
Sox10-Deficient Drug-Resistant Melanoma Cells Are Refractory to Oncolytic RNA Viruses
 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
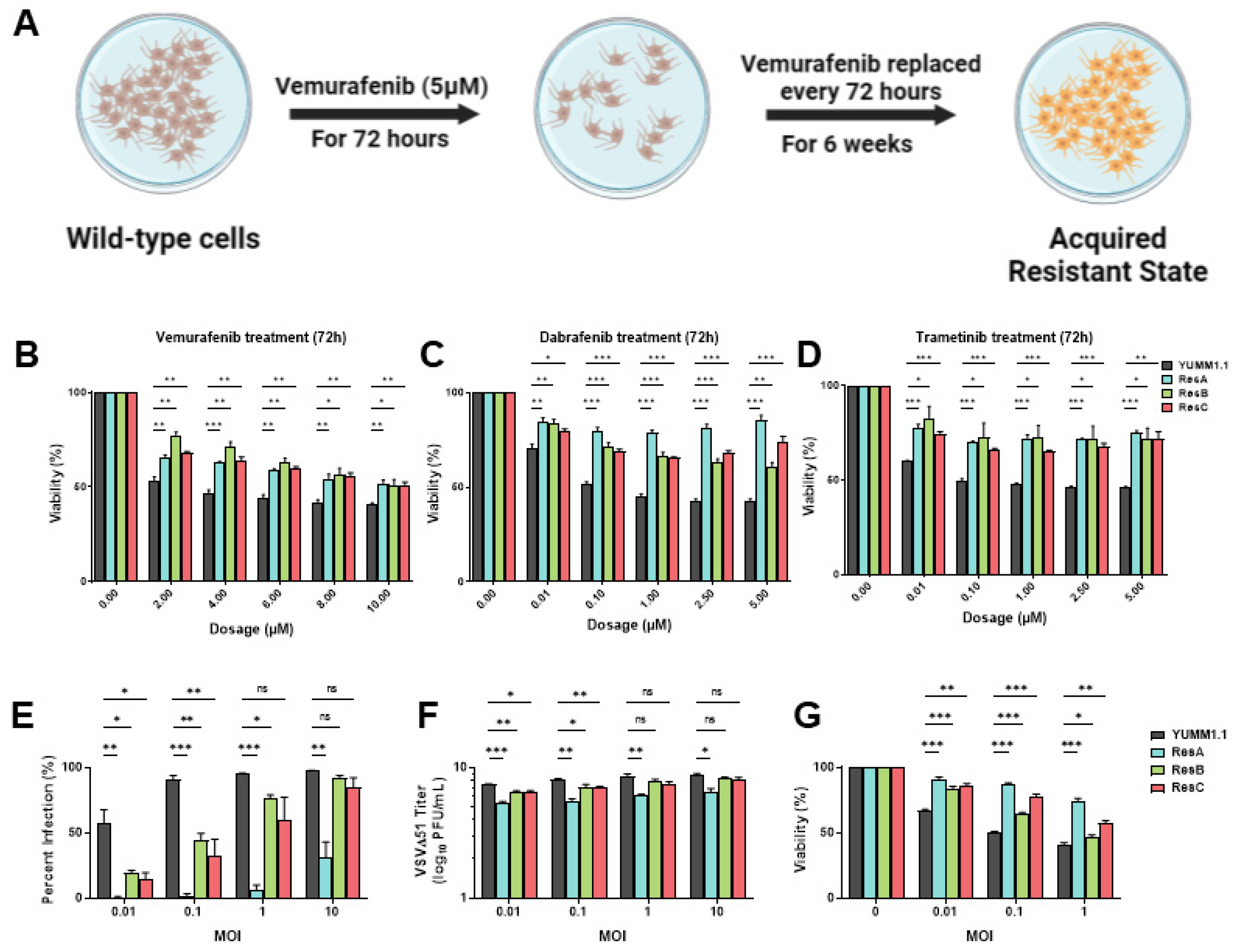
2.2. Establishment and Isolation of Vemurafenib-Resistant Cell Populations
2.3. Oncolytic Infection
2.4. Viral Titer
2.5. Resazurin Viability Assay
2.6. SiRNA Transfection and Lentivirus Production
2.7. Lentiviral-Mediated CRISPR/Cas9 Knockouts
2.8. Cell Lysis and Western Blotting
2.9. RNA-Sequencing, EnrichR Biological Pathway Analysis and TPM Analysis
2.10. Statistical Analysis
3. Results
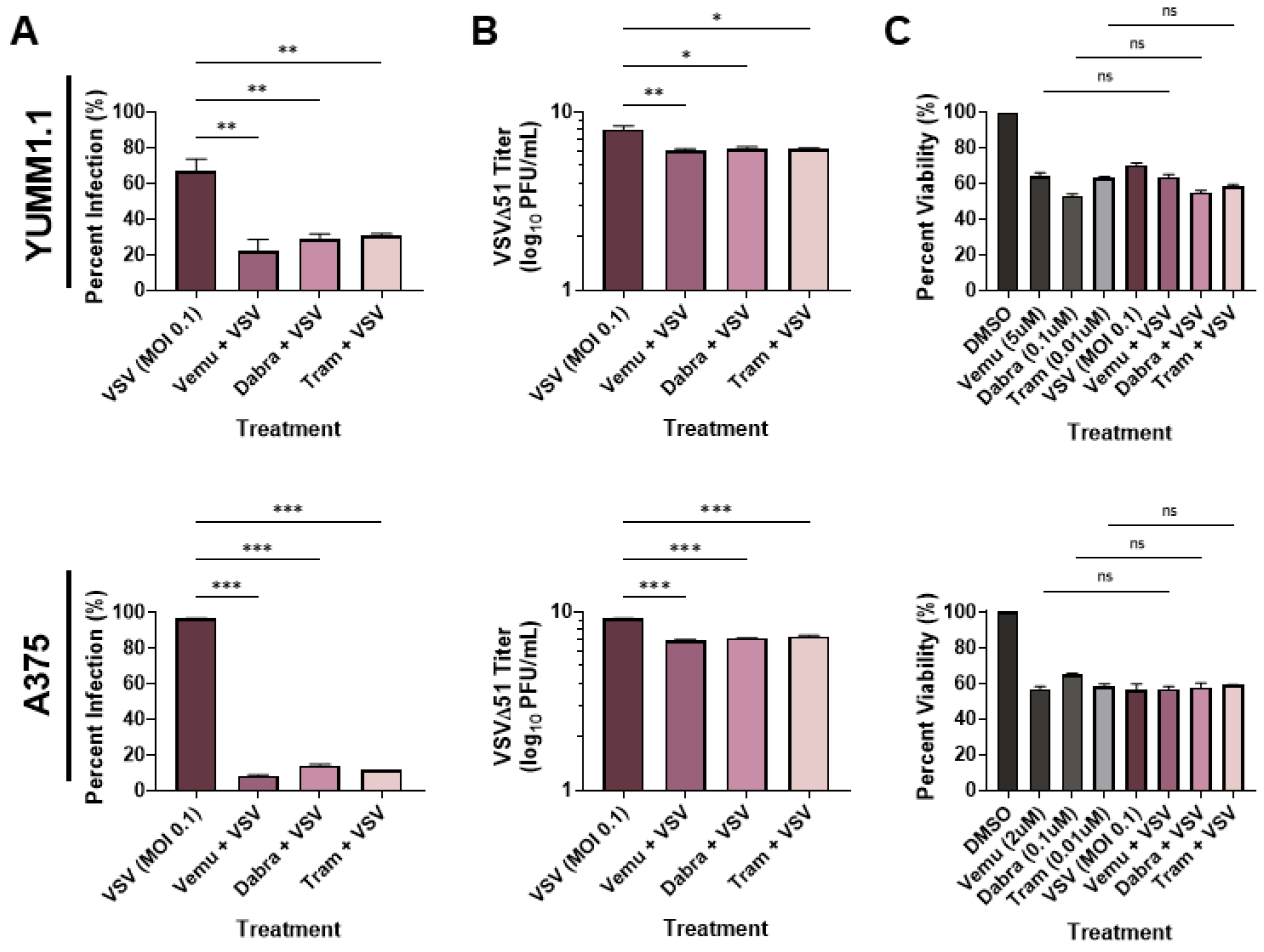
3.1. MAPKi Treatment Blocks VSV Infection in BRAFV600E Melanoma
3.2. Vemurafenib-Induced Targeted Therapy Resistance Promotes a Cross-Resistant State between Other MAPKis and VSV Infection
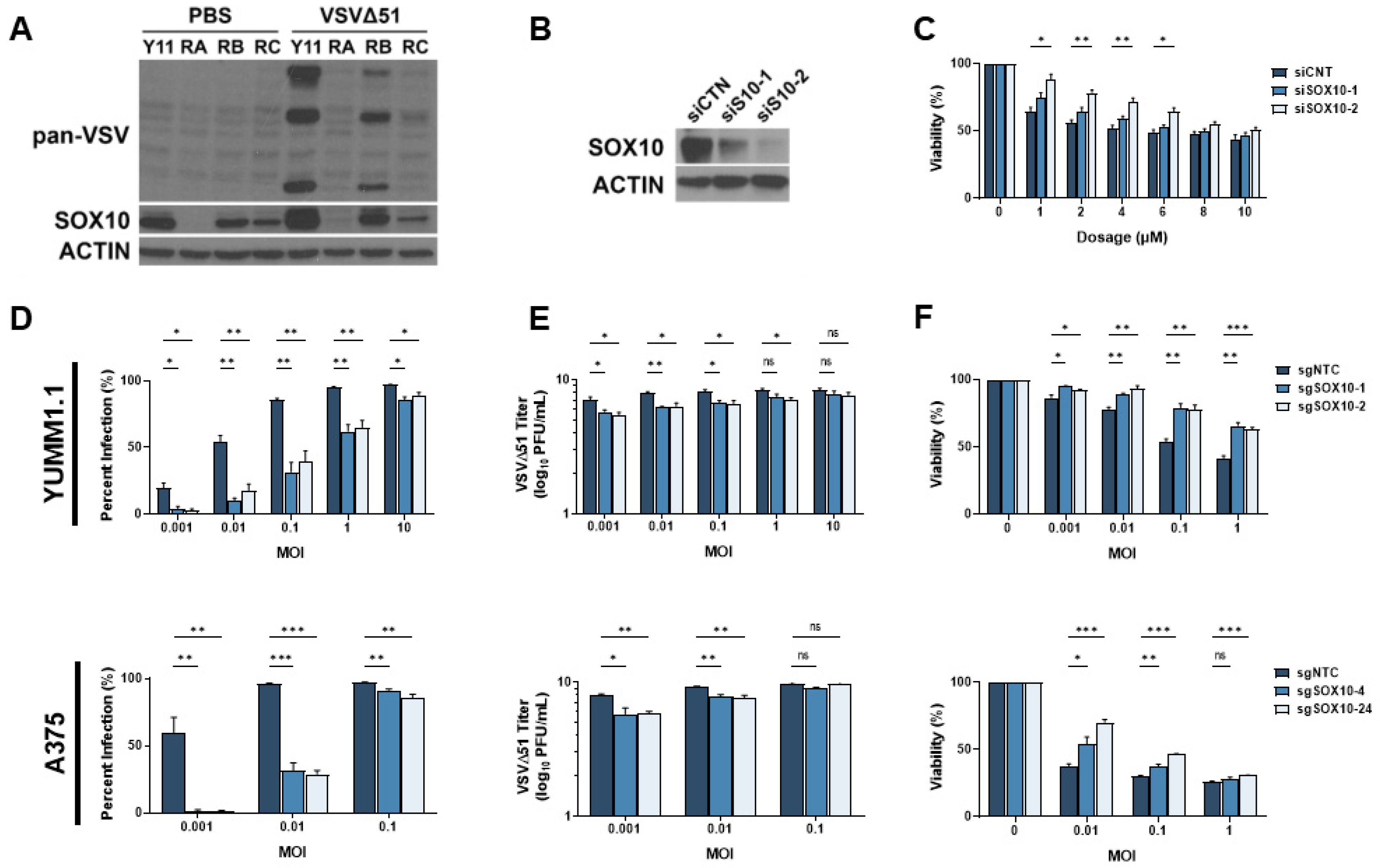
3.3. SOX10 Is Lost during Chronic Targeted Therapy Treatment and Induces a Cross-Resistant State to VSVΔ51
3.4. RNA-Sequencing Analysis of A375 SOX10 Knockout Cells Shows Enrichment of Viral Responses Gene Sets
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Go, Z.; Iyer, R.; Kolis, S.; Zhao, S.; Lee, R.; Grippo, J.F.; et al. RG7204 (PLX4032), a Selective BRAFV600E Inhibitor, Displays Potent Antitumor Activity in Preclinical Melanoma Models. Cancer Res. 2010, 70, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. An update on BREAK-3, a phase III, randomized trial: Dabrafenib (DAB) versus dacarbazine (DTIC) in patients with BRAF V600E-positive mutation metastatic melanoma (MM). J. Clin. Oncol. 2013, 31, 9013. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef]
- Graf, S.A.; Busch, C.; Bosserhoff, A.-K.; Besch, R.; Berking, C. SOX10 Promotes Melanoma Cell Invasion by Regulating Melanoma Inhibitory Activity. J. Investig. Dermatol. 2014, 134, 2212–2220. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, C.K.; Mosimann, C.; Fan, Z.P.; Yang, S.; Thomas, A.J.; Ablain, J.; Tan, J.L.; Fogley, R.D.; van Rooijen, E.; Hagedorn, E.J.; et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016, 351, aad2197. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Ren, Y.; He, W.; Liu, H.; Zhi, Z.; Zhu, X.; Yang, T.; Rong, Y.; Ma, B.; Purwin, T.J.; et al. ERK-mediated phosphorylation regulates SOX10 sumoylation and targets expression in mutant BRAF melanoma. Nat. Commun. 2018, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Purwin, T.J.; Glasheen, M.; Caksa, S.; Tiago, M.; Wilski, N.; Pomante, D.; Rosenbaum, S.; Nguyen, M.Q.; Cai, W.; et al. Targeting SOX10-deficient cells to reduce the dormant-invasive phenotype state in melanoma. Nat. Commun. 2022, 13, 1381. [Google Scholar] [CrossRef] [PubMed]
- Abou-Hamad, J.; Hodgins, J.J.; de Souza, C.T.; Garland, B.; Labrèche, C.; Marotel, M.; Gibson, C.; Delisle, S.; Pascoal, J.; Auer, R.C.; et al. CEACAM1 is a direct SOX10 target and inhibits melanoma immune infiltration and stemness. iScience 2022, 25, 105524. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1866, 165339. [Google Scholar] [CrossRef]
- Robinson, C.; Xu, M.M.; Nair, S.K.; Beasley, G.M.; Rhodin, K.E. Oncolytic viruses in melanoma. Front. Biosci. 2022, 27, 63. [Google Scholar] [CrossRef]
- Blackham, A.U.; Northrup, S.A.; Willingham, M.; D’Agostino, R.B.; Lyles, D.S.; Stewart, J.H. Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery 2012, 153, 333–343. [Google Scholar] [CrossRef]
- Wollmann, G.; Drokhlyansky, E.; Davis, J.N.; Cepko, C.; van den Pol, A.N. Lassa-Vesicular Stomatitis Chimeric Virus Safely Destroys Brain Tumors. J. Virol. 2015, 89, 6711–6724. [Google Scholar] [CrossRef] [PubMed]
- Kimpel, J.; Urbiola, C.; Koske, I.; Tober, R.; Banki, Z.; Wollmann, G.; Von Laer, D. The Oncolytic Virus VSV-GP Is Effective against Malignant Melanoma. Viruses 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Katze, M.G.; He, Y.; Gale, M., Jr. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, O.V.; Chumakov, P.M. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev. Med. Virol. 2018, 28, e2008. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; Tenoever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, W. A Review of the Molecular Pathways Involved in Resistance to BRAF Inhibitors in Patients with Advanced-Stage Melanoma. J. Pharmacol. Exp. Ther. 2020, 26, e920957-1–e920957-13. [Google Scholar] [CrossRef] [PubMed]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma—A mini review. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 2020, 1865, 129736. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Takahashi, A.; Kikuchi, R.; Nishibu, S.; Lo, J.A.; Hejna, M.; Moon, W.M.; Kato, S.; Zhou, Y.; Hodi, F.S.; et al. SOX10 Regulates Melanoma Immunogenicity through an IRF4–IRF1 Axis. Cancer Res. 2021, 81, 6131–6141. [Google Scholar] [CrossRef] [PubMed]
- Murira, A.; Lamarre, A. Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herranz, M.; Taylor, J.; Sloan, R.D. IFITM proteins: Understanding their diverse roles in viral infection, cancer, and immunity. J. Biol. Chem. 2022, 299, 102741. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Hage, A.; van Tol, S.; Rajsbaum, R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr. Clin. Microbiol. Rep. 2020, 7, 101–114. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Number of Genes | Gene List |
|---|---|---|
| Defense response to virus, Negative regulation of viral genome replication, Negative regulation of viral process, Regulation of viral entry into host cell | 4 | IFITM1, IFITM2, IFITM3, TRIM6 |
| Defense response to virus, Negative regulation of viral process, Regulation of viral entry into host cell | 5 | TRIM31, MID2, TRIM21, TRIM8, TRIM56 |
| Defense response to virus, Negative regulation of viral genome replication, Negative regulation of viral process | 14 | MAVS, APOBEC3F, ISG20, OASL, APOBEC3G, APOBEC3H, IFIH1, APOBEC3D, OAS2, ISG15, SHFL, RNASEL, APOBEC3C, BST2 |
| Defense response to virus, Negative regulation of viral process | 2 | STAT1, TRIM32 |
| Defense response to virus, Regulation of viral entry into host cell | 1 | TRIM38 |
| Negative regulation of viral genome replication, Negative regulation of viral process | 3 | LTF, SLPI, HMGA2 |
| Negative regulation of viral process, Regulation of viral entry into host cell | 1 | GSN |
| Negative regulation of viral process | 3 | SP100, ZFP36, SRPX2 |
| Regulation of viral entry into host cell | 7 | TRIM34, NECTIN2, TMPRSS2, SMPD1, FURIN, BSG, LGALS1 |
| Defense response to virus | 57 | IFI44L, DDX60, F2RL1, PTPN22, PMAIP1, CD86, IFIT1, TBK1, CREB3, DTX3L, ZDHHC12, VAMP8, TNFAIP3, CASP1, ATG7, NMB, TICAM2, KCNJ8, STAT2, UNC93B1, HTRA1, CARD9, IL6, CPTP, MOV10, TICAM1, ZC3H12A, TRIM7, MARCHF2, EXOSC4, IFNE, RIOK3, LYST, NCR1, IFNLR1, STING1, IFNA1, AIM2, PML, NLRX1, IL1B, ITGAX, GPAM, IRF1, IRF3, RNF185, UNC13D, LAMP2, SERTAD3, PARP9, IFIT3, USP20, IL10RB, NLRP3, CXADR, IRF7, ABCC9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou-Hamad, J.; Hodgins, J.J.; Yakubovich, E.; Vanderhyden, B.C.; Ardolino, M.; Sabourin, L.A. Sox10-Deficient Drug-Resistant Melanoma Cells Are Refractory to Oncolytic RNA Viruses. Cells 2024, 13, 73. https://doi.org/10.3390/cells13010073
Abou-Hamad J, Hodgins JJ, Yakubovich E, Vanderhyden BC, Ardolino M, Sabourin LA. Sox10-Deficient Drug-Resistant Melanoma Cells Are Refractory to Oncolytic RNA Viruses. Cells. 2024; 13(1):73. https://doi.org/10.3390/cells13010073
Chicago/Turabian StyleAbou-Hamad, John, Jonathan J. Hodgins, Edward Yakubovich, Barbara C. Vanderhyden, Michele Ardolino, and Luc A. Sabourin. 2024. "Sox10-Deficient Drug-Resistant Melanoma Cells Are Refractory to Oncolytic RNA Viruses" Cells 13, no. 1: 73. https://doi.org/10.3390/cells13010073
APA StyleAbou-Hamad, J., Hodgins, J. J., Yakubovich, E., Vanderhyden, B. C., Ardolino, M., & Sabourin, L. A. (2024). Sox10-Deficient Drug-Resistant Melanoma Cells Are Refractory to Oncolytic RNA Viruses. Cells, 13(1), 73. https://doi.org/10.3390/cells13010073