
Impaired Mitochondrial Function in T-Lymphocytes as a Result of Exposure to HIV and ART


 , ,
, ,  and
and 
Abstract
1. Introduction
2. HIV Pathogenesis
2.1. Acute Phase
2.2. Chronic Phase and AIDS
3. The Role of Mitochondria in Antiviral Immunity
3.1. The Role of Mitochondria in Cells
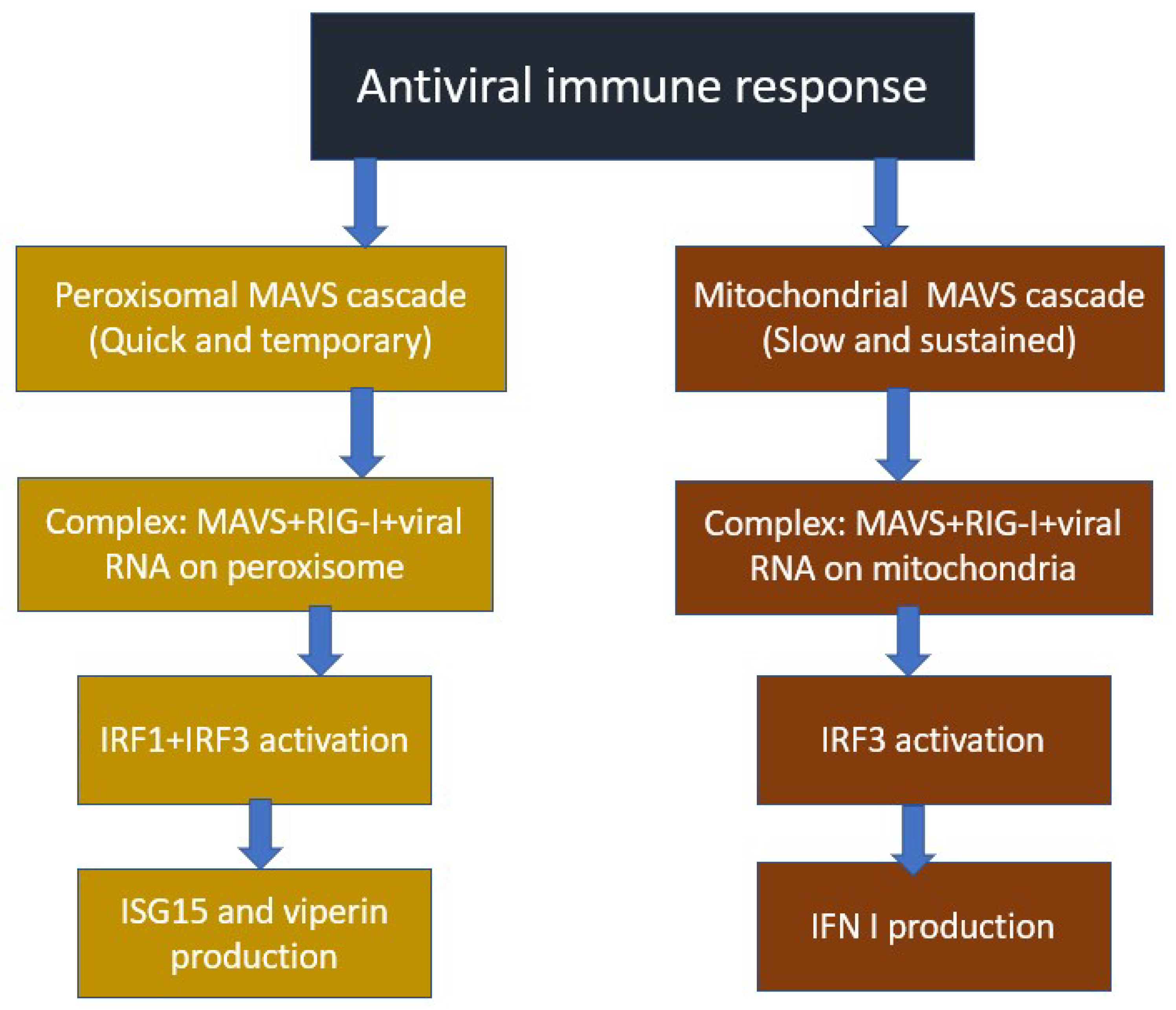
3.2. Mechanism of Antiviral Immune Response Involving Mitochondria
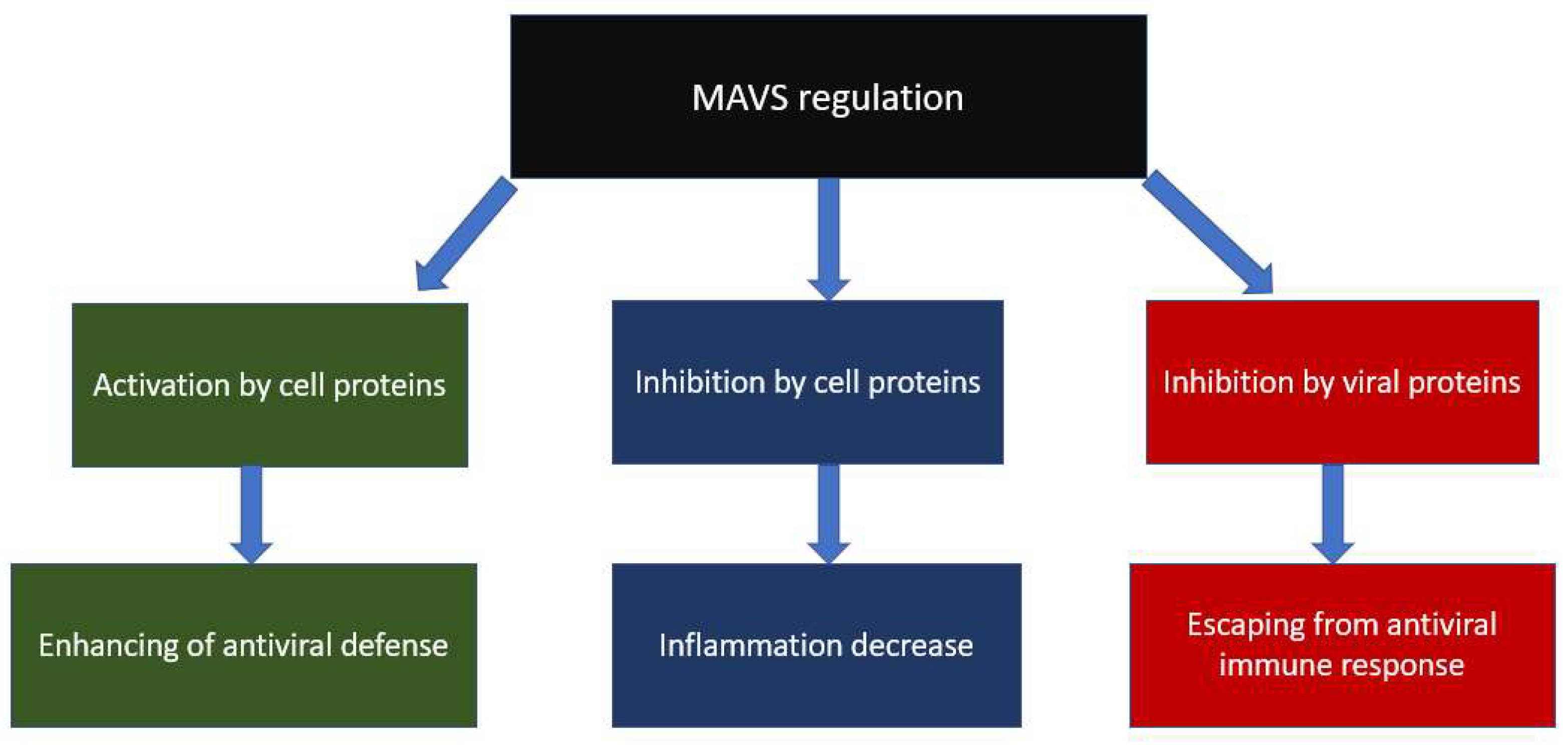
3.3. Mitochondrial and Viral Regulators of MAVS
4. Impact of HIV on the Development of Mitochondrial Dysfunction
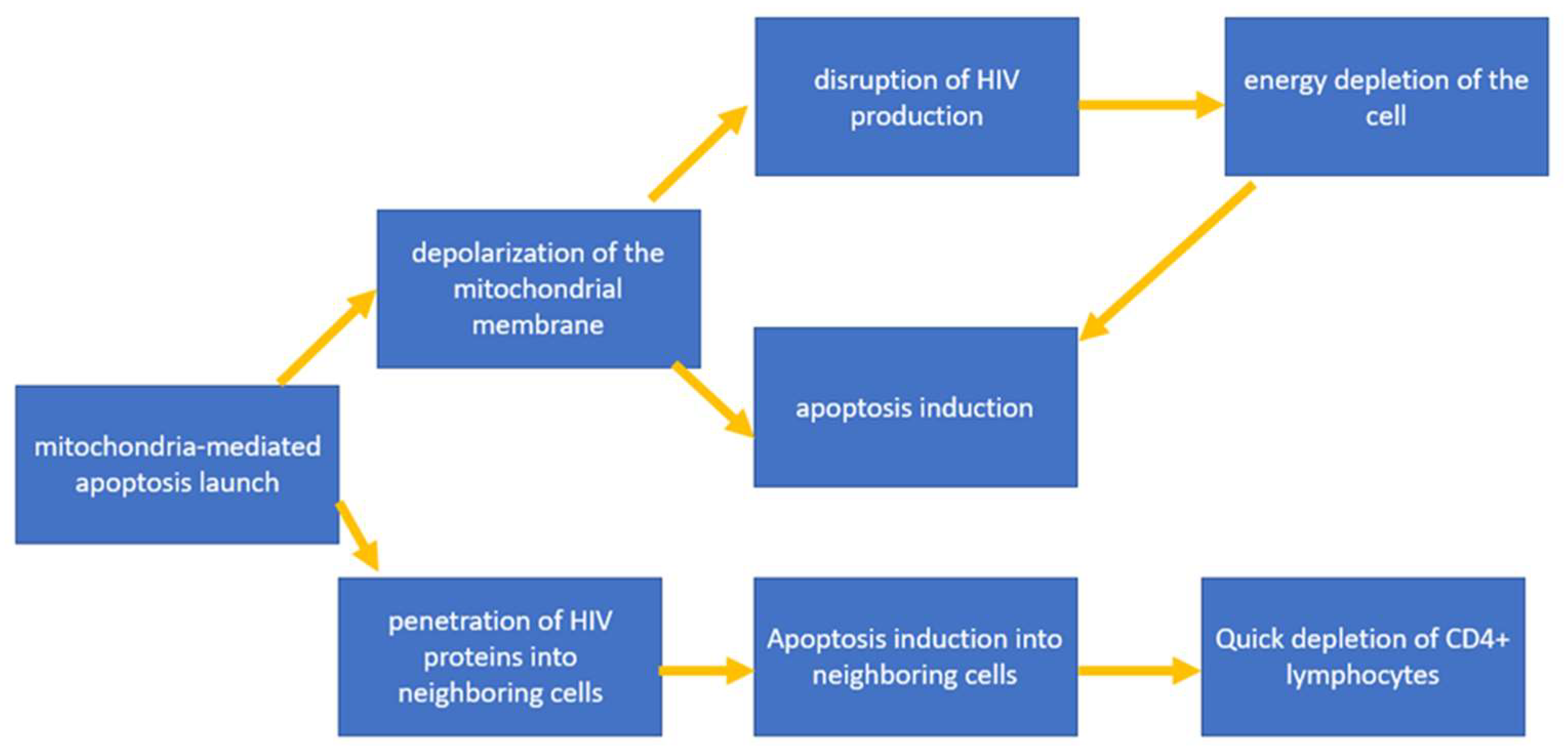
4.1. Triggering Mitochondria-Mediated Apoptosis
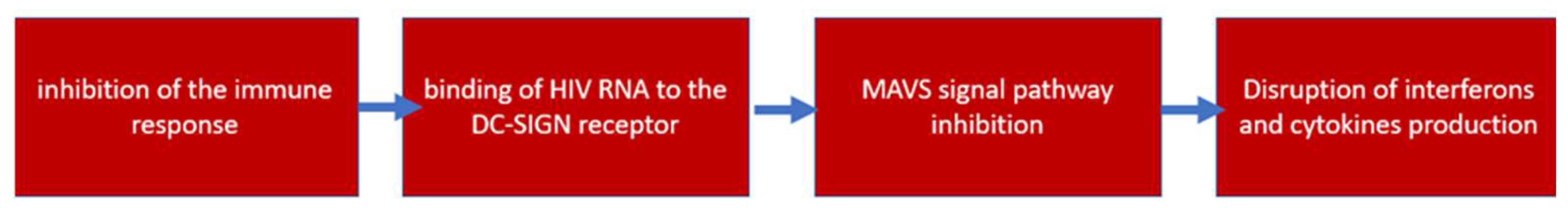
4.2. Inhibition of the Immune Response against HIV
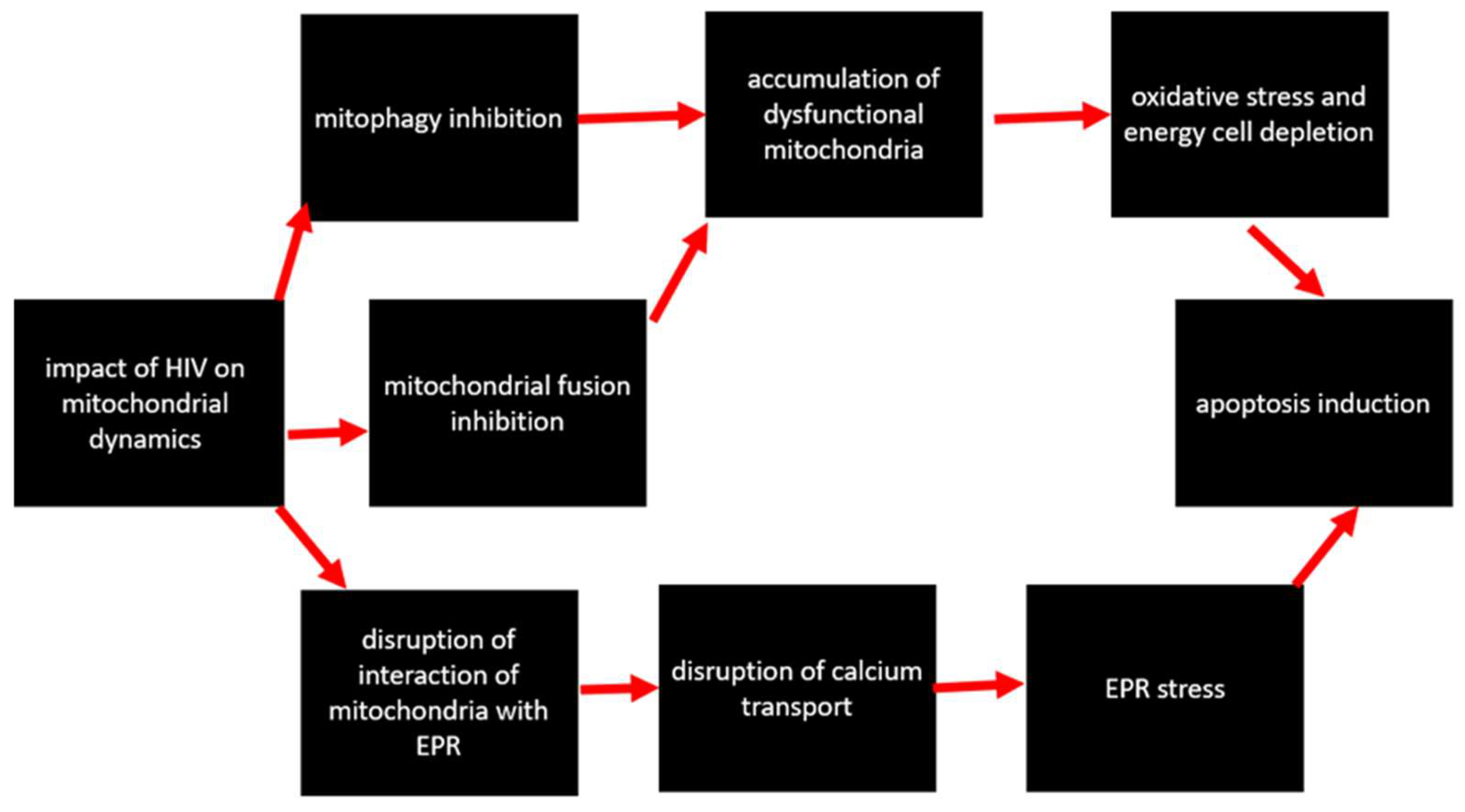
4.3. Impact on Mitochondrial Dynamics
5. Impact of ART on the Development of Mitochondrial Dysfunction
5.1. Effect of Nucleoside Reverse Transcriptase Inhibitors on Mitochondria
5.2. Effect of Non-Nucleoside Reverse Transcriptase Inhibitors on Mitochondria
5.3. Effect of Protease Inhibitors on Mitochondria
6. Future Research
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- HIV. Available online: https://www.who.int/data/gho/data/themes/hiv-aids (accessed on 28 September 2022).
- Waymack, J.R.; Sundareshan, V. Acquired Immune Deficiency Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Vaillant, A.A.J.; Naik, R. HIV-1 Associated Opportunistic Infections. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Yarchoan, R.; Uldrick, T.S. HIV-Associated Cancers and Related Diseases. N. Engl. J. Med. 2018, 378, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Poorolajal, J.; Hooshmand, E.; Mahjub, H.; Esmailnasab, N.; Jenabi, E. Survival rate of AIDS disease and mortality in HIV-infected patients: A meta-analysis. Public Health 2016, 139, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Cornett, J.K.; Kirn, T.J. Laboratory diagnosis of HIV in adults: A review of current methods. Clin. Infect. Dis. 2013, 57, 712–718. [Google Scholar] [CrossRef]
- Kemnic, T.R.; Gulick, P.G. HIV Antiretroviral Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Duewelhenke, N.; Krut, O.; Eysel, P. Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob. Agents Chemother. 2007, 51, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. Mitochondrial Dysfunction and Chronic Disease: Treatment With Natural Supplements. Integr. Med. A Clin. J. 2014, 13, 35. [Google Scholar]
- Elesela, S.; Lukacs, N.W. Role of Mitochondria in Viral Infections. Life 2021, 11, 232. [Google Scholar] [CrossRef]
- Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174. [Google Scholar] [CrossRef]
- Wallace, J.; Gonzalez, H.; Rajan, R.; Narasipura, S.D.; Virdi, A.K.; Olali, A.Z.; Naqib, A.; Arbieva, Z.; Maienschein-Cline, M.; Al-Harthi, L. Anti-HIV Drugs Cause Mitochondrial Dysfunction in Monocyte-Derived Macrophages. Antimicrob. Agents Chemother. 2022, 66, e0194121. [Google Scholar] [CrossRef]
- Moore, J.P.; Kitchen, S.G.; Pugach, P.; Zack, J.A. The CCR5 and CXCR4 coreceptors--central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retrovir. 2004, 20, 111–126. [Google Scholar] [CrossRef]
- Lackner, A.A.; Lederman, M.M.; Rodriguez, B. HIV Pathogenesis: The Host. Cold Spring Harb. Perspect. Med. 2012, 2, a007005. [Google Scholar] [CrossRef]
- Sasseville, V.G.; Du, Z.; Chalifoux, L.V.; Pauley, D.R.; Young, H.L.; Sehgal, P.K.; Desrosiers, R.C.; Lackner, A.A. Induction of lymphocyte proliferation and severe gastrointestinal disease in macaques by a nef gene variant SIVmac239. Am. J. Pathol. 1996, 149, 163. [Google Scholar] [PubMed]
- Naif, H.M. Pathogenesis of HIV Infection. Infect. Dis. Rep. 2013, 5, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.T.; Henry, K.; Zupancic, M.; Sedgewick, G.; Faust, R.A.; Melroe, H.; Cavert, W.; Gebhard, K.; Staskus, K.; Zhang, Z.Q.; et al. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 1996, 274, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 Infection. N. Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef]
- Ford, E.S.; Puronen, C.E.; Sereti, I. Immunopathogenesis of Asymptomatic Chronic HIV Infection: The Calm before the Storm. Curr. Opin. HIVAIDS 2009, 4, 206. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-Specific Immunity In Vivo by PD-1 Blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef]
- Weber, J. The pathogenesis of HIV-1 infection. Br. Med. Bull. 2001, 58, 61–72. [Google Scholar] [CrossRef]
- Romagnani, S.; Maggi, E. Th1 versus Th2 responses in AIDS. Curr. Opin. Immunol. 1994, 6, 616–622. [Google Scholar] [CrossRef]
- Koshiba, T.; Bashiruddin, N.; Kawabata, S. Mitochondria and antiviral innate immunity. Int. J Biochem. Mol. Biol. 2011, 2, 257. [Google Scholar]
- Takeuchi, O.; Akira, S. Innate Immunity to Virus Infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Kato, H.; Sato, S.; Takahashi, K.; Coban, C.; Yamamoto, M.; Uematsu, S.; Ishii, K.J.; Takeuchi, O.; et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006, 203, 1795. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef]
- Sharma, S.; Fitzgerald, K.A. Viral defense: It takes two MAVS to Tango. Cell 2010, 141, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.B.; Bergstralh, D.T.; Duncan, J.A.; Lei, Y.; Morrison, T.E.; Zimmermann, A.G.; Accavitti-Loper, M.A.; Madden, V.J.; Sun, L.; Ye, Z.; et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008, 451, 573–577. [Google Scholar] [CrossRef]
- Xu, L.; Xiao, N.; Liu, F.; Ren, H.; Gu, J. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 1530–1535. [Google Scholar] [CrossRef]
- Yasukawa, K.; Oshiumi, H.; Takeda, M.; Ishihara, N.; Yanagi, Y.; Seya, T.; Kawabata, S.I.; Koshiba, T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009, 2, ra47. [Google Scholar] [CrossRef]
- Lei, Y.; Moore, C.B.; Liesman, R.M.; O’Connor, B.P.; Bergstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P.Y. MAVS-Mediated Apoptosis and Its Inhibition by Viral Proteins. PLoS ONE 2009, 4, e5466. [Google Scholar] [CrossRef]
- Sternfeld, T.; Tischleder, A.; Schuster, M.; Bogner, J.R. Mitochondrial membrane potential and apoptosis of blood mononuclear cells in untreated HIV-1 infected patients. HIV Med. 2009, 10, 512–519. [Google Scholar] [CrossRef]
- Garg, H.; Mohl, J.; Joshi, A. HIV-1 Induced Bystander Apoptosis. Viruses 2012, 4, 3020. [Google Scholar] [CrossRef]
- Vijayan, K.K.V.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front. Immunol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed]
- Law, K.M.; Komarova, N.L.; Yewdall, A.W.; Lee, R.K.; Herrera, O.L.; Wodarz, D.; Chen, B.K. In Vivo HIV-1 Cell-to-Cell Transmission Promotes Multicopy Micro-compartmentalized Infection. Cell Rep. 2016, 15, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Termini, J.M.; Issac, B.; Guirado, E.; Stone, G.W. Constitutively Active MAVS Inhibits HIV-1 Replication via Type I Interferon Secretion and Induction of HIV-1 Restriction Factors. PLoS ONE 2016, 11, e0148929. [Google Scholar] [CrossRef] [PubMed]
- Gringhuis, S.I.; Hertoghs, N.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Sarrami-Forooshani, R.; Sprokholt, J.K.; van Teijlingen, N.H.; Kootstra, N.A.; Booiman, T.; van Dort, K.A.; et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 2017, 18, 225–235. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin Di Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef]
- Guitart-Mampel, M.; Hernandez, A.S.; Moren, C.; Catalan-Garcia, M.; Tobias, E.; Gonzalez-Casacuberta, I.; Juarez-Flores, D.L.; Gatell, J.M.; Cardellach, F.; Milisenda, J.C.; et al. Imbalance in mitochondrial dynamics and apoptosis in pregnancies among HIV-infected women on HAART with obstetric complications. J. Antimicrob. Chemother. 2017, 72, 2578–2586. [Google Scholar] [CrossRef]
- Rozzi, S.J.; Avdoshina, V.; Fields, J.A.; Mocchetti, I. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov. 2018, 4, 8. [Google Scholar] [CrossRef]
- Pinti, M.; Salomoni, P.; Cossarizza, A. Anti-HIV drugs and the mitochondria. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 700–707. [Google Scholar] [CrossRef]
- Blas-Garcia, A.; Apostolova, N.; Esplugues, J.V. Oxidative stress and mitochondrial impairment after treatment with anti-HIV drugs: Clinical implications. Curr. Pharm. Des. 2011, 17, 4076–4086. [Google Scholar] [CrossRef]
- Barile, M.; Valenti, D.; Passarella, S.; Quagliariello, E. 3′-Azido-3′-deoxythymidine uptake into isolated rat liver mitochondria and impairment of ADP/ATP translocator. Biochem. Pharmacol. 1997, 53, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Setzer, B.; Schlesier, M.; Thomas, A.K.; A Walker, U. Mitochondrial Toxicity of Nucleoside Analogues in Primary Human Lymphocytes. Antivir. Ther. 2005, 10, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Day, B.J.; Copeland, W.C. Mitochondrial toxicity of NRTI antiviral drugs: An integrated cellular perspective. Nat. Rev. Drug Discov. 2003, 2, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Pau, A.K.; George, J.M. Antiretroviral Therapy: Current Drugs. Infect. Dis. Clin. 2014, 28, 371–402. [Google Scholar] [CrossRef]
- Apostolova, N.; Gomez-Sucerquia, L.J.; Moran, A.; Alvarez, A.; Blas-Garcia, A.; Esplugues, J.V. Enhanced oxidative stress and increased mitochondrial mass during Efavirenz-induced apoptosis in human hepatic cells. Br. J. Pharmacol. 2010, 160, 2069–2084. [Google Scholar] [CrossRef]
- Korencak, M.; Byrne, M.; Richter, E.; Schultz, B.T.; Juszczak, P.; Ake, J.A.; Ganesan, A.; Okulicz, J.F.; Robb, M.L.; de Los Reyes, B.; et al. Effect of HIV infection and antiretroviral therapy on immune cellular functions. JCI Insight 2019, 4, e126675. [Google Scholar] [CrossRef]
- Reyskens, K.M.S.E.; Essop, M.F. HIV protease inhibitors and onset of cardiovascular diseases: A central role for oxidative stress and dysregulation of the ubiquitin-proteasome system. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 256–268. [Google Scholar] [CrossRef]
- Wang, X.; Mu, H.; Chai, H.; Liao, D.; Yao, Q.; Chen, C. Human Immunodeficiency Virus Protease Inhibitor Ritonavir Inhibits Cholesterol Efflux from Human Macrophage-Derived Foam Cells. Am. J. Pathol. 2007, 171, 304–314. [Google Scholar] [CrossRef]
- Estrada, V.; De Villar, N.G.P.; Larrad, M.T.M.; López, A.G.; Fernandez, C.; Serrano-Rios, M. Long-term metabolic consequences of switching from protease inhibitors to efavirenz in therapy for human immunodeficiency virus-infected patients with lipoatrophy. Clin. Infect. Dis. 2002, 35, 69–76. [Google Scholar] [CrossRef]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. eBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Group | Mechanisms of Action on HIV | Mechanisms of Development of Mitochondrial Dysfunction |
|---|---|---|
| NRTI | Disruption of the HIV reverse transcription process with integration into HIV DNA and initiation of chain break | Mutagenesis and inhibition of mitochondrial DNA synthesis; Disruption of ATP/ADP translocation; Decreased expression of cytochrome c oxidase; Development of oxidative stress; Disruption of oxidative phosphorylation. |
| NNRTI | Disruption of the HIV reverse transcription process by direct inhibition of reverse transcriptase | Decreased mitochondrial membrane potential followed by rupture; Disruption of oxidative phosphorylation; The release of cytochrome C into the cytoplasm with the initiation of apoptosis. |
| IP | Inhibition of maturation of HIV proteins by direct action on HIV integrase | Decreased mitochondrial membrane potential followed by rupture; Disruption of oxidative phosphorylation; The development of oxidative stress, both as a result of their production by mitochondria, and by an increase in the expression of NADH oxidase; Disruption of glucose transport and lipid metabolism; The release of cytochrome C into the cytoplasm with the initiation of apoptosis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagov, A.V.; Sukhorukov, V.N.; Guo, S.; Zhang, D.; Popov, M.A.; Orekhov, A.N. Impaired Mitochondrial Function in T-Lymphocytes as a Result of Exposure to HIV and ART. Cells 2023, 12, 1072. https://doi.org/10.3390/cells12071072
Blagov AV, Sukhorukov VN, Guo S, Zhang D, Popov MA, Orekhov AN. Impaired Mitochondrial Function in T-Lymphocytes as a Result of Exposure to HIV and ART. Cells. 2023; 12(7):1072. https://doi.org/10.3390/cells12071072
Chicago/Turabian StyleBlagov, Alexander V., Vasily N. Sukhorukov, Shuzhen Guo, Dongwei Zhang, Mikhail A. Popov, and Alexander N. Orekhov. 2023. "Impaired Mitochondrial Function in T-Lymphocytes as a Result of Exposure to HIV and ART" Cells 12, no. 7: 1072. https://doi.org/10.3390/cells12071072
APA StyleBlagov, A. V., Sukhorukov, V. N., Guo, S., Zhang, D., Popov, M. A., & Orekhov, A. N. (2023). Impaired Mitochondrial Function in T-Lymphocytes as a Result of Exposure to HIV and ART. Cells, 12(7), 1072. https://doi.org/10.3390/cells12071072