
A Review of the Biosynthesis and Structural Implications of Insulin Gene Mutations Linked to Human Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
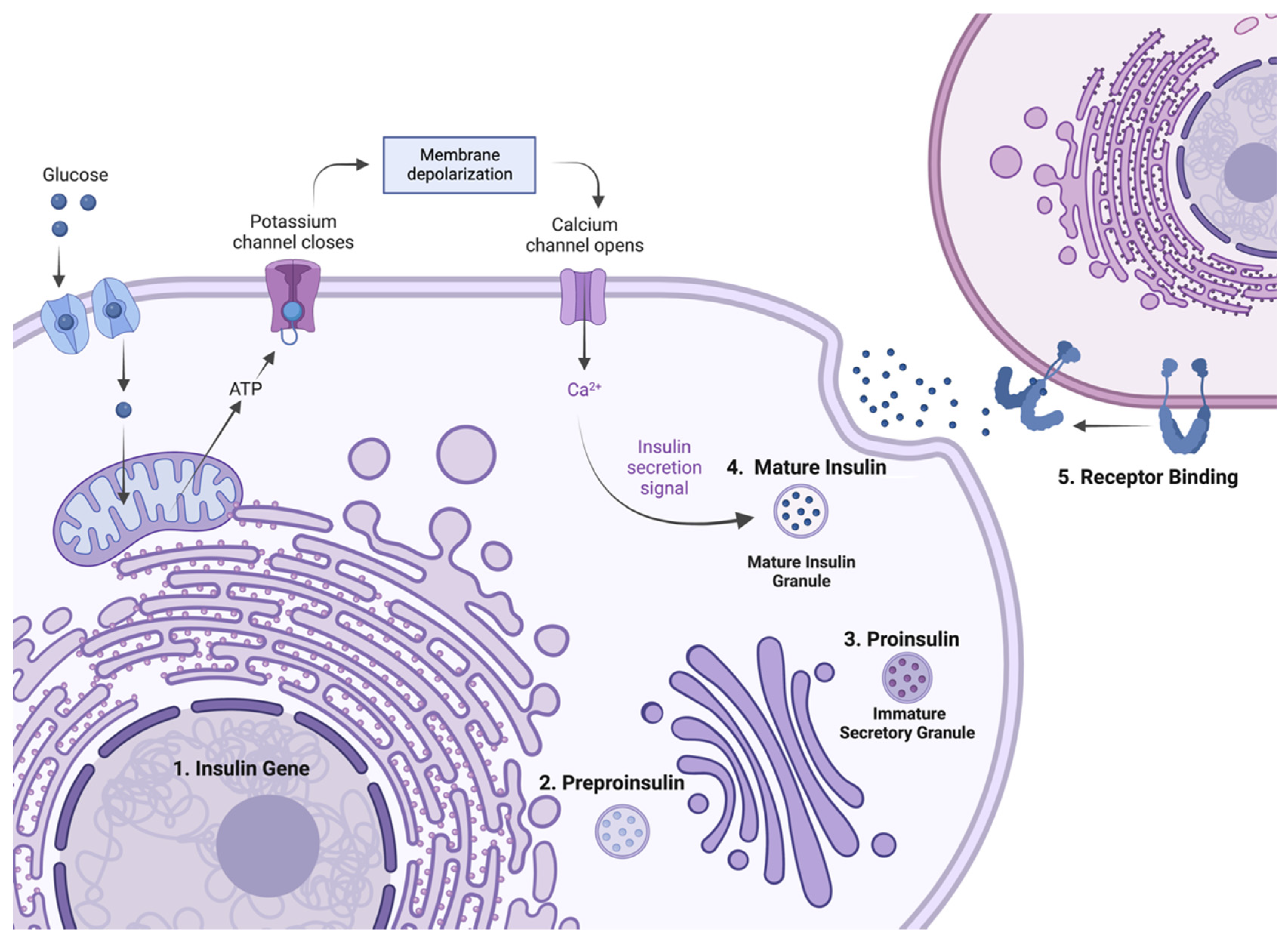
2. Insulin Expression—From Genome to Protein
3. Methods and Search Criteria
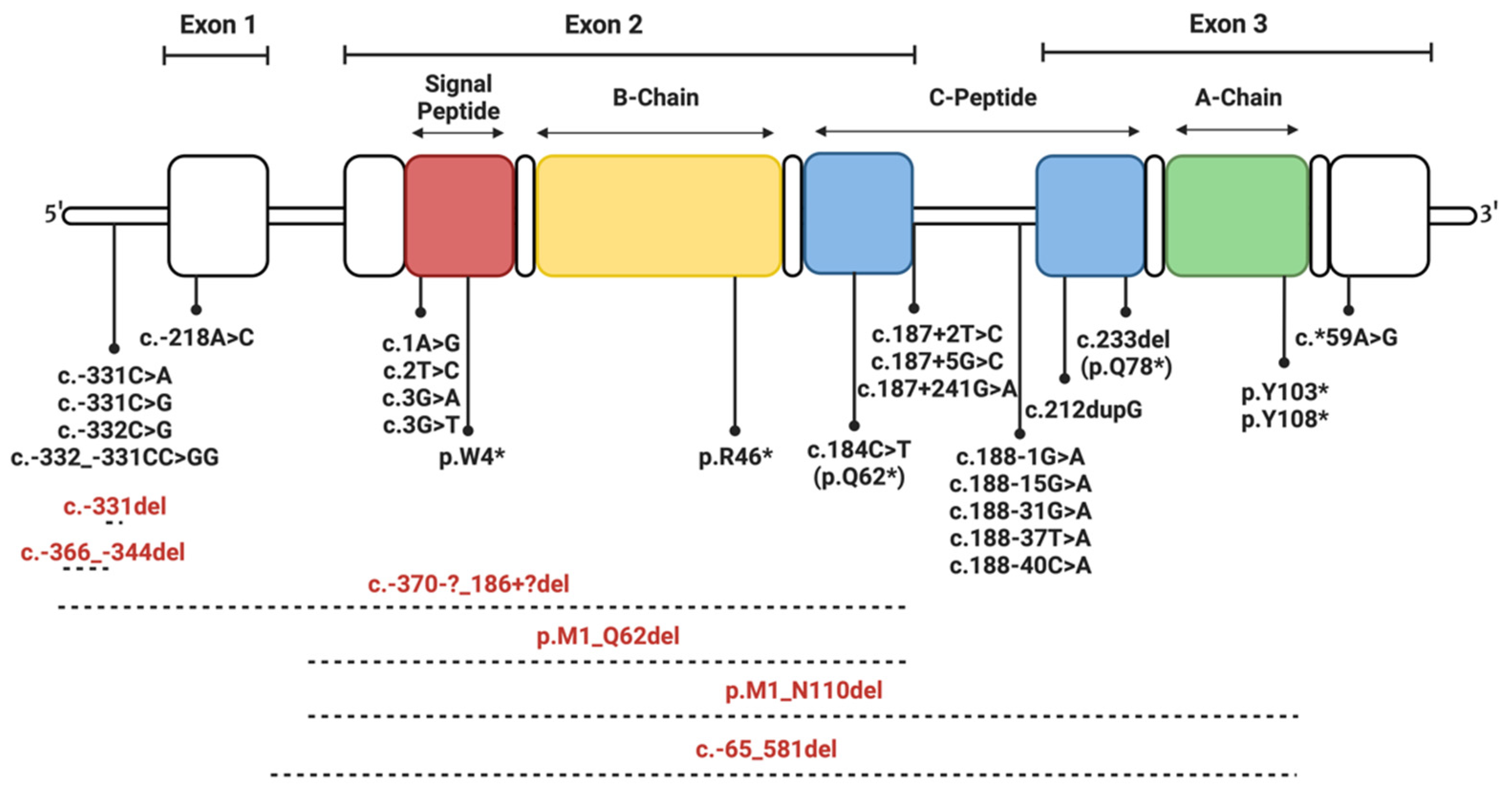
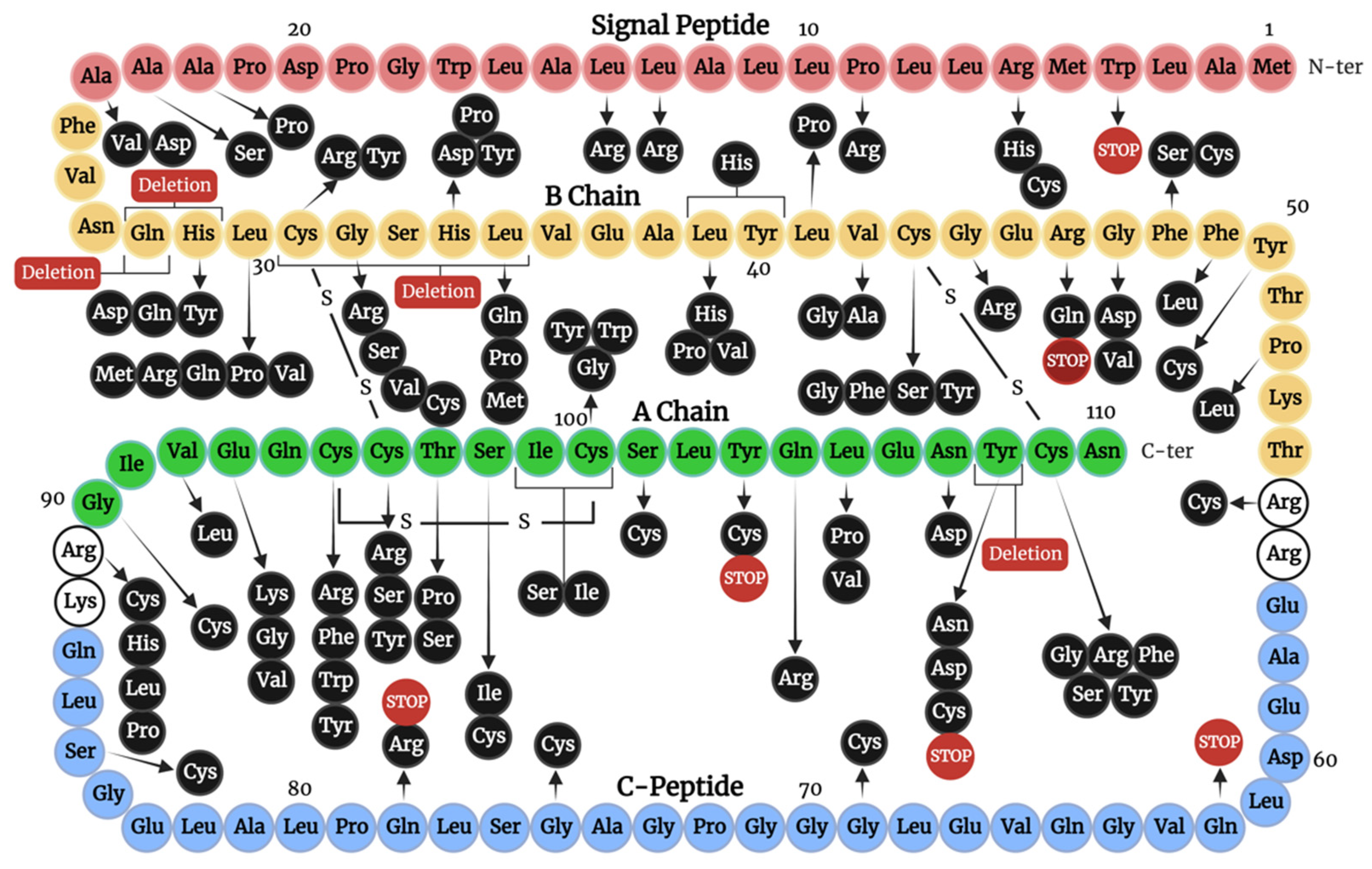
4. Insulin Gene Mutations
- Transcription and translation.
- Preproinsulin trafficking and proinsulin sorting.
- Insulin-IR interaction.
4.1. Mutations Affecting Transcription and Translation
4.2. Mutations Affecting Preproinsulin Trafficking and Proinsulin Processing
4.2.1. Signal Peptide Mutations
4.2.2. C-Peptide Mutations
4.2.3. Mutations of Cysteine Residues
4.2.4. Mutations Introducing Cysteine Residues
4.2.5. Non-Cysteine Mutations Impacting Disulfide Bonding and Structural Integrity
4.3. Mutations Affecting Insulin-IR Binding
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Støy, J.; De Franco, E.; Ye, H.; Park, S.-Y.; Bell, G.I.; Hattersley, A.T. In celebration of a century with insulin—Update of insulin gene mutations in diabetes. Mol. Metab. 2021, 52, 101280. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Owen, K.R. Genetics of Monogenic Diabetes: Present Clinical Challenges. Curr. Diabetes Rep. 2018, 18, 141. [Google Scholar] [CrossRef]
- Lawrence, M.C. Understanding insulin and its receptor from their three-dimensional structures. Mol. Metab. 2021, 52, 101255. [Google Scholar] [CrossRef]
- Trikkalinou, A.; Papazafiropoulou, A.K.; Melidonis, A. Type 2 diabetes and quality of life. World J. Diabetes 2017, 8, 120–129. [Google Scholar] [CrossRef]
- Nishi, M.; Nanjo, K. Insulin gene mutations and diabetes. J. Diabetes Investig. 2011, 2, 92–100. [Google Scholar] [CrossRef]
- Yang, Y.; Chan, L. Monogenic Diabetes: What It Teaches Us on the Common Forms of Type 1 and Type 2 Diabetes. Endocr. Rev. 2016, 37, 190–222. [Google Scholar] [CrossRef]
- Arneth, B. Insulin gene mutations and posttranslational and translocation defects: Associations with diabetes. Endocrine 2020, 70, 488–497. [Google Scholar] [CrossRef]
- Liu, M.; Sun, J.; Cui, J.; Chen, W.; Guo, H.; Barbetti, F.; Arvan, P. INS-gene mutations: From genetics and beta cell biology to clinical disease. Mol. Asp. Med. 2015, 42, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Barbetti, F.; D’Annunzio, G. Genetic causes and treatment of neonatal diabetes and early childhood diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kastner, K.; Abdul-Wahid, B.; Izaguirre, J.A. Evaluation of conformational changes in diabetes-associated mutation in insulin a chain: A molecular dynamics study. Proteins 2015, 83, 662–669. [Google Scholar] [CrossRef]
- Iafusco, D.; Zanfardino, A.; Bonfanti, R.; Rabbone, I.; Tinto, N.; Iafusco, F.; Meola, S.; Gicchino, M.F.; Ozen, G.; Casaburo, F.; et al. Congenital diabetes mellitus. Minerva Pediatr. 2020, 72, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hodish, I.; Haataja, L.; Lara-Lemus, R.; Rajpal, G.; Wright, J.; Arvan, P. Proinsulin misfolding and diabetes: Mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 2010, 21, 652–659. [Google Scholar] [CrossRef]
- Naylor, R. Economics of Genetic Testing for Diabetes. Curr. Diabetes Rep. 2019, 19, 23. [Google Scholar] [CrossRef]
- Weis, F.; Menting, J.G.; Margetts, M.B.; Chan, S.J.; Xu, Y.; Tennagels, N.; Wohlfart, P.; Langer, T.; Müller, C.W.; Dreyer, M.K.; et al. The signalling conformation of the insulin receptor ectodomain. Nat. Commun. 2018, 9, 4420. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef]
- Hua, Q.-x.; Chu, Y.-C.; Jia, W.; Phillips, N.F.B.; Wang, R.-y.; Katsoyannis, P.G.; Weiss, M.A. Mechanism of Insulin Chain Combination: ASYMMETRIC ROLES OF A-CHAIN α-HELICES IN DISULFIDE PAIRING*. J. Biol. Chem. 2002, 277, 43443–43453. [Google Scholar] [CrossRef]
- Chang, S.G.; Choi, K.D.; Jang, S.H.; Shin, H.C. Role of disulfide bonds in the structure and activity of human insulin. Mol. Cells 2003, 16, 323–330. [Google Scholar]
- Oslowski, C.M.; Urano, F. A switch from life to death in endoplasmic reticulum stressed β-cells. Diabetes Obes. Metab. 2010, 12 Suppl 2, 58–65. [Google Scholar] [CrossRef]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 28–50. [Google Scholar] [CrossRef] [PubMed]
- Lang, J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur. J. Biochem. 1999, 259, 3–17. [Google Scholar] [CrossRef]
- Hall, C.; Yu, H.; Choi, E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp. Mol. Med. 2020, 52, 911–920. [Google Scholar] [CrossRef]
- Di Camillo, B.; Carlon, A.; Eduati, F.; Toffolo, G.M. A rule-based model of insulin signalling pathway. BMC Syst. Biol. 2016, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Tager, H.; Given, B.; Baldwin, D.; Mako, M.; Markese, J.; Rubenstein, A.; Olefsky, J.; Kobayashi, M.; Kolterman, O.; Poucher, R. A structurally abnormal insulin causing human diabetes. Nature 1979, 281, 122–125. [Google Scholar] [CrossRef]
- Garin, I.; Edghill, E.L.; Akerman, I.; Rubio-Cabezas, O.; Rica, I.; Locke, J.M.; Maestro, M.A.; Alshaikh, A.; Bundak, R.; del Castillo, G.; et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3105–3110. [Google Scholar] [CrossRef]
- Demiral, M.; Demirbilek, H.; Çelik, K.; Okur, N.; Hussain, K.; Ozbek, M.N. Neonatal diabetes due to homozygous INS gene promoter mutations: Highly variable phenotype, remission and early relapse during the first 3 years of life. Pediatr. Diabetes 2020, 21, 1169–1175. [Google Scholar] [CrossRef]
- Yildiz, M.; Akcay, T.; Aydin, B.; Akgun, A.; Dogan, B.B.; De Franco, E.; Ellard, S.; Onal, H. Emergence of insulin resistance following empirical glibenclamide therapy: A case report of neonatal diabetes with a recessive INS gene mutation. J. Pediatr. Endocrinol. Metab. 2018, 31, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.W.; Docherty, K. Comparative analysis of insulin gene promoters: Implications for diabetes research. Diabetes 2006, 55, 3201–3213. [Google Scholar] [CrossRef]
- Noguchi, H.; Miyagi-Shiohira, C.; Nakashima, Y.; Kinjo, T.; Saitoh, I.; Watanabe, M. Mutations in the C1 element of the insulin promoter lead to diabetic phenotypes in homozygous mice. Commun. Biol. 2020, 3, 309. [Google Scholar] [CrossRef]
- De León, D.D.; Stanley, C.A. Permanent Neonatal Diabetes Mellitus. In GeneReviews(®); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Carmody, D.; Park, S.Y.; Ye, H.; Perrone, M.E.; Alkorta-Aranburu, G.; Highland, H.M.; Hanis, C.L.; Philipson, L.H.; Bell, G.I.; Greeley, S.A. Continued lessons from the INS gene: An intronic mutation causing diabetes through a novel mechanism. J. Med. Genet. 2015, 52, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Dusatkova, L.; Dusatkova, P.; Vosahlo, J.; Vesela, K.; Cinek, O.; Lebl, J.; Pruhova, S. Frameshift mutations in the insulin gene leading to prolonged molecule of insulin in two families with Maturity-Onset Diabetes of the Young. Eur. J. Med. Genet. 2015, 58, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, S.; Furuta, H.; Kosaka, K.; Doi, A.; Yorifuji, T.; Fukuda, T.; Senmaru, T.; Uraki, S.; Matsutani, N.; Furuta, M.; et al. Identification of a variant associated with early-onset diabetes in the intron of the insulin gene with exome sequencing. J. Diabetes Investig. 2019, 10, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Liu, L.; Xiao, Y.; Xie, Z.; Li, L.; Zhou, H.; Tang, W.; Liu, S.; Zhou, Z. Novel frameshift mutation in the insulin (INS) gene in a family with maturity onset diabetes of the young (MODY). J. Diabetes 2019, 11, 83–86. [Google Scholar] [CrossRef]
- Støy, J.; Steiner, D.F.; Park, S.-Y.; Ye, H.; Philipson, L.H.; Bell, G.I. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev. Endocr. Metab. Disord. 2010, 11, 205–215. [Google Scholar] [CrossRef]
- Raile, K.; O’Connell, M.; Galler, A.; Werther, G.; Kühnen, P.; Krude, H.; Blankenstein, O. Diabetes caused by insulin gene (INS) deletion: Clinical characteristics of homozygous and heterozygous individuals. Eur. J. Endocrinol. 2011, 165, 255–260. [Google Scholar] [CrossRef]
- Keeling, K.M.; Bedwell, D.M. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip. Rev. RNA 2011, 2, 837–852. [Google Scholar] [CrossRef]
- Yang, Y.; Shu, H.; Hu, J.; Li, L.; Wang, J.; Chen, T.; Zhen, J.; Sun, J.; Feng, W.; Xiong, Y.; et al. A Novel Nonsense INS Mutation Causes Inefficient Preproinsulin Translocation Into the Endoplasmic Reticulum. Front. Endocrinol. 2022, 12. [Google Scholar] [CrossRef]
- Rajan, S.; Eames, S.C.; Park, S.Y.; Labno, C.; Bell, G.I.; Prince, V.E.; Philipson, L.H. In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E403–E410. [Google Scholar] [CrossRef]
- Hussain, S.; Mohd Ali, J.; Jalaludin, M.Y.; Harun, F. Permanent neonatal diabetes due to a novel insulin signal peptide mutation. Pediatr. Diabetes 2013, 14, 299–303. [Google Scholar] [CrossRef]
- Boesgaard, T.W.; Pruhova, S.; Andersson, E.A.; Cinek, O.; Obermannova, B.; Lauenborg, J.; Damm, P.; Bergholdt, R.; Pociot, F.; Pisinger, C.; et al. Further evidence that mutations in INS can be a rare cause of Maturity-Onset Diabetes of the Young (MODY). BMC Med. Genet. 2010, 11, 42. [Google Scholar] [CrossRef]
- Edghill, E.L.; Flanagan, S.E.; Patch, A.M.; Boustred, C.; Parrish, A.; Shields, B.; Shepherd, M.H.; Hussain, K.; Kapoor, R.R.; Malecki, M.; et al. Insulin mutation screening in 1,044 patients with diabetes: Mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008, 57, 1034–1042. [Google Scholar] [CrossRef]
- Meur, G.; Simon, A.; Harun, N.; Virally, M.; Dechaume, A.; Bonnefond, A.; Fetita, S.; Tarasov, A.I.; Guillausseau, P.-J.; Boesgaard, T.W.; et al. Insulin gene mutations resulting in early-onset diabetes: Marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 2010, 59, 653–661. [Google Scholar] [CrossRef]
- Laurenzano, S.E.; McFall, C.; Nguyen, L.; Savla, D.; Coufal, N.G.; Wright, M.S.; Tokita, M.; Dimmock, D.; Kingsmore, S.F.; Newfield, R.S. Neonatal diabetes mellitus due to a novel variant in the INS gene. Cold Spring Harb. Mol. Case Stud. 2019, 5. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Li, M.; Ge, X.; Wang, Y.; Huang, X.; Yang, D.; Zhang, R.; Chen, Y.; Lu, M.; et al. Identification of Ala2Thr mutation in insulin gene from a Chinese MODY10 family. Mol. Cell Biochem. 2020, 470, 77–86. [Google Scholar] [CrossRef]
- Bonfanti, R.; Colombo, C.; Nocerino, V.; Massa, O.; Lampasona, V.; Iafusco, D.; Viscardi, M.; Chiumello, G.; Meschi, F.; Barbetti, F. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes autoantibodies. Diabetes Care 2009, 32, 123–125. [Google Scholar] [CrossRef]
- Dimova, R.; Tankova, T.; Gergelcheva, I.; Tournev, I.; Konstantinova, M. A family with permanent neonatal diabetes due to a novel mutation in INS gene. Diabetes Res. Clin. Pract. 2015, 108, e28–e30. [Google Scholar] [CrossRef]
- Liu, M.; Lara-Lemus, R.; Shan, S.-o.; Wright, J.; Haataja, L.; Barbetti, F.; Guo, H.; Larkin, D.; Arvan, P. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes 2012, 61, 828–837. [Google Scholar] [CrossRef]
- Wahren, J.; Ekberg, K.; Johansson, J.; Henriksson, M.; Pramanik, A.; Johansson, B.-L.; Rigler, R.; Jörnvall, H. Role of C-peptide in human physiology. Am. J. Physiol.-Endocrinol. Metab. 2000, 278, E759–E768. [Google Scholar] [CrossRef]
- Moritani, M.; Yokota, I.; Tsubouchi, K.; Takaya, R.; Takemoto, K.; Minamitani, K.; Urakami, T.; Kawamura, T.; Kikuchi, N.; Itakura, M.; et al. Identification of INS and KCNJ11 gene mutations in type 1B diabetes in Japanese children with onset of diabetes before 5 years of age. Pediatr. Diabetes 2013, 14, 112–120. [Google Scholar] [CrossRef]
- Polak, M.; Dechaume, A.; Cavé, H.; Nimri, R.; Crosnier, H.; Sulmont, V.; de Kerdanet, M.; Scharfmann, R.; Lebenthal, Y.; Froguel, P.; et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: A report from the French ND (Neonatal Diabetes) Study Group. Diabetes 2008, 57, 1115–1119. [Google Scholar] [CrossRef]
- Fu, J.; Wang, T.; Li, M.; Xiao, X. Identification of insulin gene variants in patients with neonatal diabetes in the Chinese population. J. Diabetes Investig. 2020, 11, 578–584. [Google Scholar] [CrossRef]
- Dhayalan, B.; Chatterjee, D.; Chen, Y.-S.; Weiss, M.A. Structural Lessons From the Mutant Proinsulin Syndrome. Front. Endocrinol. 2021, 12, 754693. [Google Scholar] [CrossRef]
- Sieber, P.; Eisler, K.; Kamber, B.; Riniker, B.; Rittel, W.; Märki, F.; de Gasparo, M. Synthesis and biological activity of two disulphide bond isomers of human insulin: [A7-A11,A6-B7-cystine]- and [A6-A7,A11-B7-cystine]insulin (human). Biol. Chem. 1978, 359, 113–123. [Google Scholar] [CrossRef]
- Hua, Q.X.; Jia, W.; Frank, B.H.; Phillips, N.F.; Weiss, M.A. A protein caught in a kinetic trap: Structures and stabilities of insulin disulfide isomers. Biochemistry 2002, 41, 14700–14715. [Google Scholar] [CrossRef]
- Hua, Q.X.; Mayer, J.P.; Jia, W.; Zhang, J.; Weiss, M.A. The folding nucleus of the insulin superfamily: A flexible peptide model foreshadows the native state. J. Biol. Chem. 2006, 281, 28131–28142. [Google Scholar] [CrossRef]
- Dai, Y.; Tang, J.G. Characteristic, activity and conformational studies of [A6-Ser, A11-Ser]-insulin. Biochim. Biophys. Acta 1996, 1296, 63–68. [Google Scholar] [CrossRef]
- Weiss, M.A.; Hua, Q.X.; Jia, W.; Chu, Y.C.; Wang, R.Y.; Katsoyannis, P.G. Hierarchical protein “un-design”: Insulin’s intrachain disulfide bridge tethers a recognition alpha-helix. Biochemistry 2000, 39, 15429–15440. [Google Scholar] [CrossRef]
- Guo, Z.Y.; Feng, Y.M. Effects of cysteine to serine substitutions in the two inter-chain disulfide bonds of insulin. Biol. Chem. 2001, 382, 443–448. [Google Scholar] [CrossRef]
- Liu, M.; Li, Y.; Cavener, D.; Arvan, P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 13209–13212. [Google Scholar] [CrossRef]
- Can, N.T.B.; Vu, D.C.; Bui, T.P.; Nguyen, K.N.; Nguyen, D.P.; Craig, M.; Ellard, S.; Nguyen, H.T. AB125. Neonatal diabetes mellitus due to insulin gene mutation. Ann. Transl. Med. 2015, 3 (Suppl. 2), AB125. [Google Scholar] [CrossRef]
- Bowman, P.; Day, J.; Torrens, L.; Shepherd, M.H.; Knight, B.A.; Ford, T.J.; Flanagan, S.E.; Chakera, A.; Hattersley, A.T.; Zeman, A. Cognitive, Neurological, and Behavioral Features in Adults With KCNJ11 Neonatal Diabetes. Diabetes Care 2019, 42, 215–224. [Google Scholar] [CrossRef]
- Ozturk, M.A.; Kurtoglu, S.; Bastug, O.; Korkmaz, L.; Daar, G.; Memur, S.; Halis, H.; Günes, T.; Hussain, K.; Ellard, S. Neonatal diabetes in an infant of diabetic mother: Same novel INS missense mutation in the mother and her offspring. J. Pediatr. Endocrinol. Metab. 2014, 27, 745–748. [Google Scholar] [CrossRef]
- Amirruddin, N.S.; Tan, Y.S.; Verma, C.S.; Gardner, D.; Bee, Y.M.; Hoon, S.; Teo, A. 1784-P: Studying the Impact of Heterozygous Human INS Gene Mutation on Pancreatic ß Cell. Diabetes 2019, 68 (Suppl. 1), 1784-P. [Google Scholar] [CrossRef]
- Gopi, S.; Gowri, P.; Panda, J.K.; Sathyanarayana, S.O.; Gupta, S.; Chandru, S.; Chandni, R.; Raghupathy, P.; Dayal, D.; Mohan, V.; et al. Insulin gene mutations linked to permanent neonatal diabetes mellitus in Indian population. J. Diabetes Its Complicat. 2021, 35, 108022. [Google Scholar] [CrossRef] [PubMed]
- Fredheim, S.; Svensson, J.; Pørksen, S.; Hansen, L.; Hansen, T.; Pedersen, O.B.; Mortensen, H.B.; Barbetti, F.; Nielsen, L.B. Intrafamilial Variability of Early-Onset Diabetes due to an INS Mutation. Case Rep. Genet. 2011, 2011, 258978. [Google Scholar] [CrossRef][Green Version]
- Fendler, W.; Borowiec, M.; Antosik, K.; Jaroszewska-Swiatek, B.; Szwalkiewicz-Warowicka, E.; Malecki, M.; Mysliwiec, M.; Mlynarski, W. Paternally Inherited Proinsulin Mutations May Result in Earlier Onset of Monogenic Diabetes Mutation Identity Effect in Monogenic Diabetes. Diabetes Care 2011, 34, e9. [Google Scholar] [CrossRef][Green Version]
- Park, S.-Y.; Ye, H.; Steiner, D.F.; Bell, G.I. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem. Biophys. Res. Commun. 2010, 391, 1449–1454. [Google Scholar] [CrossRef]
- Molven, A.; Ringdal, M.; Nordbø, A.M.; Raeder, H.; Støy, J.; Lipkind, G.M.; Steiner, D.F.; Philipson, L.H.; Bergmann, I.; Aarskog, D.; et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes 2008, 57, 1131–1135. [Google Scholar] [CrossRef]
- Rege, N.K.; Liu, M.; Yang, Y.; Dhayalan, B.; Wickramasinghe, N.P.; Chen, Y.-S.; Rahimi, L.; Guo, H.; Haataja, L.; Sun, J.; et al. Evolution of insulin at the edge of foldability and its medical implications. Proc. Natl. Acad. Sci. USA 2020, 117, 29618–29628. [Google Scholar] [CrossRef] [PubMed]
- Sohma, Y.; Hua, Q.X.; Liu, M.; Phillips, N.B.; Hu, S.Q.; Whittaker, J.; Whittaker, L.J.; Ng, A.; Roberts, C.T., Jr.; Arvan, P.; et al. Contribution of residue B5 to the folding and function of insulin and IGF-I: Constraints and fine-tuning in the evolution of a protein family. J. Biol. Chem. 2010, 285, 5040–5055. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Porzio, O.; Liu, M.; Massa, O.; Vasta, M.; Salardi, S.; Beccaria, L.; Monciotti, C.; Toni, S.; Pedersen, O.; et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Investig. 2008, 118, 2148–2156. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Edghill, E.L.; Argente, J.; Hattersley, A.T. Testing for monogenic diabetes among children and adolescents with antibody-negative clinically defined Type 1 diabetes. Diabet Med. 2009, 26, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Moritani, M.; Yokota, I.; Horikawa, R.; Urakami, T.; Nishii, A.; Kawamura, T.; Kikuchi, N.; Kikuchi, T.; Ogata, T.; Sugihara, S.; et al. Identification of monogenic gene mutations in Japanese subjects diagnosed with type 1B diabetes between >5 and 15.1 years of age. J. Pediatr. Endocrinol. Metab. JPEM 2016, 29, 1047–1054. [Google Scholar] [CrossRef]
- Wasserman, H.; Hufnagel, R.B.; Miraldi Utz, V.; Zhang, K.; Valencia, C.A.; Leslie, N.D.; Crimmins, N.A. Bilateral cataracts in a 6-yr-old with new onset diabetes: A novel presentation of a known INS gene mutation. Pediatr. Diabetes 2016, 17, 535–539. [Google Scholar] [CrossRef]
- Nakagawa, S.H.; Zhao, M.; Hua, Q.X.; Hu, S.Q.; Wan, Z.L.; Jia, W.; Weiss, M.A. Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry 2005, 44, 4984–4999. [Google Scholar] [CrossRef]
- Gross, D.J.; Halban, P.A.; Kahn, C.R.; Weir, G.C.; Villa-Komaroff, L. Partial diversion of a mutant proinsulin (B10 aspartic acid) from the regulated to the constitutive secretory pathway in transfected AtT-20 cells. Proc. Natl. Acad. Sci. USA 1989, 86, 4107–4111. [Google Scholar] [CrossRef]
- Schwartz, G.P.; Burke, G.T.; Katsoyannis, P.G. A superactive insulin: [B10-aspartic acid]insulin(human). Proc. Natl. Acad. Sci. USA 1987, 84, 6408–6411. [Google Scholar] [CrossRef]
- Wang, T.; Ding, S.; Li, S.; Guo, H.; Chen, X.; Huang, Y.; Huang, J.; Wu, J.; Hu, C.; Fang, C.; et al. A novel mutation in INS gene linked to permanent neonatal diabetes mellitus. Endocrine 2019, 64, 719–723. [Google Scholar] [CrossRef]
- Globa, E.; Zelinska, N.; Mackay, D.J.G.; Temple, K.I.; Houghton, J.A.L.; Hattersley, A.T.; Flanagan, S.E.; Ellard, S. Neonatal diabetes in Ukraine: Incidence, genetics, clinical phenotype and treatment. J. Pediatr. Endocrinol. Metab. JPEM 2015, 28, 1279–1286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Piccini, B.; Artuso, R.; Lenzi, L.; Guasti, M.; Braccesi, G.; Barni, F.; Casalini, E.; Giglio, S.; Toni, S. Clinical and molecular characterization of a novel INS mutation identified in patients with MODY phenotype. Eur. J. Med. Genet. 2016, 59, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Du, W.; Ma, J.; Gu, M.; Wang, J.; Zhu, H.; Song, H.; Gao, G. A Novel c.125 T>G (p.Val42Gly) Mutation in The Human INS Gene Leads to Neonatal Diabetes Mellitus via a Decrease in Insulin Synthesis. Exp. Clin. Endocrinol. Diabetes 2020, 128, 182–189. [Google Scholar] [CrossRef]
- Nakagawa, S.H.; Hua, Q.X.; Hu, S.Q.; Jia, W.; Wang, S.; Katsoyannis, P.G.; Weiss, M.A. Chiral mutagenesis of insulin. Contribution of the B20-B23 beta-turn to activity and stability. J. Biol. Chem. 2006, 281, 22386–22396. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Saint-Martin, C.; Xu, J.; Ding, L.; Wang, R.; Feng, W.; Liu, M.; Shu, H.; Fan, Z.; Haataja, L.; et al. Biological behaviors of mutant proinsulin contribute to the phenotypic spectrum of diabetes associated with insulin gene mutations. Mol. Cell Endocrinol. 2020, 518, 111025. [Google Scholar] [CrossRef]
- Støy, J.; Olsen, J.; Park, S.Y.; Gregersen, S.; Hjørringgaard, C.U.; Bell, G.I. In vivo measurement and biological characterisation of the diabetes-associated mutant insulin p.R46Q (GlnB22-insulin). Diabetologia 2017, 60, 1423–1431. [Google Scholar] [CrossRef]
- Wan, Z.; Xu, B.; Huang, K.; Chu, Y.-C.; Li, B.; Nakagawa, S.H.; Qu, Y.; Hu, S.-Q.; Katsoyannis, P.G.; Weiss, M.A. Enhancing the Activity of Insulin at the Receptor Interface: Crystal Structure and Photo-Cross-Linking of A8 Analogues. Biochemistry 2004, 43, 16119–16133. [Google Scholar] [CrossRef]
- Lal, R.A.; Moeller, H.P.; Thomson, E.A.; Horton, T.M.; Lee, S.; Freeman, R.; Prahalad, P.; Poon, A.S.Y.; Annes, J.P. Novel Pathogenic De Novo INS p.T97P Variant Presenting With Severe Neonatal DKA. Endocrinology 2022, 163. [Google Scholar] [CrossRef]
- Cao, B.; Gong, C.; Wu, D.; Lu, C.; Liu, F.; Liu, X.; Zhang, Y.; Gu, Y.; Qi, Z.; Li, X.; et al. Genetic Analysis and Follow-Up of 25 Neonatal Diabetes Mellitus Patients in China. J. Diabetes Res. 2016, 2016, 6314368. [Google Scholar] [CrossRef]
- Kshirsagar, V.Y.; Ahmed, M.; Colaco, S.M.; Houghton, J.A.; Ellard, S. Permanent neonatal diabetes due to a novel L105P (c.314T>C; p.Leu105Pro) heterozygous mutation in insulin gene. Int. J. Diabetes Dev. Ctries. 2013, 33, 226–228. [Google Scholar] [CrossRef]
- Liu, M.; Wan, Z.L.; Chu, Y.C.; Aladdin, H.; Klaproth, B.; Choquette, M.; Hua, Q.X.; Mackin, R.B.; Rao, J.S.; De Meyts, P.; et al. Crystal structure of a “nonfoldable” insulin: Impaired folding efficiency despite native activity. J. Biol. Chem. 2009, 284, 35259–35272. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P. Insulin/receptor binding: The last piece of the puzzle? What recent progress on the structure of the insulin/receptor complex tells us (or not) about negative cooperativity and activation. Bioessays 2015, 37, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Menting, J.G.; Whittaker, J.; Margetts, M.B.; Whittaker, L.J.; Kong, G.K.W.; Smith, B.J.; Watson, C.J.; Záková, L.; Kletvíková, E.; Jiráček, J.; et al. How insulin engages its primary binding site on the insulin receptor. Nature 2013, 493, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Glendorf, T.; Sørensen, A.R.; Nishimura, E.; Pettersson, I.; Kjeldsen, T. Importance of the solvent-exposed residues of the insulin B chain alpha-helix for receptor binding. Biochemistry 2008, 47, 4743–4751. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Polonsky, K.S.; Zeidler, A.; Rubenstein, A.H.; Tager, H.S. Human insulin B24 (Phe----Ser). Secretion and metabolic clearance of the abnormal insulin in man and in a dog model. J. Clin. Investig. 1984, 73, 1351–1358. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin Degradation: Progress and Potential*. Endocr. Rev. 1998, 19, 608–624. [Google Scholar] [CrossRef]
- Wan, Z.L.; Huang, K.; Xu, B.; Hu, S.Q.; Wang, S.; Chu, Y.C.; Katsoyannis, P.G.; Weiss, M.A. Diabetes-associated mutations in human insulin: Crystal structure and photo-cross-linking studies of a-chain variant insulin Wakayama. Biochemistry 2005, 44, 5000–5016. [Google Scholar] [CrossRef]
- Liu, M.; Haataja, L.; Wright, J.; Wickramasinghe, N.P.; Hua, Q.X.; Phillips, N.F.; Barbetti, F.; Weiss, M.A.; Arvan, P. Mutant INS-gene induced diabetes of youth: Proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS ONE 2010, 5, e13333. [Google Scholar] [CrossRef]
- Johnson, S.R.; McGown, I.; Oppermann, U.; Conwell, L.S.; Harris, M.; Duncan, E.L. A novel INS mutation in a family with maturity-onset diabetes of the young: Variable insulin secretion and putative mechanisms. Pediatr. Diabetes 2018, 19, 905–909. [Google Scholar] [CrossRef]
- Shoelson, S.; Fickova, M.; Haneda, M.; Nahum, A.; Musso, G.; Kaiser, E.T.; Rubenstein, A.H.; Tager, H. Identification of a mutant human insulin predicted to contain a serine-for-phenylalanine substitution. Proc. Natl. Acad. Sci. USA 1983, 80, 7390–7394. [Google Scholar] [CrossRef]
- Kwok, S.C.; Steiner, D.F.; Rubenstein, A.H.; Tager, H.S. Identification of a point mutation in the human insulin gene giving rise to a structurally abnormal insulin (insulin Chicago). Diabetes 1983, 32, 872–875. [Google Scholar] [CrossRef]
- Islam, M.A.; Bhayye, S.; Adeniyi, A.A.; Soliman, M.E.; Pillay, T.S. Diabetes mellitus caused by mutations in human insulin: Analysis of impaired receptor binding of insulins Wakayama, Los Angeles and Chicago using pharmacoinformatics. J. Biomol. Struct Dyn 2017, 35, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Haneda, M.; Chan, S.J.; Kwok, S.C.; Rubenstein, A.H.; Steiner, D.F. Studies on mutant human insulin genes: Identification and sequence analysis of a gene encoding [SerB24]insulin. Proc. Natl. Acad. Sci. USA 1983, 80, 6366–6370. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.; Haneda, M.; Blix, P.; Nanjo, A.; Sanke, T.; Inouye, K.; Steiner, D.; Rubenstein, A.; Tager, H. Three mutant insulins in man. Nature 1983, 302, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Avital-Shmilovici, M.; Whittaker, J.; Weiss, M.A.; Kent, S.B. Deciphering a molecular mechanism of neonatal diabetes mellitus by the chemical synthesis of a protein diastereomer, [D-AlaB8]human proinsulin. J. Biol. Chem. 2014, 289, 23683–23692. [Google Scholar] [CrossRef]
- Gutmann, T.; Schäfer, I.B.; Poojari, C.; Brankatschk, B.; Vattulainen, I.; Strauss, M.; Coskun, Ü. Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Weiss, M.A.; Wan, Z.; Zhao, M.; Chu, Y.C.; Nakagawa, S.H.; Burke, G.T.; Jia, W.; Hellmich, R.; Katsoyannis, P.G. Non-standard insulin design: Structure-activity relationships at the periphery of the insulin receptor. J. Mol. Biol. 2002, 315, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Gauguin, L.; Klaproth, B.; Sajid, W.; Andersen, A.S.; McNeil, K.A.; Forbes, B.E.; De Meyts, P. Structural basis for the lower affinity of the insulin-like growth factors for the insulin receptor. J. Biol. Chem. 2008, 283, 2604–2613. [Google Scholar] [CrossRef]
- Kaarsholm, N.C.; Norris, K.; Jørgensen, R.J.; Mikkelsen, J.; Ludvigsen, S.; Olsen, O.H.; Sørensen, A.R.; Havelund, S. Engineering stability of the insulin monomer fold with application to structure-activity relationships. Biochemistry 1993, 32, 10773–10778. [Google Scholar] [CrossRef]
- Simon, J.; Freychet, P.; Rosselin, G.; DeMeyts, P. Enhanced binding affinity of chicken insulin in rat liver membranes and human lymphocytes: Relationship tothe kinetic properties of the hormone- receptor interaction. Endocrinology 1977, 100, 115–121. [Google Scholar] [CrossRef]
- Glendorf, T.; Knudsen, L.; Stidsen, C.E.; Hansen, B.F.; Hegelund, A.C.; Sørensen, A.R.; Nishimura, E.; Kjeldsen, T. Systematic evaluation of the metabolic to mitogenic potency ratio for B10-substituted insulin analogues. PLoS ONE 2012, 7, e29198. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ataie-Ashtiani, S.; Forbes, B. A Review of the Biosynthesis and Structural Implications of Insulin Gene Mutations Linked to Human Disease. Cells 2023, 12, 1008. https://doi.org/10.3390/cells12071008
Ataie-Ashtiani S, Forbes B. A Review of the Biosynthesis and Structural Implications of Insulin Gene Mutations Linked to Human Disease. Cells. 2023; 12(7):1008. https://doi.org/10.3390/cells12071008
Chicago/Turabian StyleAtaie-Ashtiani, Sara, and Briony Forbes. 2023. "A Review of the Biosynthesis and Structural Implications of Insulin Gene Mutations Linked to Human Disease" Cells 12, no. 7: 1008. https://doi.org/10.3390/cells12071008
APA StyleAtaie-Ashtiani, S., & Forbes, B. (2023). A Review of the Biosynthesis and Structural Implications of Insulin Gene Mutations Linked to Human Disease. Cells, 12(7), 1008. https://doi.org/10.3390/cells12071008