoyama S, Nagai R. A Potent PDK4 Inhibitor for Treatment of Heart Failure with Reduced Ejection Fraction. Cells. 2024; 13(1):87. https://doi.org/10.3390/cells13010087
Chicago/Turabian StyleAizawa, Kenichi, Akari Ikeda, Shota Tomida, Koki Hino, Yuuki Sugita, Tomoyasu Hirose, Toshiaki Sunazuka, Hiroshi Kido, Shigeyuki Yokoyama, and Ryozo Nagai. 2024. "A Potent PDK4 Inhibitor for Treatment of Heart Failure with Reduced Ejection Fraction" Cells 13, no. 1: 87. https://doi.org/10.3390/cells13010087
APA StyleAizawa, K., Ikeda, A., Tomida, S., Hino, K., Sugita, Y., Hirose, T., Sunazuka, T., Kido, H., Yokoyama, S., & Nagai, R. (2024). A Potent PDK4 Inhibitor for Treatment of Heart Failure with Reduced Ejection Fraction. Cells, 13(1), 87. https://doi.org/10.3390/cells13010087