
Nuclei on the Rise: When Nuclei-Based Methods Meet Next-Generation Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
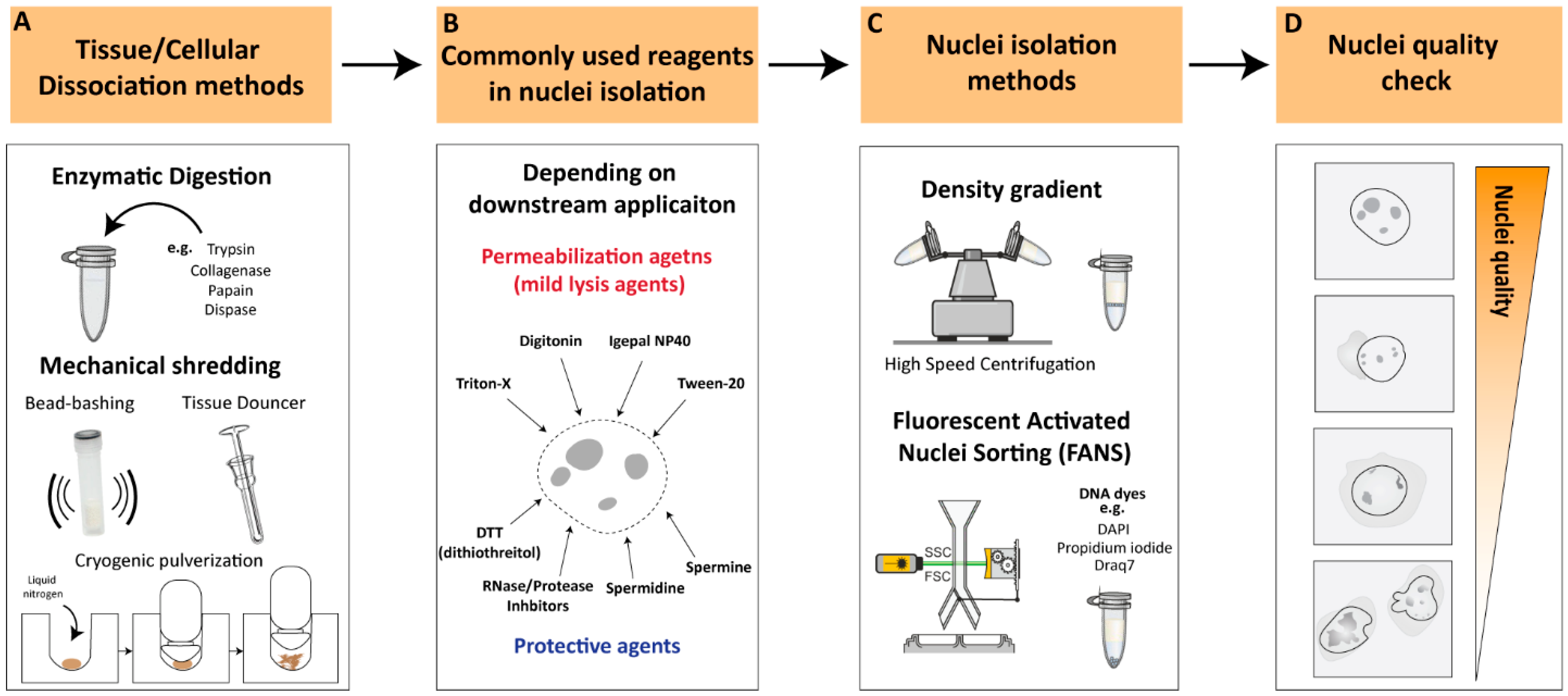
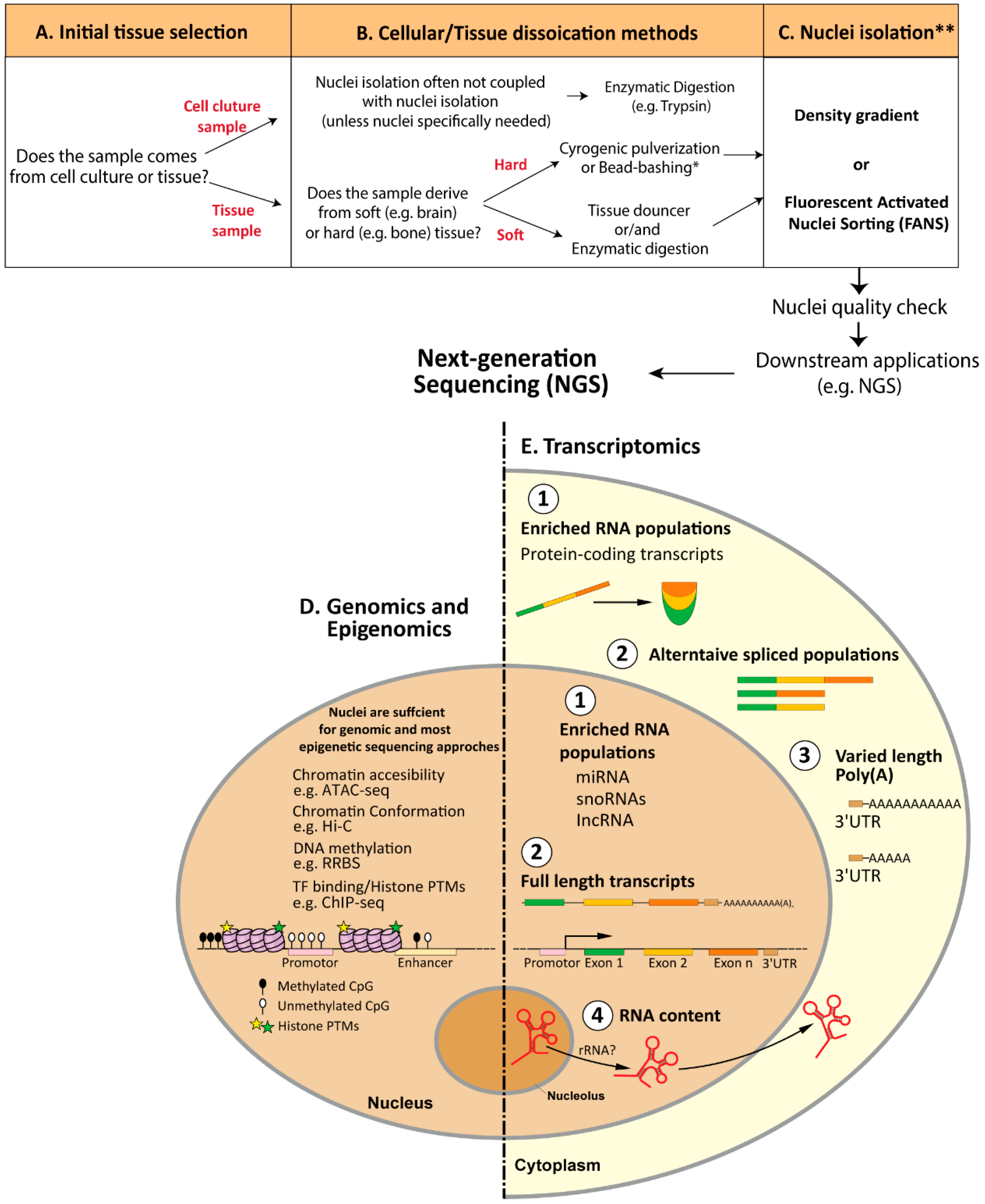
2. Nuclei-Isolation Procedures: From Cellular Dissociation to Nuclei Quality Check
2.1. Cellular Dissociation Methods
2.1.1. Enzymatic Digestion
2.1.2. Mechanical Dissociation
2.2. Nuclear Permeabilization and Protective Reagents
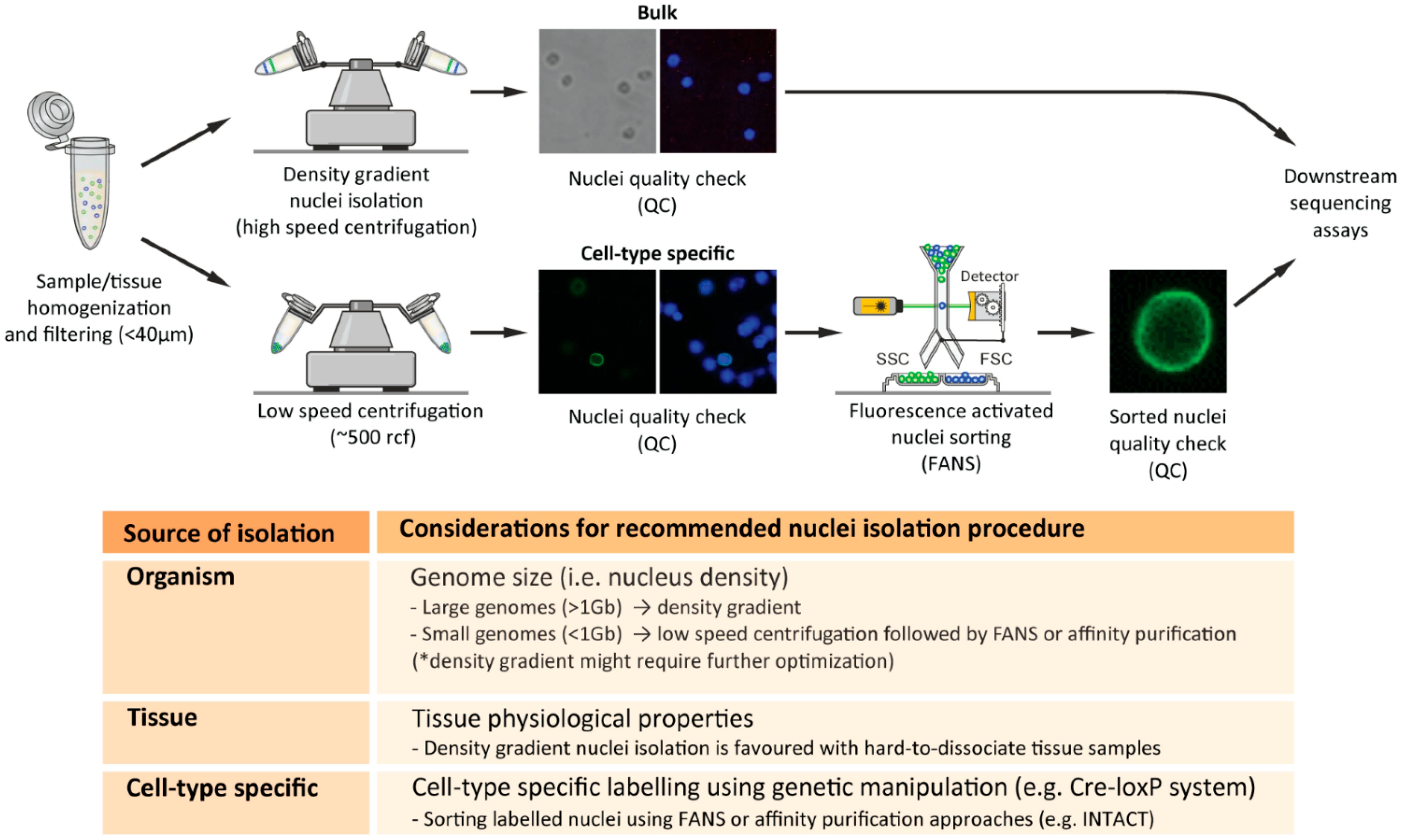
2.3. Nuclei-Isolation Methodologies
2.4. Nuclei Quality Check
2.5. Critical Considerations for Efficient Nuclei Isolation from Diverse Biological Sources
2.5.1. Nuclei Isolation from Distinct Organisms
2.5.2. Nuclei Isolation from Distinct Tissues
2.5.3. Cell-Type-Specific Nuclei Isolation
3. Association of Nuclei with Next-Generation Sequencing
3.1. Genomics and Epigenomics
3.2. Transcriptomics
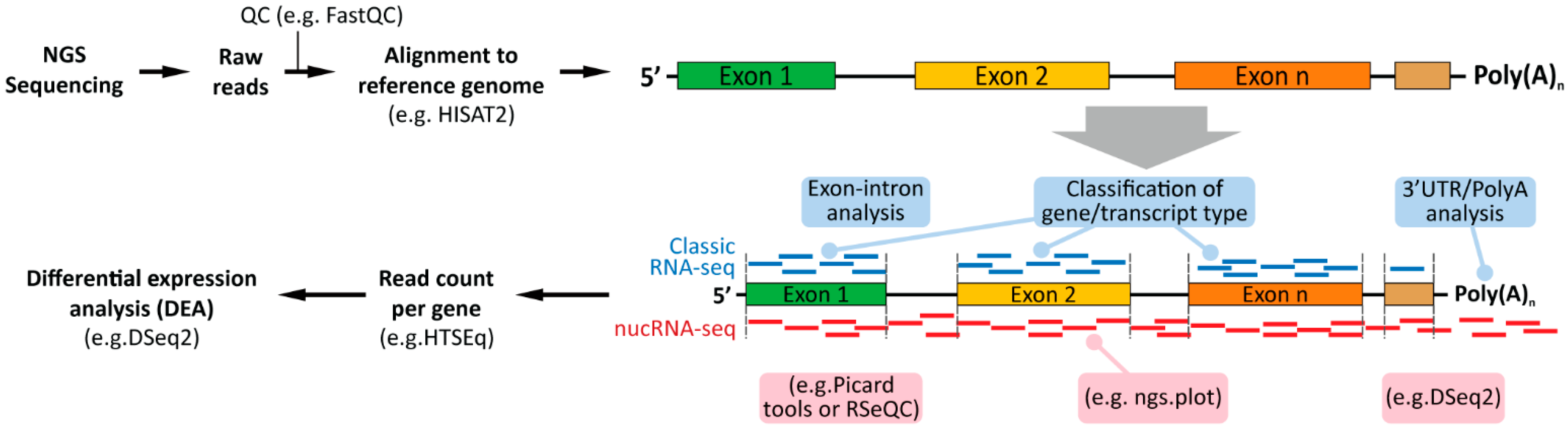
3.2.1. RNA-seq
3.2.2. Nuclear RNA-seq
3.2.3. Association of nucRNA-seq in ‘Multi-Omics’ Studies
3.3. Single-Cell and Single-Nucleus Sequencing Studies
Lessons from Single-Cell/-Nucleus Sequencing Analyses
4. Limitations of Nuclei-Based Studies
4.1. RNA Content-RNA Population Bias between Nuclei and Whole Cell
4.2. Experimental Design of Nuclear RNA-seq-Sequencing Depth and Analysis of Exon Versus Intron Reads
4.3. Nuclear RNA Quality and Library Preparation Strategies
4.3.1. Nuclear RNA Quality
4.3.2. RNA Library Preparation Strategies
5. Computational Analyses of Classical (Whole Cells) and Nuclear RNA-seq
6. Discussion and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Devos, D.P.; Gräf, R.; Field, M.C. Evolution of the nucleus. Curr. Opin. Cell Biol. 2014, 28, 8–15. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Pederson, T. The nucleus introduced. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–16. [Google Scholar] [CrossRef]
- Mekhail, K.; Moazed, D. The nuclear envelope in genome organization, expression and stability. Nat. Rev. Mol. Cell Biol. 2010, 11, 317–328. [Google Scholar] [CrossRef]
- Köhler, A.; Hurt, E. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 2007, 8, 761–773. [Google Scholar] [CrossRef]
- Illumina. An Introduction to Next-Generation Sequencing Technology. Available online: http://www.illumina.com/technology/next-generation-sequencing.html (accessed on 3 August 2022).
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, M.J.; Tomlinson, S.; Yang, X.B.; Kirkham, J. Cell separation: Terminology and practical considerations. J. Tissue Eng. 2013, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Garcia-Montero, A.C.; Orfao, A. Cell Purification: A New Challenge for Biobanks. Pathobiology 2015, 81, 261–275. [Google Scholar] [CrossRef]
- Volovitz, I.; Shapira, N.; Ezer, H.; Gafni, A.; Lustgarten, M.; Alter, T.; Ben-Horin, I.; Barzilai, O.; Shahar, T.; Kanner, A.; et al. A non-aggressive, highly efficient, enzymatic method for dissociation of human brain-tumors and brain-tissues to viable single-cells. BMC Neurosci. 2016, 17, 30. [Google Scholar] [CrossRef]
- Reichard, A.; Asosingh, K. Best Practices for Preparing a Single Cell Suspension from Solid Tissues for Flow Cytometry. Cytom. Part A 2019, 95, 219. [Google Scholar] [CrossRef]
- Mendibil, U.; Ruiz-Hernandez, R.; Retegi-Carrion, S.; Garcia-Urquia, N.; Olalde-Graells, B.; Abarrategi, A. Tissue-Specific Decellularization Methods: Rationale and Strategies to Achieve Regenerative Compounds. Int. J. Mol. Sci. 2020, 21, 5447. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Burattini, S.; Ciacci, C.; Ambrogini, P.; Carloni, S.; Balduini, W.; Lopez, D.; Panza, G.; Papa, S.; Canonico, B. Automated–Mechanical Procedure Compared to Gentle Enzy-matic Tissue Dissociation in Cell Function Studies. Biomolecules 2022, 12, 701. [Google Scholar] [CrossRef] [PubMed]
- Miersch, C.; Stange, K.; Röntgen, M. Effects of trypsinization and of a combined trypsin, collagenase, and DNase digestion on liberation and in vitro function of satellite cells isolated from juvenile porcine muscles. Vitr. Cell. Dev. Biol. Animal 2018, 54, 406. [Google Scholar] [CrossRef]
- Yousef, H.; Czupalla, C.J.; Lee, D.; Butcher, E.C.; Wyss-Coray, T. Papain-based Single Cell Isolation of Primary Murine Brain Endothelial Cells Using Flow Cytometry. Bio-Protocol 2018, 8, e3091. [Google Scholar] [CrossRef]
- Gao, M.; Guo, P.; Liu, X.; Zhang, P.; He, Z.; Wen, L.; Liu, S.; Zhou, Z.; Zhu, W. Systematic study of single-cell isolation from musculoskeletal tissues for single-sell sequencing. BMC Mol. Cell Biol. 2022, 23, 32. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef]
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; Kock, L.; Lassmann, T.; Poppe, D.; Clement, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef] [PubMed]
- Nott, A.; Schlachetzki, J.C.M.; Fixsen, B.R.; Glass, C.K. Nuclei isolation of multiple brain cell types for omics interrogation. Nat. Protoc. 2021, 16, 1629–1646. [Google Scholar] [CrossRef]
- Wei, S.; Levy, B.; Hoffman, N.; Cujar, C.; Rodney-Sandy, R.; Wapner, R.; D’Alton, M.; Williams, Z. A rapid and simple bead-bashing-based method for genomic DNA extraction from mammalian tissue. Biotechniques 2020, 68, 240–244. [Google Scholar] [CrossRef]
- Bead Beating Guide|MP Biomedicals. Available online: https://www.mpbio.com/bs/bead-beating-technology-explained (accessed on 12 October 2022).
- Givens, R.M.; Mesner, L.D.; Hamlin, J.L.; Buck, M.J.; Huberman, J.A. Integrity of chromatin and replicating DNA in nuclei released from fission yeast by semi-automated grinding in liquid nitrogen. BMC Res. Notes 2011, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zerpa-Catanho, D.; Zhang, X.; Song, J.; Hernandez, A.G.; Ming, R. Ultra-long DNA molecule isolation from plant nuclei for ultra-long read genome sequencing. STAR Protoc. 2021, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, F.; Douglas, C.; Lega, B.C.; Konopka, G. Nuclei isolation from surgically resected human hippocampus. STAR Protoc. 2021, 2, 100844. [Google Scholar] [CrossRef] [PubMed]
- Loft, A.; Herzig, S.; Schmidt, S.F. Purification of GFP-tagged nuclei from frozen livers of INTACT mice for RNA- and ATAC-sequencing. STAR Protoc. 2021, 2, 100805. [Google Scholar] [CrossRef] [PubMed]
- Linke, D. Detergents: An Overview. Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2009; Chapter 34; pp. 603–617. [Google Scholar] [CrossRef]
- Krishnaswami, S.R.; Grindberg, R.V.; Novotny, M.; Venepally, P.; Lacar, B.; Bhutani, K.; Linker, S.B.; Pham, S.; Erwin, J.A.; Miller, J.A.; et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat. Protoc. 2016, 11, 499–524. [Google Scholar] [CrossRef] [PubMed]
- Maitra, M.; Nagy, C.; Chawla, A.; Wang, Y.C.; Nascimento, C.; Suderman, M.; Théroux, J.F.; Mechawar, N.; Ragoussis, J.; Turecki, G. Extraction of nuclei from archived postmortem tissues for single-nucleus sequencing applications. Nat. Protoc. 2021, 16, 2788–2801. [Google Scholar] [CrossRef]
- Khan, A.U.; Mei, Y.H.; Wilson, T. A proposed function for spermine and spermidine: Protection of replicating DNA against damage by singlet oxygen. Proc. Natl. Acad. Sci. USA 1992, 89, 11426–11427. [Google Scholar] [CrossRef]
- Chauveau, J.; Moulé, Y.; Rouiller, C. Isolation of pure and unaltered liver nuclei morphology and biochemical composition. Exp. Cell Res. 1956, 11, 317–321. [Google Scholar] [CrossRef]
- Hadjiolov, A.A.; Tencheva, Z.S.; Bojadjieva-Mikhailova, A.G. Isolation and some characteristics of cell nuclei from brain cortex of adult cat. J. Cell Biol. 1965, 26, 383–393. [Google Scholar] [CrossRef]
- Blobel, G.; Potter, V.R. Nuclei from Rat Liver: Isolation Method That Combines Purity with High Yield. Science 1966, 154, 1662–1665. [Google Scholar] [CrossRef] [PubMed]
- Katholnig, K.; Poglitsch, M.; Hengstschläger, M.; Weichhart, T. Lysis gradient centrifugation: A flexible method for the isolation of nuclei from primary cells. Methods Mol. Biol. 2015, 1228, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Guck, J. The Relative Densities of Cytoplasm and Nuclear Compartments Are Robust against Strong Perturbation. Biophys. J. 2020, 119, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Chongtham, M.C.; Butto, T.; Mungikar, K.; Gerber, S.; Winter, J. INTACT vs. FANS for Cell-Type-Specific Nuclei Sorting: A Comprehensive Qualitative and Quantitative Comparison. Int. J. Mol. Sci. 2021, 22, 5335. [Google Scholar] [CrossRef] [PubMed]
- Eremenko, E.; Golova, A.; Stein, D.; Einav, M.; Khrameeva, E.; Toiber, D. FACS-based isolation of fixed mouse neuronal nuclei for ATAC-seq and Hi-C. STAR Protoc. 2021, 2, 3. [Google Scholar] [CrossRef]
- Ligasová, A.; Koberna, K. DNA Dyes—Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods. Molecules 2021, 26, 5515. [Google Scholar] [CrossRef]
- Orchard, P.; Manickam, N.; Ventresca, C.; Vadlamudi, S.; Varshney, A.; Rai, V.; Kaplan, J.; Lalancette, C.; Mohlke, K.L.; Gallagher, K.; et al. Human and rat skeletal muscle single-nuclei multi-omic integrative analyses nominate causal cell types, regulatory elements, and SNPs for complex traits. Genome Res. 2021, 31, 2258–2275. [Google Scholar] [CrossRef]
- Lovtrup-Rein, H.; McEwen, B.S. Isolation and fractionation of rat brain nuclei. J. Cell Biol. 1966, 30, 405–415. [Google Scholar] [CrossRef]
- Cui, M.; Wang, Z.; Chen, K.; Shah, A.M.; Tan, W.; Duan, L.; Sanchez-Ortiz, E.; Li, H.; Xu, L.; Liu, N.; et al. Dynamic Transcriptional Responses to Injury of Regenerative and Non-regenerative Cardiomyocytes Revealed by Single-Nucleus RNA Sequencing. Dev. Cell 2020, 53, 102–116. [Google Scholar] [CrossRef]
- Gupta, V.; Lazzaro, B.P. A robust method to isolate Drosophila fat body nuclei for transcriptomic analysis. Fly 2022, 16, 62–67. [Google Scholar] [CrossRef]
- Steiner, F.A.; Talbert, P.B.; Kasinathan, S.; Deal, R.B.; Henikoff, S. Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genome Res. 2012, 22, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Wei, G.; McManus, C.E.; Hillier, L.W.; Reinke, V.; Isolated, C. elegans germ nuclei exhibit distinct genomic profiles of histone modification and gene expression. BMC Genom. 2019, 20, 500. [Google Scholar] [CrossRef]
- Annunziata, R.; Balestra, C.; Marotta, P.; Ruggiero, A.; Manfellotto, F.; Benvenuto, G.; Biffali, E.; Ferrante, M.I. An optimised method for intact nuclei isolation from diatoms. Sci. Rep. 2021, 11, 1681. [Google Scholar] [CrossRef] [PubMed]
- Folta, K.M.; Kaufman, L.S. Isolation of Arabidopsis nuclei and measurement of gene transcription rates using nuclear run-on assays. Nat. Protoc. 2006, 1, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Sikorskaite, S.; Rajamäki, M.L.; Baniulis, D.; Stanys, V.; Valkonen, J.P.T. Protocol: Optimised methodology for isolation of nuclei from leaves of species in the Solanaceae and Rosaceae families. Plant Methods 2013, 9, 31. [Google Scholar] [CrossRef]
- Moro, B.; Kisielow, M.; Borrero, V.B.; Bouet, A.; Brosnan, C.A.; Bologna, N.G. Nuclear RNA purification by flow cytometry to study nuclear processes in plants. STAR Protoc. 2021, 2, 1. [Google Scholar] [CrossRef]
- Wu, H.; Kirita, Y.; Donnelly, E.L.; Humphreys, B.D. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol. 2019, 30, 23–32. [Google Scholar] [CrossRef]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- Tang J C, Y.; Rudolph, S.; Dhande O, S.; Abraira, V.E.; Choi, S.; Lapan, S.W.; Drew, I.R.; Drokhlyansky, E.; Huberman, A.D.; Regher, W.G. Cell type–specific manipulation with GFP-dependent Cre recombinase. Nat. Neurosci. 2015, 18, 1334–1341. [Google Scholar] [CrossRef]
- Deal, R.B.; Henikoff, S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev. Cell 2010, 18, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Deal, R.B.; Henikoff, S. The INTACT method for cell typeg-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Henry, G.L.; Davis, F.P.; Picard, S.; Eddy, S.R. Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res. 2012, 40, 9691–9704. [Google Scholar] [CrossRef] [PubMed]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef]
- Mo, A.; Luo, C.; Davis, F.P.; Mukamel, E.A.; Henry, G.L.; Nery, J.R.; Urich, M.A.; Picard, S.; Lister, R.; Eddy, S.R.; et al. Epigenomic landscapes of retinal rods and cones. eLife 2016, 5, e11613. [Google Scholar] [CrossRef]
- Handley, A.; Schauer, T.; Ladurner, A.G.; Margulies, C.E. Designing Cell-Type-Specific Genome-wide Experiments. Mol. Cell 2015, 58, 621–631. [Google Scholar] [CrossRef]
- Jiang, Y.; Matevossian, A.; Huang, H.S.; Straubhaar, J.; Akbarian, S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008, 9, 42. [Google Scholar] [CrossRef]
- Haenni, S.; Ji, Z.; Hoque, M.; Rust, N.; Sharpe, H.; Eberhard, R.; Browne, C.; Hengartner, M.O.; Mellor, J.; Tian, B.; et al. Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3′-end-seq. Nucleic Acids Res. 2012, 40, 6304–6318. [Google Scholar] [CrossRef]
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 2018, 24, 335–341. [Google Scholar] [CrossRef]
- Wang, Y.C.; Peterson, S.E.; Loring, J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014, 2, 143–160. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Mitchell J, A.; Clay, I.; Umlauf, D.; Chen, C.; Moir, C.A.; Eskiw, C.H.; Schoenfelder, S.; Chakalova, L.; Nagano, T.; Fraser, P. Nuclear RNA sequencing of the mouse erythroid cell transcriptome. PLoS ONE 2012, 7, e49274. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Pinu, F.R.; Beale, D.J.; Paten, A.M.; Kouremenos, K.; Swarup, S.; Schirra, H.J.; Wishart, D. Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 2019, 9, 76. [Google Scholar] [CrossRef]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2017, 36, 70–80. [Google Scholar] [CrossRef]
- Lacar, B.; Linker, S.B.; Jaeger, B.N.; Krishnaswami, S.; Barron, J.; Kelder, M.; Parylak, S.; Paquola, A.; Venepally, P.; Novotny, M.; et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 2016, 7, 11022. [Google Scholar] [CrossRef]
- Bakken, T.E.; Hodge, R.D.; Miller, J.A.; Yao, Z.; Nguyen, T.N.; Aevermann, B.; Barkan, E.; Bertagnolli, D.; Casper, T.; Dee, N.; et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 2018, 13, e0209648. [Google Scholar] [CrossRef] [PubMed]
- Korrapati, S.; Taukulis, I.; Olszewski, R.; Pyle, M.; Gu, S.; Singh, R.; Griffiths, C.; Martin, D.; Boger, E.; Morell, R.J.; et al. Single Cell and Single Nucleus RNA-Seq Reveal Cellular Heterogeneity and Homeostatic Regulatory Networks in Adult Mouse Stria Vascularis. Front. Mol. Neurosci. 2019, 12, 316. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Subramaniam, M.; Targ, S.; Nguyen, M.; Maliskova, L.; McCarthy, E.; Wan, E.; Wong, S.; Byrnes, L.; Lanata, C.M.; et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018, 36, 89–94. [Google Scholar] [CrossRef]
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M.; Smibert, P.; Satija, R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018, 19, 224. [Google Scholar] [CrossRef] [PubMed]
- van den Brink, S.C.; Sage, F.; Vértesy, Á.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; van Oudenaarden, A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017, 14, 935–936. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.; Um, J.W. Synapse development organized by neuronal activity-regulated immediate-early genes. Exp. Mol. Med. 2018, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thrupp, N.; Sala Frigerio, C.; Wolfs, L.; Skene, N.G.; Fattorelli, N.; Poovathingal, S.; Fourne, Y.; Matthews, P.M.; Theys, T.; Mancuso, R.; et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020, 32, 108189. [Google Scholar] [CrossRef]
- Del-Aguila, J.L.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Fernandez, M.V.; Ibanez, L.; Bradley, J.; Wang, F.; Bergmann, K.; et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimer’s Res. Ther. 2019, 11, 71. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Theis, F.J.; Yang, H.; Zelikovsky, A.; McHardy, A.C.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Ayers, T. Single nucleus RNA-sequencing: How it’s done, applications and limitations. Emerg. Top. Life Sci. 2021, 5, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Barthelson, R.A.; Lambert, G.M.; Vanier, C.; Lynch, R.M.; Galbraith, D.W. Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells. BMC Genom. 2007, 8, 340. [Google Scholar] [CrossRef]
- Solnestam, B.W.; Stranneheim, H.; Hällman, J.; Käller, M.; Lundberg, E.; Lundeberg, J.; Akan, P. Comparison of total and cytoplasmic mRNA reveals global regulation by nuclear retention and miRNAs. BMC Genom. 2012, 13, 574. [Google Scholar] [CrossRef]
- Price, A.J.; Hwang, T.; Tao, R.; Burke, E.E.; Rajpurohit, A.; Shin, J.H.; Hyde, T.M.; Kleinman, J.E.; Jaffe, A.E.; Weinberger, D.R. Characterizing the nuclear and cytoplasmic transcriptomes in developing and mature human cortex uncovers new insight into psychiatric disease gene regulation. Genome Res. 2020, 30, 1–11. [Google Scholar] [CrossRef]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Zhang, A.Y.; Su, S.; Ng, A.P.; Holik, A.Z.; Asselin-Labat, M.L.; Ritchie, M.E.; Law, C.W. Covering all your bases: Incorporating intron signal from RNA-seq data. NAR Genom. Bioinform. 2020, 2, lqaa073. [Google Scholar] [CrossRef]
- Bahar Halpern, K.; Caspi, I.; Lemze, D.; Levy, M.; Landen, S.; Elinav, E.; Ulitsky, I.; Itzkovitz, S. Nuclear Retention of mRNA in Mammalian Tissues. Cell Rep. 2015, 13, 2653–2662. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Pulido, T.H.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef]
- Zaghlool, S.B.; Kühnel, B.; Elhadad, M.A.; Kader, S.; Halama, A.; Thareja, G.; Engelke, R.; Sarwath, H.; Al-Dous, E.K.; Mohamoud, Y.A.; et al. Epigenetics meets proteomics in an epigenome-wide association study with circulating blood plasma protein traits. Nat. Commun. 2020, 11, 15. [Google Scholar] [CrossRef]
- Fernandez-Albert, J.; Lipinski, M.; Lopez-Cascales, M.T.; Rowley, M.J.; Martin-Gonzalez, A.M.; del Blanco, B.; Corces, V.G.; Barco, A. Immediate and deferred epigenomic signatures of in vivo neuronal activation in mouse hippocampus. Nat. Neurosci. 2019, 22, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Ohta, Y.; Xu, M.; Tsutsumi, S.; Minami, T.; Inoue, K.; Komura, D.; Kitakami, J.; Oshida, N.; Papantonis, A.; et al. A wave of nascent transcription on activated human genes. Proc. Natl. Acad. Sci. USA 2009, 106, 18357–18361. [Google Scholar] [CrossRef] [PubMed]
- Georgomanolis, T.; Sofiadis, K.; Papantonis, A. Cutting a long intron short: Recursive splicing and its implications. Front. Physiol. 2016, 7, 598. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef]
- Adiconis, X.; Borges-Rivera, D.; Satija, R.; Deluca, D.S.; Busby, M.A.; Berlin, A.M.; Sivachenko, A.; Thompson, D.A.; Wysoker, A.; Fennell, T.; et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 2013, 10, 623–629. [Google Scholar] [CrossRef]
- Gallego Romero, I.; Pai, A.A.; Tung, J.; Gilad, Y. RNA-seq: Impact of RNA degradation on transcript quantification. BMC Biol. 2014, 12, 42. [Google Scholar] [CrossRef]
- Schuierer, S.; Carbone, W.; Knehr, J.; Petitjean, V.; Fernandez, A.; Sultan, M.; Roma, G. A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples. BMC Genom. 2017, 18, 442. [Google Scholar] [CrossRef]
- Reiman, M.; Laan, M.; Rull, K.; Sõber, S. Effects of RNA integrity on transcript quantification by total RNA sequencing of clinically collected human placental samples. FASEB J. 2017, 31, 3298–3308. [Google Scholar] [CrossRef]
- Rouquette, J.; Choesmel, V.; Gleizes, P.E. Nuclear export and cytoplasmic processing of precursors to the 40S ribosomal subunits in mammalian cells. EMBO J. 2005, 24, 2862–2872. [Google Scholar] [CrossRef]
- Mueller, O.; Lightfoot, S.; Schroeder, A. RNA Integrity Number (RIN)-Standardization of RNA Quality Control. Agil. Appl. Note Publ. 2004, 1, 1–8. [Google Scholar]
- Kaul, T.; Morales, M.E.; Sartor, A.O.; Belancio, V.P.; Deininger, P. Comparative analysis on the expression of L1 loci using various RNA-Seq preparations. Mob. DNA 2020, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.J.; Heller, E.A. Single sample sequencing (S3EQ) of epigenome and transcriptome in nucleus accumbens. J. Neurosci. Methods 2018, 308, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Lodato, N.J.; Melia, T.; Rampersaud, A.; Waxman, D.J. Sex-Differential Responses of Tumor Promotion-Associated Genes and Dysregulation of Novel Long Noncoding RNAs in Constitutive Androstane Receptor-Activated Mouse Liver. Toxicol. Sci. 2017, 159, 25–41. [Google Scholar] [CrossRef]
- Kenyon, A.; Gavriouchkina, D.; Zorman, J.; Napolitani, G.; Cerundolo, V.; Sauka-Spengler, T. Active nuclear transcriptome analysis reveals inflammasome-dependent mechanism for early neutrophil response to Mycobacterium marinum. Sci. Rep. 2017, 7, 6505. [Google Scholar] [CrossRef]
- Vilborg, A.; Sabath, N.; Wiesel, Y.; Nathans, J.; Levy-Adam, F.; Yario, T.A.; Steitz, J.A.; Shalgi, R. Comparative analysis reveals genomic features of stress-induced transcriptional readthrough. Proc. Natl. Acad. Sci. USA 2017, 114, E8362–E8371. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef]
- Ma, F.; Fuqua, B.K.; Hasin, Y.; Yukhtman, C.; Vulpe, C.D.; Lusis, A.J.; Pellegrini, M. A comparison between whole transcript and 3′ RNA sequencing methods using Kapa and Lexogen library preparation methods 06 Biological Sciences 0604 Genetics. BMC Genom. 2019, 20, 9. [Google Scholar] [CrossRef]
- Herbert, Z.T.; Kershner, J.P.; Butty, V.L.; Thimmapuram, J.; Choudhari, S.; Alekseyev, Y.O.; Fan, J.; Podnar, J.W.; Wilcox, E.; Gipson, J.; et al. Cross-site comparison of ribosomal depletion kits for Illumina RNAseq library construction. BMC Genom. 2018, 19, 199. [Google Scholar] [CrossRef]
- Haile, S.; Corbett, R.D.; Bilobram, S.; Mungall, K.; Grande, B.M.; Kirk, H.; Pandoh, P.; MacLeod, T.; McDonald, H.; Bala, M.; et al. Evaluation of protocols for rRNA depletion-based RNA sequencing of nanogram inputs of mammalian total RNA. PLoS ONE 2019, 14, e0224578. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. hisAt: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef]
- Gaidatzis, D.; Burger, L.; Florescu, M.; Stadler, M.B. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat. Biotechnol. 2015, 33, 722–729. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Shen, L.; Shao, N.; Liu, X.; Nestler, E. Ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genom. 2014, 15, 284. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson M, D.; McCarthy D, J.; Smyth G, K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butto, T.; Mungikar, K.; Baumann, P.; Winter, J.; Lutz, B.; Gerber, S. Nuclei on the Rise: When Nuclei-Based Methods Meet Next-Generation Sequencing. Cells 2023, 12, 1051. https://doi.org/10.3390/cells12071051
Butto T, Mungikar K, Baumann P, Winter J, Lutz B, Gerber S. Nuclei on the Rise: When Nuclei-Based Methods Meet Next-Generation Sequencing. Cells. 2023; 12(7):1051. https://doi.org/10.3390/cells12071051
Chicago/Turabian StyleButto, Tamer, Kanak Mungikar, Peter Baumann, Jennifer Winter, Beat Lutz, and Susanne Gerber. 2023. "Nuclei on the Rise: When Nuclei-Based Methods Meet Next-Generation Sequencing" Cells 12, no. 7: 1051. https://doi.org/10.3390/cells12071051
APA StyleButto, T., Mungikar, K., Baumann, P., Winter, J., Lutz, B., & Gerber, S. (2023). Nuclei on the Rise: When Nuclei-Based Methods Meet Next-Generation Sequencing. Cells, 12(7), 1051. https://doi.org/10.3390/cells12071051