
Characterization of a Preclinical In Vitro Model Derived from a SMARCA4-Mutated Sinonasal Teratocarcinosarcoma
 , , , , , ,
, , , , , ,  ,
, 
Abstract
:1. Introduction
2. Material and Methods
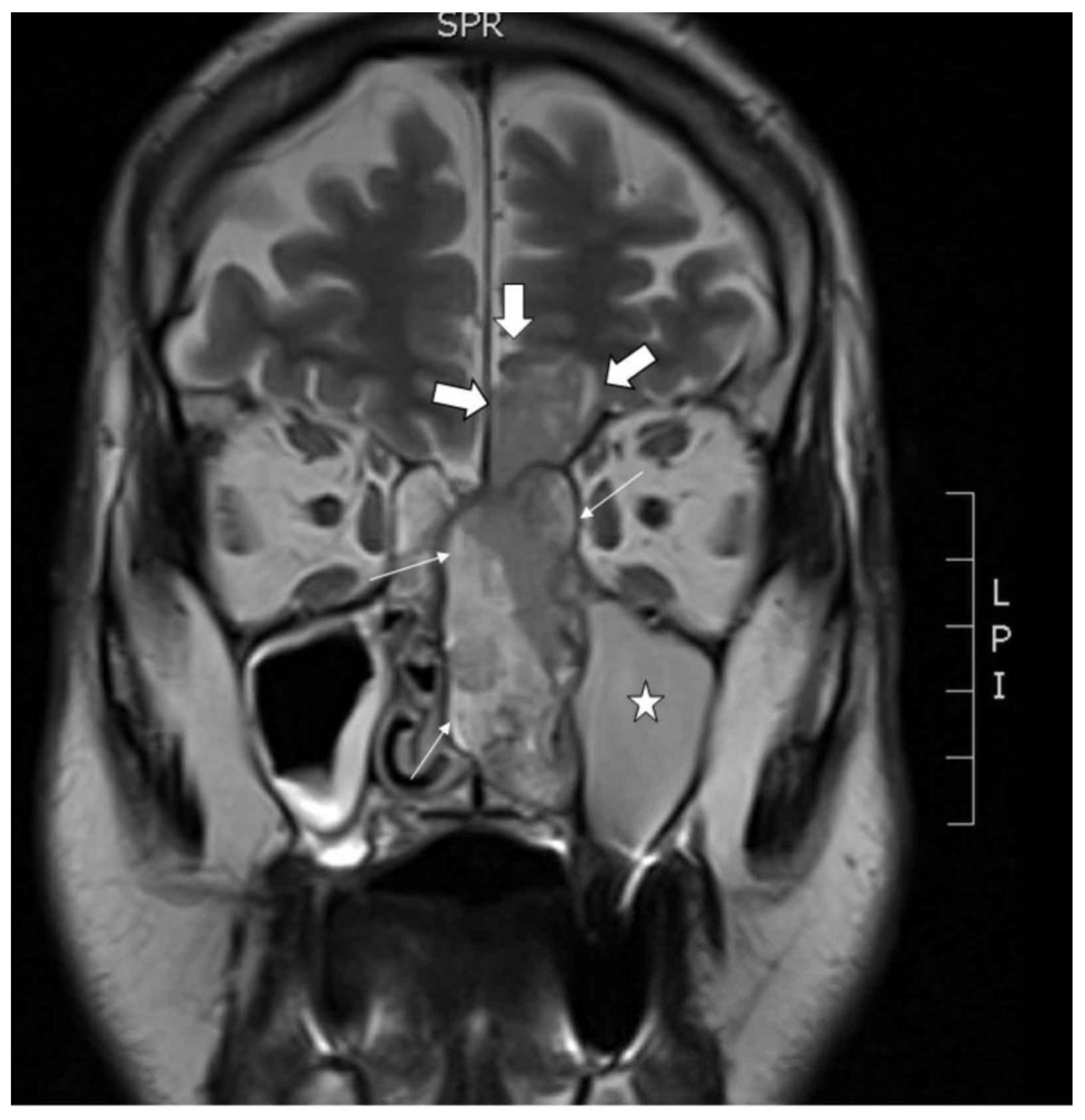
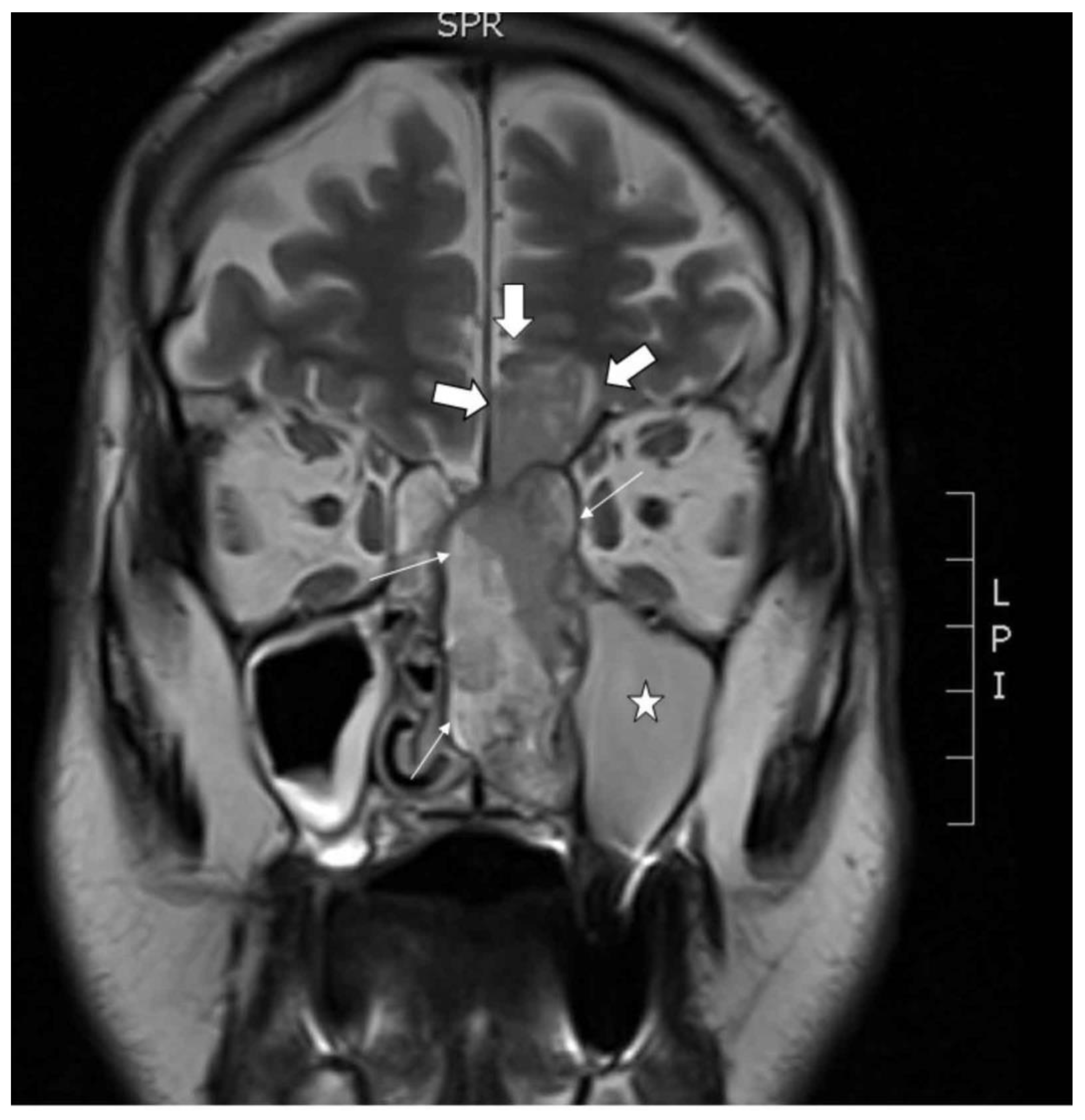
2.1. Clinical Description
2.2. Establishment of Cell Line TCS627
2.3. DNA Extraction and Cell Line Authentication
2.4. Genetic Characterization
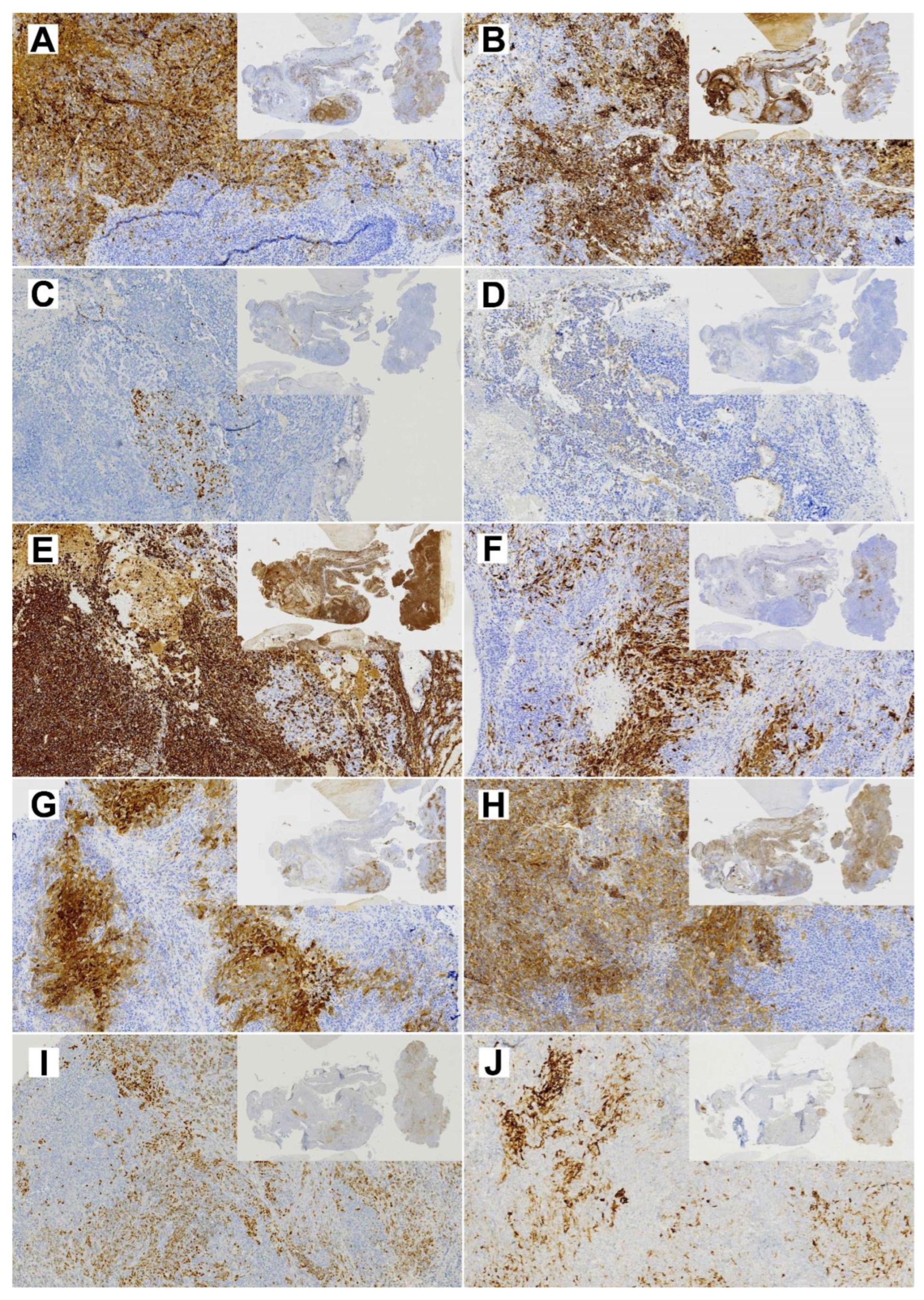
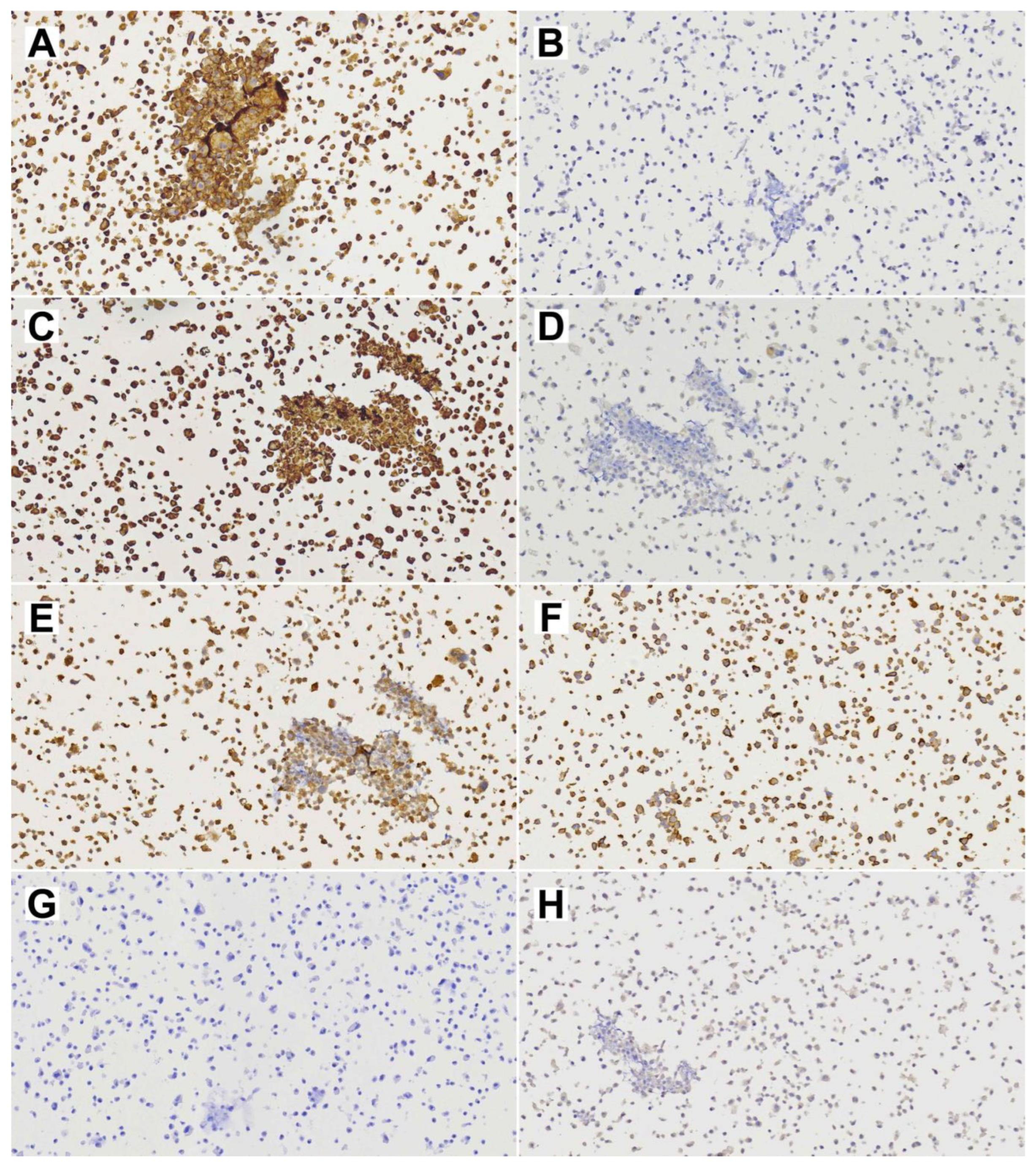
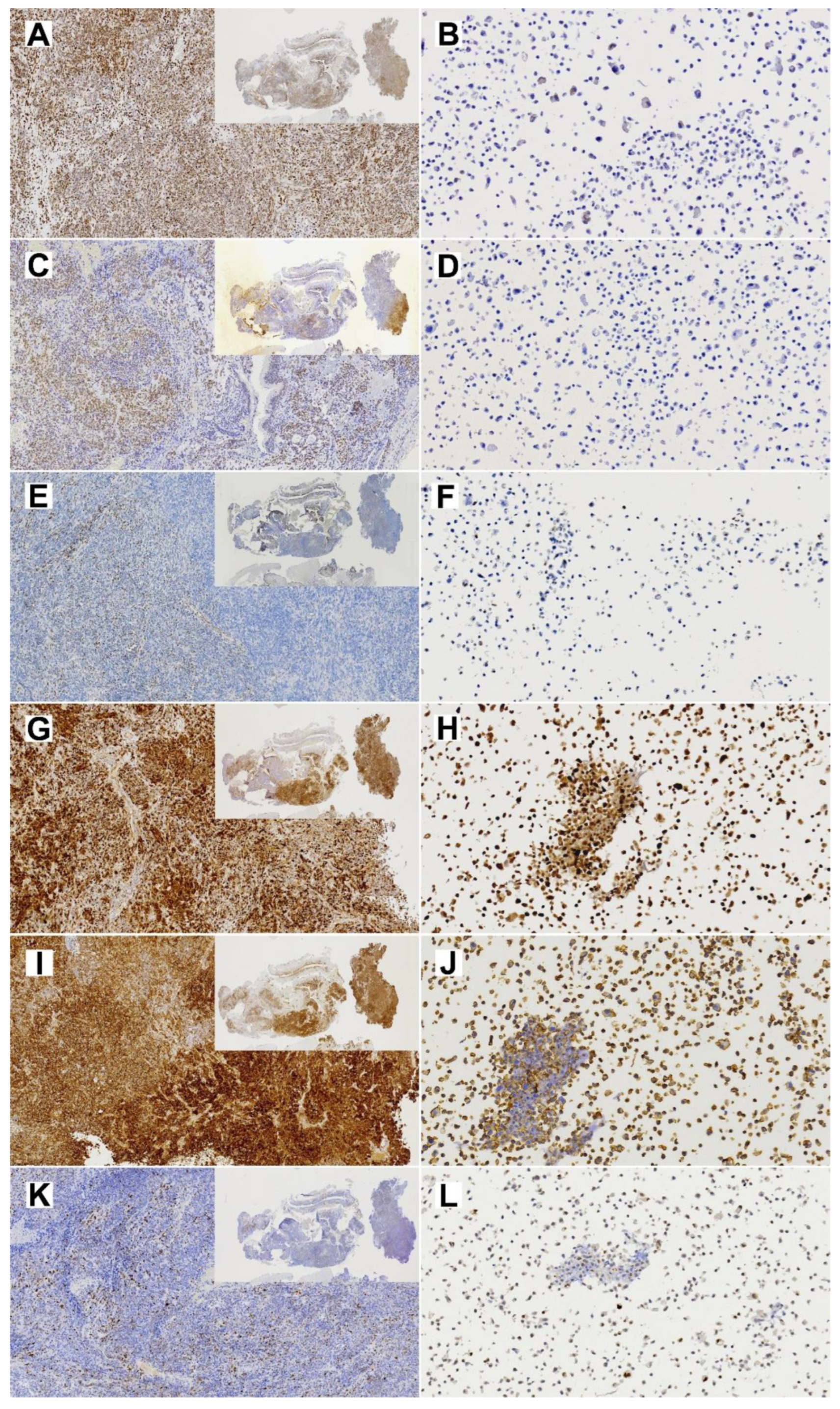
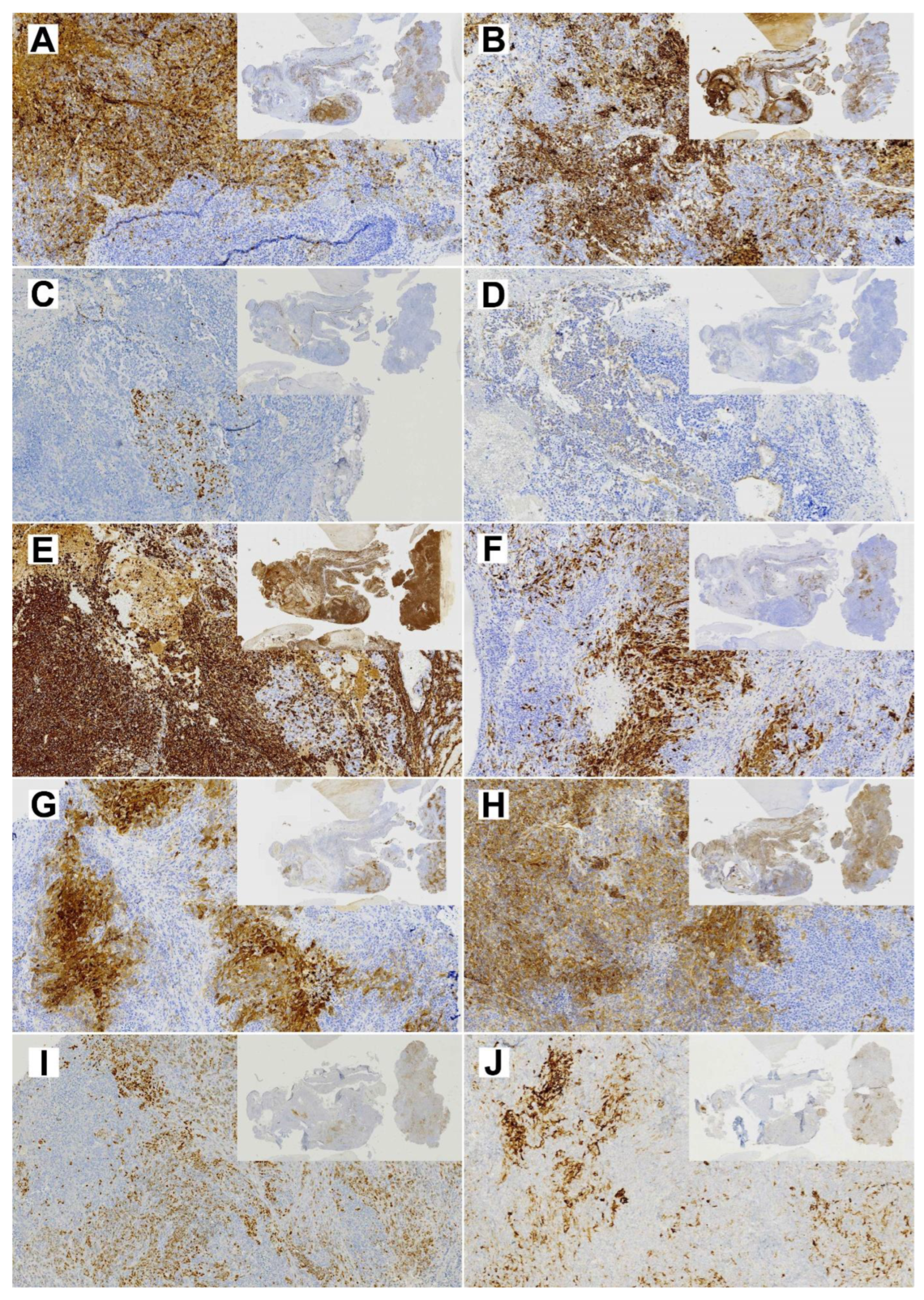
2.5. Immunohistochemistry
2.6. Cell Proliferation and Drug Sensitivity Assay
3. Results
3.1. TCS627 Cell Morphology, Differentiation and Proliferation Rate
3.2. TCS627 Authentication
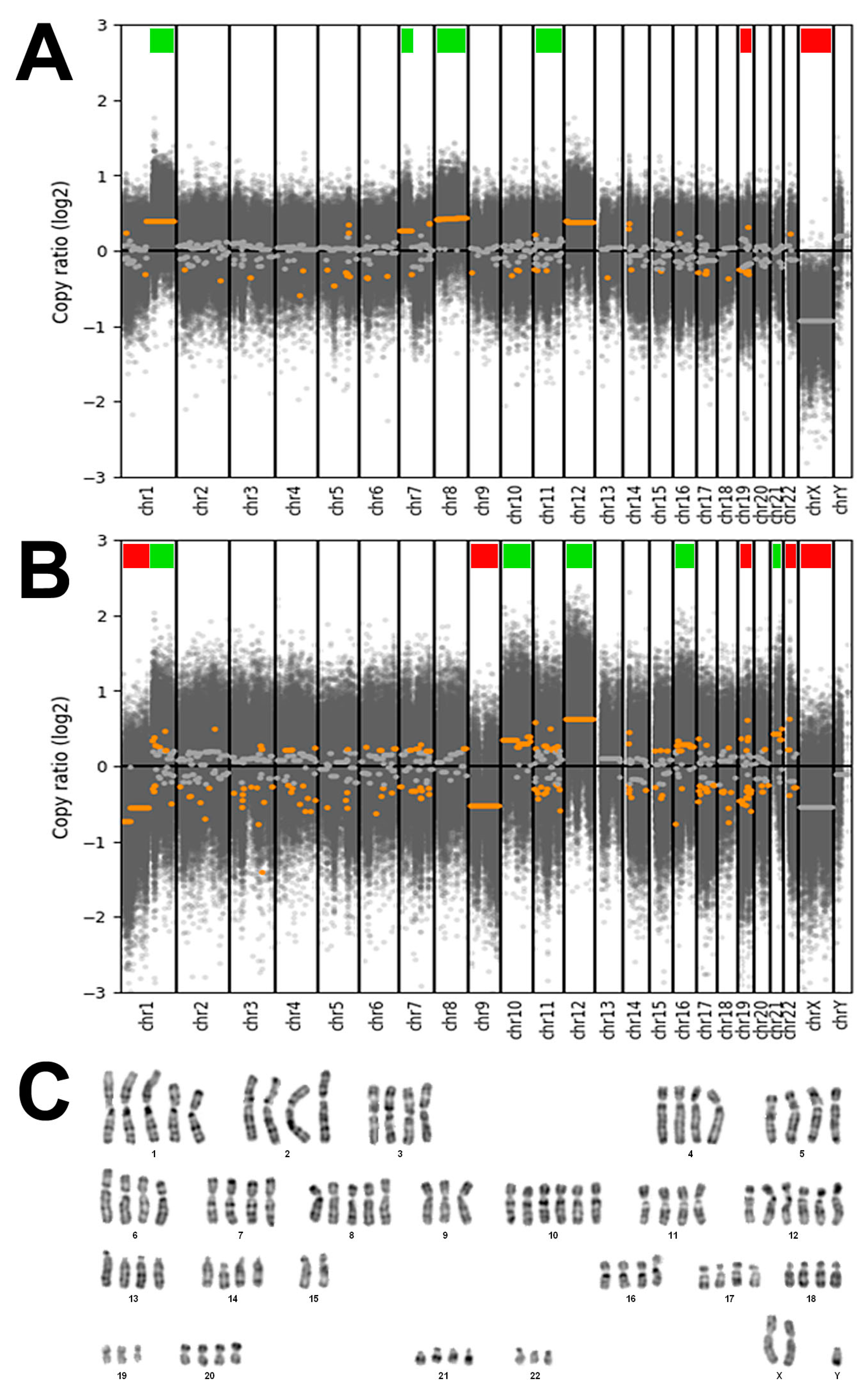
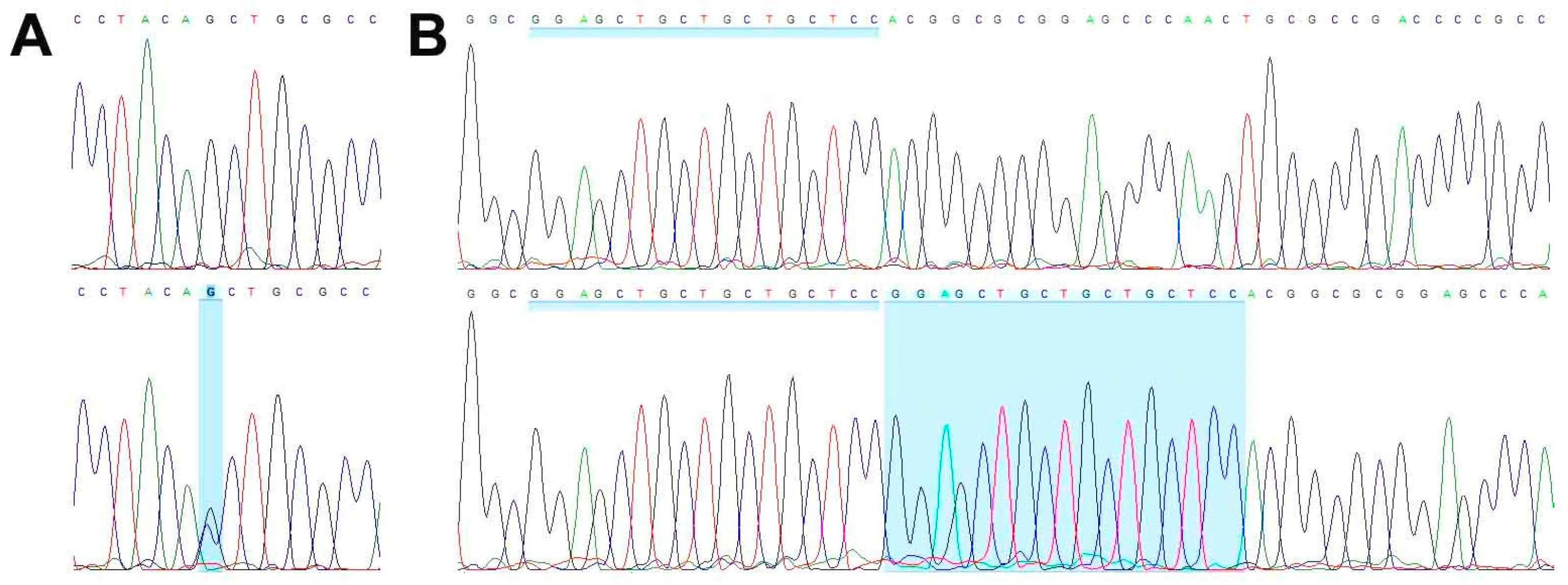
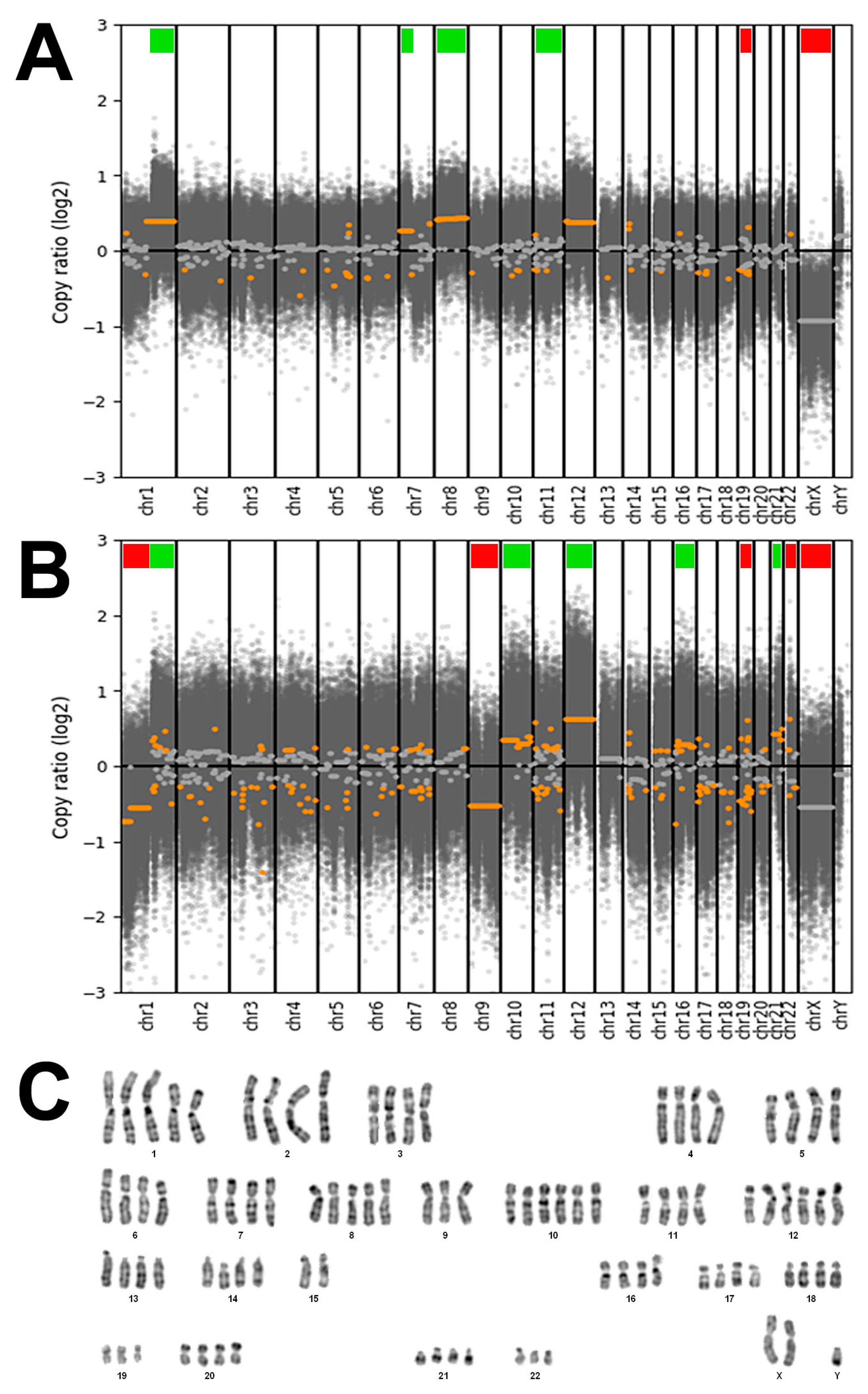
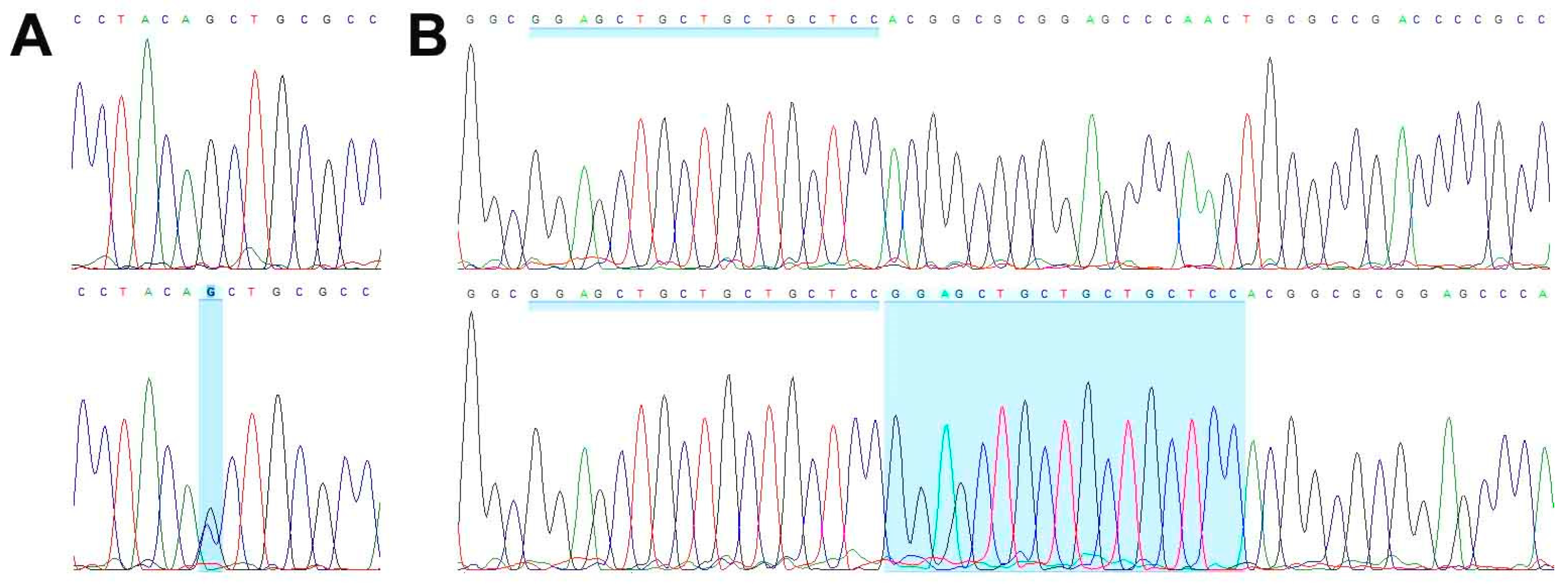
3.3. Genetic Characterization
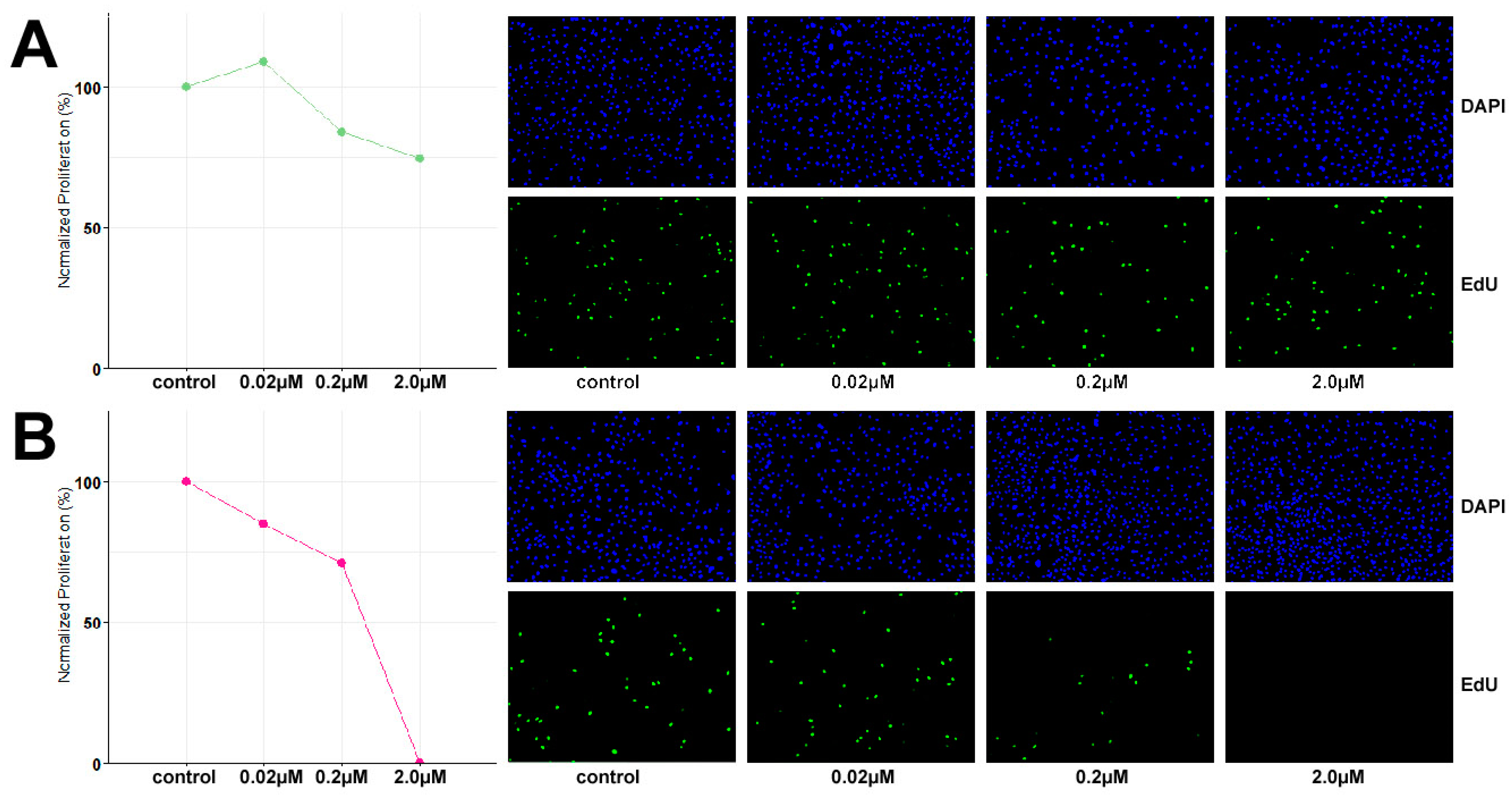
3.4. TCS627 Growth Inhibition Assays
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bishop, J.A.; Loney, E.L.; Thompson, L.D.R. (Eds.) Nasal, paranasal, and skull base tumours. In WHO Classification of Head and Neck Tumours; International Agency for Research on Cancer: Lyon, France, 2022. [Google Scholar]
- Franchi, A. (Ed.) Pathology of Sinonasal Tumors and Tumor-Like Lesions; Springer Nature: Cham, Switzerland, 2020. [Google Scholar]
- Agarwal, S.; van Zante, A.; Granados, M.L. Combined Neuroendocrine and Squamous Cell Carcinoma of the Sinonasal Tract: A Morphologic and Immunohistochemical Analysis and Review of Literature. Head Neck Pathol. 2022, 16, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Barham, H.P.; Said, S.; Ramakrishnan, V.R. Colliding Tumor of the Paranasal Sinus. Allergy Rhinol. 2013, 4, e13–e16. [Google Scholar] [CrossRef] [PubMed]
- Franchi, A.; Palomba, A.; Miligi, L.; Ranucci, V.; Innocenti, D.R.; Simoni, A.; Pepi, M.; Santucci, M. Primary combined neuroendocrine and squamous cell carcinoma of the maxillary sinus: Report of a case with immunohistochemical and molecular characterization. Head Neck Pathol. 2015, 9, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sugianto, I.; Yanagi, Y.; Hisatomi, M.; Okada, S.; Takeshita, Y.; Bamgbose, B.O.; Asaumi, J. Collision tumor of small cell carcinoma and squamous cell carcinoma of the maxillary sinus: Case report. Mol. Clin. Oncol. 2022, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Gramigna, V.; Sanchez-Marull, R.; Perez-Ordoñez, B. Composite intestinal-type adenocarcinoma and small cell carcinoma of sinonasal tract. J. Clin. Pathol. 2009, 62, 634–637. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, S.; Furlan, D.; Franzi, F.; Battaglia, P.; Frattini, M.; Zanellato, E.; Marando, A.; Sahnane, N.; Turri-Zanoni, M.; Castelnuovo, P.; et al. Mixed exocrine-neuroendocrine carcinoma of the nasal cavity: Clinico-pathologic and molecular study of a case and review of the literature. Head Neck Pathol. 2013, 7, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Attwood, J.E.; Jeyaretna, D.S.; Sheerin, F.; Shah, K.A. Mixed Olfactory Neuroblastoma and Adenocarcinoma with In Situ Neuroendocrine Hyperplasia. Head Neck Pathol. 2020, 14, 792–798. [Google Scholar] [CrossRef]
- Lao, W.P.; Thompson, J.M.; Evans, L.; Kim, Y.; Denham, L.; Lee, S.C. Mixed olfactory neuroblastoma and neuroendocrine carcinoma: An unusual case report and literature review. Surg. Neurol. Int. 2020, 11, 97. [Google Scholar] [CrossRef]
- Kang, S.Y.; McHugh, J.B.; Sullivan, S.E.; Marentette, L.J.; McKean, E.L. Sinonasal undifferentiated carcinoma and esthesioneuroblastoma recurring as nonintestinal adenocarcinoma. Laryngoscope 2013, 123, 1121–1124. [Google Scholar] [CrossRef]
- Valentini, V.; Giovannetti, F.; Cassoni, A.; Terenzi, V.; Priore, P.; Raponi, I.; Bosco, S.; Alesini, F.; Mezi, S.; Musio, D.; et al. Sinonasal Undifferentiated Carcinoma in a Patient Previously Treated for an Intestinal-Type Adenocarcinoma: Metachronous Neoplasms or Recurrence of a Different Tumor Type? Indian J. Otolaryngol. Head Neck Surg. 2019, 71 (Suppl. 3), 1779–1781. [Google Scholar] [CrossRef]
- Chapurin, N.; Totten, D.J.; Morse, J.C.; Khurram, M.S.; Louis, P.C.; Sinard, R.J.; Chowdhury, N.I. Treatment of Sinonasal Teratocarcinosarcoma: A Systematic Review and Survival Analysis. Am. J. Rhinol. Allergy 2021, 35, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Misra, P.; Husain, Q.; Svider, P.F.; Sanghvi, S.; Liu, J.K.; Eloy, J.A. Management of sinonasal teratocarcinosarcoma: A systematic review. Am. J. Otolaryngol. 2014, 35, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Belotti, A.; Carpenito, L.; Bulfamante, A.M.; Maccari, A.; Bulfamante, G. Sinonasal teratocarcinosarcoma treated with surgery and proton beam therapy: Clinical, histological aspects and differential diagnosis of a new case. Pathologica 2021, 113, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Suarez, G.P.; Dibs, K.; Carrau, R.L.; Schoenfield, L.; Blakaj, D.M.; Tinoco, G. Sinonasal teratocarcinosarcoma: A therapeutic dilemma. BMJ Case Rep. 2022, 15, e252429. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Ramirez-Garcia, R.; Ginat, D.T.; Agrawal, N.; Seible, D.; Cipriani, N.A.; Horowitz, P.; Pinto, J.M.; Chiu, B.L. Multimodality management of sinonasal teratocarcinosarcoma in a 76-year-old Alaska Native female during the COVID-19 pandemic. Clin. Case Rep. 2022, 10, e05635. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, A.C.; Burgin, S.J.; Yanik, M.; Scott, M.V.; Bradford, C.R.; McHugh, J.B.; McLean, S.A.; Sullivan, S.E.; Nor, J.E.; McKean, E.L.; et al. Pathogenetic analysis of sinonasal teratocarcinosarcomas reveal actionable β-catenin overexpression and a β-catenin mutation. J. Neurol. Surg. B Skull Base 2017, 78, 346–352. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, T.; He, X.; Ge, M. Targeted next-generation sequencing reveals activating CTNNB1 mutations in SMARCA4/BRG1-deficient sinonasal carcinomas: A report of two new cases and a brief review of the literature with an emphasis on histogenesis. Virchows Arch. 2023, 482, 453–460. [Google Scholar] [CrossRef]
- Almarzooqi, S.; Reyes-Múgica, M.; Ali, B.R.; Habbal, A.; Asha, M.J.; AlShamsi, E.T. Congenital Teratocarcinosarcoma with CTNNB1 Gene Mutation Presenting as an Ocular Mass. Pediatr. Dev. Pathol. 2022, 25, 562–567. [Google Scholar] [CrossRef]
- Compton, M.L.; Lewis, J.S., Jr.; Faquin, W.C.; Cipriani, N.A.; Shi, Q.; Ely, K.A. SALL-4 and Beta-Catenin Expression in Sinonasal Teratocarcinosarcoma. Head Neck Pathol. 2022, 16, 229–235. [Google Scholar] [CrossRef]
- Kakkar, A.; Ashraf, S.F.; Rathor, A.; Adhya, A.K.; Mani, S.; Sikka, K.; Jain, D. SMARCA4/BRG1-Deficient Sinonasal Carcinoma. Arch. Pathol. Lab. Med. 2022, 146, 1122–1130. [Google Scholar] [CrossRef]
- Belardinilli, F.; De Vincentiis, L.; D’Ecclesia, A.; Giannini, G.; Giangaspero, F.; Corsi, A. PIK3CA somatic mutation in sinonasal teratocarcinosarcoma. Auris Nasus Larynx 2021, 48, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Rooper, L.M.; Uddin, N.M.; Gagan, J.; Brosens, L.A.; Magliocca, K.R.D.; Edgar, M.A.; Thompson, L.D.; Agaimy, A.; Bishop, J.A. Recurrent Loss of SMARCA4 in Sinonasal Teratocarcinosarcoma. Am. J. Surg. Pathol. 2020, 44, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Rooper, L.M.; Agaimy, A.; Gagan, J.; Simpson, R.H.W.; Thompson, L.D.R.; Trzcinska, A.M.; Ud Din, N.; Bishop, J.A. Comprehensive Molecular Profiling of Sinonasal Teratocarcinosarcoma Highlights Recurrent SMARCA4 Inactivation and CTNNB1 Mutations. Am. J. Surg. Pathol. 2023, 47, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.A.; Tansey, W.P.; Weissmiller, A.M. Emerging Themes in Mechanisms of Tumorigenesis by SWI/SNF Subunit Mutation. Epigenet Insights 2022, 15, 25168657221115656. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Morel, D.; Postel-Vinay, S. Exploiting epigenetic vulnerabilities in solid tumors: Novel therapeutic opportunities in the treatment of SWI/SNF-defective cancers. Semin. Cancer Biol. 2020, 61, 180–198. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, P.; Riobello, C.; Costales, M.; Vivanco, B.; Cabal, V.N.; García-Marín, R.; Suárez-Fernández, L.; López, F.; Cabanillas, R.; Hermsen, M.A.; et al. Next-generation sequencing for identification of actionable gene mutations in intestinal-type sinonasal adenocarcinoma. Sci. Rep. 2021, 11, 2247. [Google Scholar] [CrossRef] [PubMed]
- Sjöstedt, S.; Schmidt, A.Y.; Vieira, F.G.; Willemoe, G.L.; Agander, T.K.; Olsen, C.; Nielsen, F.C.; von Buchwald, C. Major driver mutations are shared between sinonasal intestinal-type adenocarcinoma and the morphologically identical colorectal adenocarcinoma. J. Cancer Res. Clin. Oncol. 2021, 147, 1019–1027. [Google Scholar] [CrossRef]
- Riobello, C.; Sánchez-Fernández, P.; Cabal, V.N.; García-Marín, R.; Suárez-Fernández, L.; Vivanco, B.; Blanco-Lorenzo, V.; Marcos, C.; López, F.; Llorente, J.L.; et al. Aberrant Signaling Pathways in Sinonasal Intestinal-Type Adenocarcinoma. Cancers 2021, 13, 5022. [Google Scholar] [CrossRef]
- Rooper, L.M. Proceedings of the 2023 North American Society of Head and Neck Pathology Companion Meeting, New Orleans, LA, March 12, 2023: Navigating New Developments in High Grade Sinonasal Neuroendocrine and Neuroectodermal Neoplasms. Head Neck Pathol. 2023, 17, 299–312. [Google Scholar] [CrossRef]
- Bell, D.; Brandea, A.I.; Hanna, E.Y. Olfactory Neuroblastoma: Morphological Reappraisal and Molecular Insights with Quantum Leap in Clinical Perspectives. Curr. Oncol. Rep. 2023, 25, 11–18. [Google Scholar] [CrossRef]
- Díaz-Molina, J.P.; Llorente, J.L.; Vivanco, B.; Martínez-Camblor, P.; Fresno, M.F.; Pérez-Escuredo, J.; Álvarez-Marcos, C.; Hermsen, M.A. Wnt-pathway activation in intestinal-type sinonasal adenocarcinoma. Rhinology 2011, 49, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Almouzni, G.; Soria, J.C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2017, 28, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Rialdi, A.; Duffy, M.; Scopton, A.P.; Fonseca, F.; Zhao, J.N.; Schwarz, M.; Molina-Sanchez, P.; Mzoughi, S.; Arceci, E.; Abril-Fornaguera, J.; et al. WNTinib is a multi-kinase inhibitor with specificity against β-catenin mutant hepatocellular carcinoma. Nat. Cancer 2023, 4, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Targ. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Codina-Martínez, H.; Lorenzo-Guerra, S.L.; Cabal, V.N.; García-Marín, R.; Riobello, C.; Suárez-Fernández, L.; Vivanco, B.; Blanco-Lorenzo, V.; López, F.; Sánchez-Fernández, P.; et al. Experimental Models of Sinonasal Tumors for Preclinical Testing of Candidate Targeted Therapies. Curr. Otorhinolaryngol. Rep. 2023. [Google Scholar] [CrossRef]
- Vranic, S.; Caughron, S.K.; Djuricic, S.; Bilalovic, N.; Zaman, S.; Suljevic, I.; Lydiatt, W.M.; Emanuel, J.; Gatalica, Z. Hamartomas, teratomas and teratocarcinosarcomas of the head and neck: Report of 3 new cases with clinico-pathologic correlation, cytogenetic analysis, and review of the literature. BMC Ear Nose Throat Disord. 2008, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Adegboyega, P.; Iloabachie, K.; Mooring, J.W.; Lian, T. Sinonasal teratocarcinosarcoma with yolk sac elements: A neoplasm of somatic or germ cell origin? Ann. Diagn. Pathol. 2011, 15, 135–139. [Google Scholar] [CrossRef]
- Jo, V.Y.; Chau, N.G.; Hornick, J.L.; Krane, J.F.; Sholl, L.M. Recurrent IDH2 R172X mutations in sinonasal undifferentiated carcinoma. Mod. Pathol. 2017, 30, 650–659. [Google Scholar] [CrossRef]
- Dogan, S.; Chute, D.J.; Xu, B.; Ptashkin, R.N.; Chandramohan, R.; Casanova-Murphy, J.; Nafa, K.; Bishop, J.A.; Chiosea, S.I.; Stelow, E.B.; et al. Frequent IDH2 R172 mutations in undifferentiated and poorly-differentiated sinonasal carcinomas. J. Pathol. 2017, 242, 400–408. [Google Scholar] [CrossRef]
- Dogan, S.; Vasudevaraja, V.; Xu, B.; Serrano, J.; Ptashkin, R.N.; Jung, H.J.; Chiang, S.; Jungbluth, A.A.; Cohen, M.A.; Ganly, I.; et al. DNA methylation-based classification of sinonasal undifferentiated carcinoma. Mod. Pathol. 2019, 32, 1447–1459. [Google Scholar] [CrossRef]
- Classe, M.; Yao, H.; Mouawad, R.; Creighton, C.J.; Burgess, A.; Allanic, F.; Wassef, M.; Leroy, X.; Verillaud, B.; Mortuaire, G.; et al. Integrated Multi-omic Analysis of Esthesioneuroblastomas Identifies Two Subgroups Linked to Cell Ontogeny. Cell Rep. 2018, 25, 811.e5–821.e5. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Engel, N.W.; Stichel, D.; Lechner, M.; Glöss, S.; Schmid, S.; Koelsche, C.; Schrimpf, D.; Niesen, J.; Wefers, A.K.; et al. DNA methylation-based reclassification of olfactory neuroblastoma. Acta Neuropathol. 2018, 136, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Jurmeister, P.; Glöß, S.; Roller, R.; Leitheiser, M.; Schmid, S.; Mochmann, L.H.; Capilla, E.P.; Fritz, R.; Dittmayer, C.; Friedrich, C.; et al. DNA methylation-based classification of sinonasal tumors. Nat. Commun. 2022, 13, 7148. [Google Scholar] [CrossRef] [PubMed]
- Agaimy, A. Proceedings of the North American Society of Head and Neck Pathology, Los Angeles, CA, March 20, 2022: SWI/SNF-deficient Sinonasal Neoplasms: An Overview. Head Neck Pathol. 2022, 16, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; He, K.; Zhang, Y.; Song, J.; Shi, Z.; Chen, W.; Shao, Y. Inactivation of SMARCA2 by promoter hypermethylation drives lung cancer development. Gene 2019, 687, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wan, Z.; Zhang, E.; Piao, Y. Genomic profiling and immune landscape of olfactory neuroblastoma in China. Front. Oncol. 2023, 13, 1226494. [Google Scholar] [CrossRef] [PubMed]
- Rooper, L.M.; Gagan, J.; Nishino, M.; Bishop, J. Sinonasal Tumors with Overlapping Neuroblastic and Epithelial Features Have Recurrent CTNNB1, SWI/SNF, and PPP2R1A Mutations: Validating the Concept of a True Olfactory Carcinoma? (abs 836). USCAP Head Neck Pathol. Lab. Investig. 2022, 102 (Suppl. 1), 901–902. [Google Scholar]
- Griffin, C.T.; Curtis, C.D.; Davis, R.B.; Muthukumar, V.; Magnuson, T. The chromatin-remodeling enzyme BRG1 modulates vascular Wnt signaling at two levels. Proc. Natl. Acad. Sci. USA 2011, 108, 2282–2287. [Google Scholar] [CrossRef]
- Xue, W.; Cai, L.; Li, S.; Hou, Y.; Wang, Y.D.; Yang, D.; Xia, Y.; Nie, X. WNT ligands in non-small cell lung cancer: From pathogenesis to clinical practice. Discov. Oncol. 2023, 14, 136. [Google Scholar] [CrossRef]
- Lan, L.; Wang, W.; Huang, Y.; Bu, X.; Zhao, C. Roles of Wnt7a in embryo development, tissue homeostasis, and human diseases. J. Cell. Biochem. 2019, 120, 18588–18598. [Google Scholar] [CrossRef]
- Sun, M.; Wang, J.; Zhang, Q.; Lin, X. SMARCA4-deficient Sinonasal Malignant Tumor with Striated Muscle and Neuroendocrine Differentiation: A Case Report and Letter to the Editor. Am. J. Surg. Pathol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Dobreva, G.; Chahrour, M.; Dautzenberg, M.; Chirivella, L.; Kanzler, B.; Fariñas, I.; Karsenty, G.; Grosschedl, R. SATB2 Is a Multifunctional Determinant of Craniofacial Patterning and Osteoblast Differentiation. Cell 2006, 125, 971–986. [Google Scholar] [CrossRef] [PubMed]
- Brocato, J.; Costa, M. SATB1 and 2 in colorectal cancer. Carcinogenesis 2015, 36, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Skalova, A.; Sar, A.; Laco, J.; Metelkova, A.; Miesbauerova, M.; Steiner, P.; Švajdler, M.; Michal, M. The Role of SATB2 as a Diagnostic Marker of Sinonasal Intestinal-type Adenocarcinoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Mondal, G.; Stevers, M.; Goode, B.; Ashworth, A.; Solomon, D.A. A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat. Commun. 2019, 10, 1686. [Google Scholar] [CrossRef] [PubMed]
- Mardinian, K.; Adashek, J.J.; Botta, G.P.; Kato, S.; Kurzrock, R. SMARCA4: Implications of an Altered Chromatin-Remodeling Gene for Cancer Development and Therapy. Mol. Cancer Ther. 2021, 20, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef] [PubMed]
- Januario, T.; Ye, X.; Bainer, R.; Alicke, B.; Smith, T.; Haley, B.; Modrusan, Z.; Gould, S.; Yauch, R.L. PRC2-mediated repression of SMARCA2 predicts EZH2 inhibitor activity in SWI/SNF mutant tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 12249–12254. [Google Scholar] [CrossRef]
- Chan-Penebre, E.; Armstrong, K.; Drew, A.; Grassian, A.R.; Feldman, I.; Knutson, S.K.; Kuplast-Barr, K.; Roche, M.; Campbell, J.; Ho, P.; et al. Selective Killing of SMARCA2- and SMARCA4-deficient Small Cell Carcinoma of the Ovary, Hypercalcemic Type Cells by Inhibition of EZH2: In Vitro and In Vivo Preclinical Models. Mol Cancer Ther. 2017, 16, 850–860. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Macdonald, E.; Venneti, S.; Wang, X.Q.D.; Witkowski, L.; Jelinic, P.; Kong, T.; Martinez, D.; Morin, G.; et al. CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat. Commun. 2019, 10, 558. [Google Scholar] [CrossRef]
- Gao, Y.; Zheng, K.; Kang, M.; Xu, J.; Ning, Y.; Hu, W.; Li, K.; Kang, Y.; Xu, C. Establishment and characterization of a novel cell line (SCCOHT-CH-1) and PDX models derived from Chinese patients of small cell ovarian carcinoma of the hypercalcemic type. Hum. Cell 2023, 36, 2214–2227. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | c.Hgvs | p.Hgvs | Variant Reads Normal | Variant Reads Primary Tumor | Variant Reads TCS627 | Variant Frequency Normal | Variant Frequency Primary Tumor | Variant Frequency TCS627 |
|---|---|---|---|---|---|---|---|---|
| ARID2 | c.1518_1519del | p.Gln507Alafs*13 | 0/133 | 38/161 | 73/184 | 0 | 0.24 | 0.4 |
| CDKN2A | c.223_239dupGGAGCTGCTGCTGCTCC | p.Arg81Glufs*? | 0/369 | 63/250 | 40/104 | 0 | 0.25 | 0.38 |
| NOTCH3 | c.1057G>A | p.Asp353Asn | 0/343 | 112/310 | 154/229 | 0 | 0.36 | 0.67 |
| SATB2 | c.1741-12C>G | 0/77 | 21/75 | 31/52 | 0 | 0.28 | 0.6 | |
| SMARCA4 | c.1246-1G>C | 0/222 | 68/203 | 133/205 | 0 | 0.33 | 0.65 | |
| STAG2 | c.2537G>T | p.Gly846Val | 0/57 | 39/59 | 53/53 | 0 | 0.66 | 1 |
| TET2 | c.863C>T | p.Pro288Leu | 0/141 | 37/131 | 69/177 | 0 | 0.28 | 0.39 |
| WNT7A | c.1043G>T | p.Cys348Phe | 0/175 | 83/193 | 148/211 | 0 | 0.43 | 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzo-Guerra, S.L.; Codina-Martínez, H.; Suárez-Fernández, L.; Cabal, V.N.; García-Marín, R.; Riobello, C.; Vivanco, B.; Blanco-Lorenzo, V.; Sánchez-Fernández, P.; López, F.; et al. Characterization of a Preclinical In Vitro Model Derived from a SMARCA4-Mutated Sinonasal Teratocarcinosarcoma. Cells 2024, 13, 81. https://doi.org/10.3390/cells13010081
Lorenzo-Guerra SL, Codina-Martínez H, Suárez-Fernández L, Cabal VN, García-Marín R, Riobello C, Vivanco B, Blanco-Lorenzo V, Sánchez-Fernández P, López F, et al. Characterization of a Preclinical In Vitro Model Derived from a SMARCA4-Mutated Sinonasal Teratocarcinosarcoma. Cells. 2024; 13(1):81. https://doi.org/10.3390/cells13010081
Chicago/Turabian StyleLorenzo-Guerra, Sara Lucila, Helena Codina-Martínez, Laura Suárez-Fernández, Virginia N. Cabal, Rocío García-Marín, Cristina Riobello, Blanca Vivanco, Verónica Blanco-Lorenzo, Paula Sánchez-Fernández, Fernando López, and et al. 2024. "Characterization of a Preclinical In Vitro Model Derived from a SMARCA4-Mutated Sinonasal Teratocarcinosarcoma" Cells 13, no. 1: 81. https://doi.org/10.3390/cells13010081
APA StyleLorenzo-Guerra, S. L., Codina-Martínez, H., Suárez-Fernández, L., Cabal, V. N., García-Marín, R., Riobello, C., Vivanco, B., Blanco-Lorenzo, V., Sánchez-Fernández, P., López, F., Llorente, J. L., & Hermsen, M. A. (2024). Characterization of a Preclinical In Vitro Model Derived from a SMARCA4-Mutated Sinonasal Teratocarcinosarcoma. Cells, 13(1), 81. https://doi.org/10.3390/cells13010081