
In Vivo Efficacy and Safety Evaluations of Therapeutic Splicing Correction Using U1 snRNA in the Mouse Retina

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation and Cloning of Mutation-Adapted U1/U6 Cassettes
2.2. Generation and Purification of AAV2/8
2.3. Mice and Subretinal Injection
2.4. Analysis of Transcript Levels with Semi-Quantitative RT-PCR
2.5. Sanger Sequencing of Vectors and Opa1 Transcripts
2.6. Western-Blot Analysis
2.7. Histological Analyses of Retinal Slices
2.8. Electroretinogram (ERG) Measurements
2.9. Off-Target Gene Analyses
2.10. Statistical Analysis
3. Results
3.1. Engineered U1 Treatment Rescues Opa1 Splice Defect
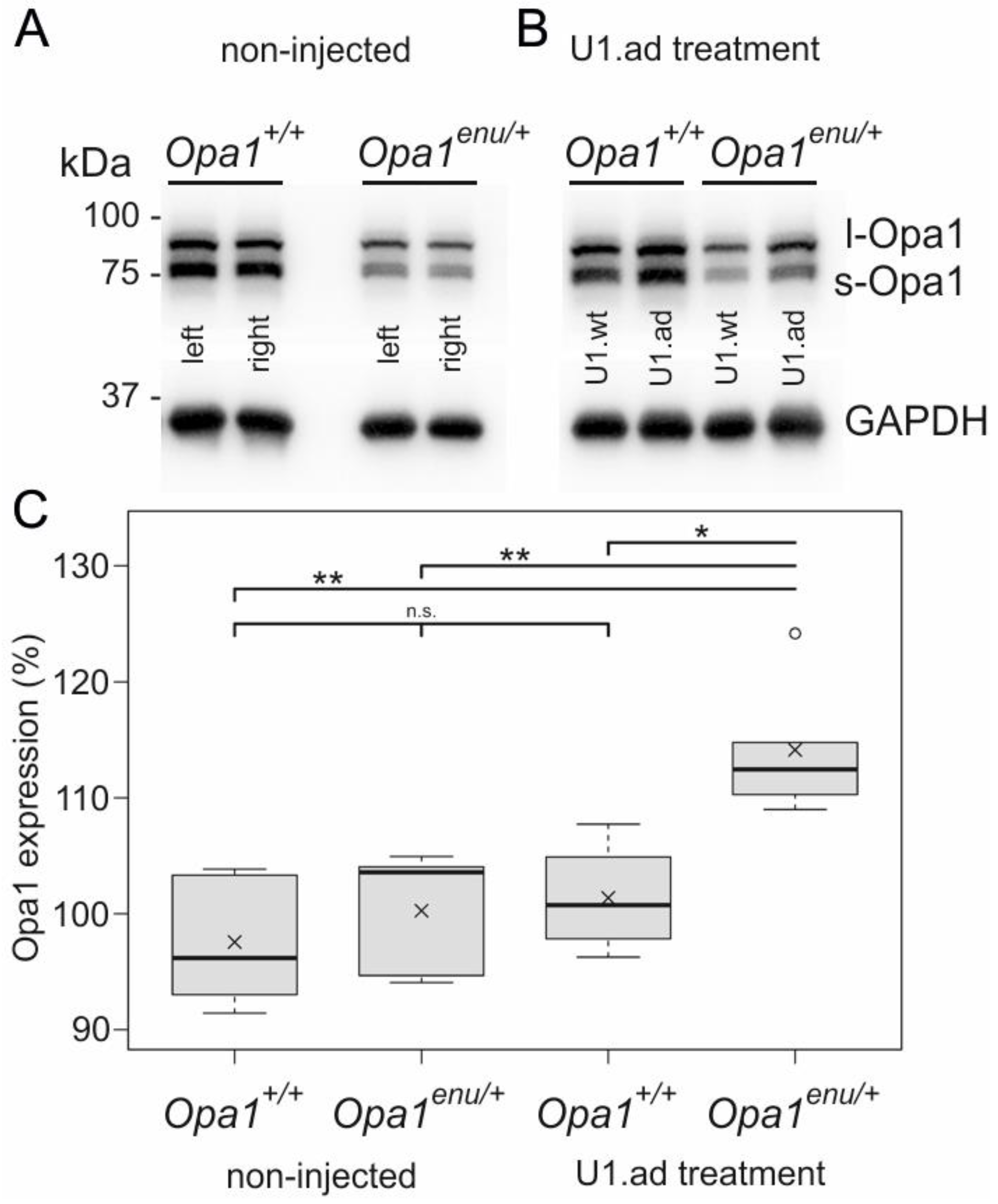
3.2. Opa1 Protein Levels Are Increased upon U1 Therapy
3.3. Off-Target Splicing Is Not Affected by the U1 Treatment
3.4. Retinal Morphology Remains Inconspicuous after Subretinal AAV2/8 Injection
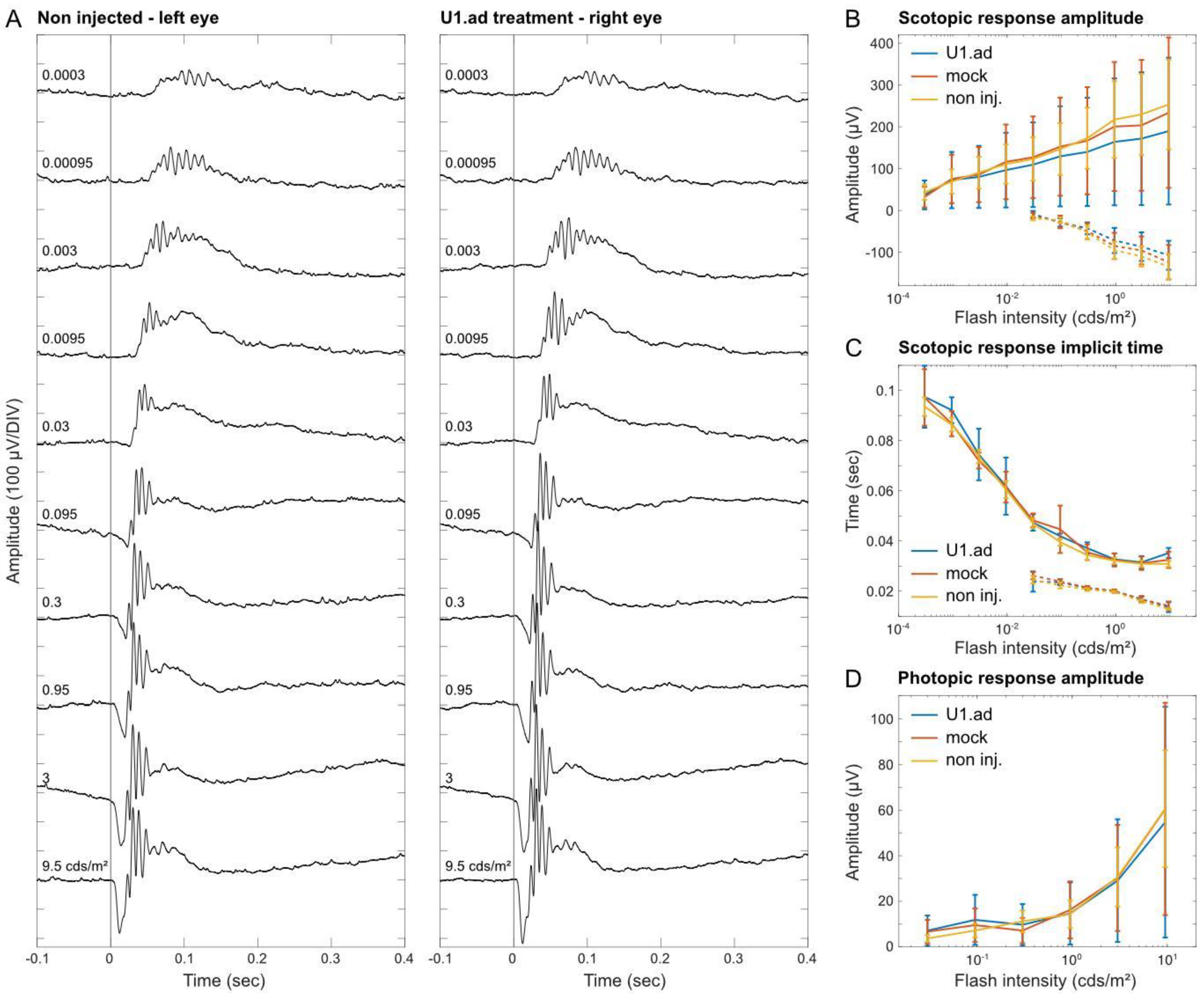
3.5. Electroretinogram Responses Are Unaltered after U1 Treatments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.; Kjems, J. Defining a 5′ splice site by functional selection in the presence and absence of U1 snRNA 5′ end. RNA 2002, 8, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Kandels-Lewis, S.; Seraphin, B. Involvement of U6 snRNA in 5′ splice site selection. Science 1993, 262, 2035–2039. [Google Scholar] [CrossRef]
- Lesser, C.F.; Guthrie, C. Mutations in U6 snRNA that alter splice site specificity: Implications for the active site. Science 1993, 262, 1982–1988. [Google Scholar] [CrossRef]
- Krawczak, M.; Reiss, J.; Cooper, D.N. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: Causes and consequences. Hum. Genet. 1992, 90, 41–54. [Google Scholar] [CrossRef]
- Zhuang, Y.; Weiner, A.M. A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell 1986, 46, 827–835. [Google Scholar] [CrossRef]
- Breuel, S.; Vorm, M.; Brauer, A.U.; Owczarek-Lipska, M.; Neidhardt, J. Combining Engineered U1 snRNA and Antisense Oligonucleotides to Improve the Treatment of a BBS1 Splice Site Mutation. Mol. Ther. Nucleic Acids 2019, 18, 123–130. [Google Scholar] [CrossRef]
- Balestra, D.; Giorgio, D.; Bizzotto, M.; Fazzari, M.; Ben Zeev, B.; Pinotti, M.; Landsberger, N.; Frasca, A. Splicing Mutations Impairing CDKL5 Expression and Activity Can be Efficiently Rescued by U1snRNA-Based Therapy. Int. J. Mol. Sci. 2019, 20, 4130. [Google Scholar] [CrossRef]
- Matos, L.; Canals, I.; Dridi, L.; Choi, Y.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Perez, B.; Pshezhetsky, A.V.; Grinberg, D.; et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014, 9, 180. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Hiller, T.; Korner, G.; Glaus, E.; Berger, W.; Neidhardt, J. A gene therapeutic approach to correct splice defects with modified U1 and U6 snRNPs. Hum. Gene Ther. 2013, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- van der Woerd, W.L.; Mulder, J.; Pagani, F.; Beuers, U.; Houwen, R.H.; van de Graaf, S.F. Analysis of aberrant pre-messenger RNA splicing resulting from mutations in ATP8B1 and efficient in vitro rescue by adapted U1 small nuclear RNA. Hepatology 2015, 61, 1382–1391. [Google Scholar] [CrossRef]
- Pinotti, M.; Rizzotto, L.; Balestra, D.; Lewandowska, M.A.; Cavallari, N.; Marchetti, G.; Bernardi, F.; Pagani, F. U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency. Blood 2008, 111, 2681–2684. [Google Scholar] [CrossRef]
- Tanner, G.; Glaus, E.; Barthelmes, D.; Ader, M.; Fleischhauer, J.; Pagani, F.; Berger, W.; Neidhardt, J. Therapeutic strategy to rescue mutation-induced exon skipping in rhodopsin by adaptation of U1 snRNA. Hum. Mutat. 2009, 30, 255–263. [Google Scholar] [CrossRef]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef]
- Schmid, F.; Glaus, E.; Barthelmes, D.; Fliegauf, M.; Gaspar, H.; Nurnberg, G.; Nurnberg, P.; Omran, H.; Berger, W.; Neidhardt, J. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum. Mutat. 2011, 32, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Boussaad, I.; Obermaier, C.D.; Hanss, Z.; Bobbili, D.R.; Bolognin, S.; Glaab, E.; Wolynska, K.; Weisschuh, N.; De Conti, L.; May, C.; et al. A patient-based model of RNA mis-splicing uncovers treatment targets in Parkinson′s disease. Sci. Transl. Med. 2020, 12, eaau3960. [Google Scholar] [CrossRef]
- Balestra, D.; Faella, A.; Margaritis, P.; Cavallari, N.; Pagani, F.; Bernardi, F.; Arruda, V.R.; Pinotti, M. An engineered U1 small nuclear RNA rescues splicing defective coagulation F7 gene expression in mice. J. Thromb. Haemost. 2014, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Balestra, D.; Scalet, D.; Ferrarese, M.; Lombardi, S.; Ziliotto, N.; C, C.C.; Petersen, N.; Bosma, P.; Riccardi, F.; Pagani, F.; et al. A Compensatory U1snRNA Partially Rescues FAH Splicing and Protein Expression in a Splicing-Defective Mouse Model of Tyrosinemia Type I. Int. J. Mol. Sci. 2020, 21, 2136. [Google Scholar] [CrossRef]
- Lee, N.C.; Lee, Y.M.; Chen, P.W.; Byrne, B.J.; Hwu, W.L. Mutation-adapted U1 snRNA corrects a splicing error of the dopa decarboxylase gene. Hum. Mol. Genet. 2016, 25, 5142–5147. [Google Scholar] [CrossRef] [PubMed]
- Kjer, P.; Jensen, O.A.; Klinken, L. Histopathology of eye, optic nerve and brain in a case of dominant optic atrophy. Acta Ophthalmol. 1983, 61, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef]
- Kjer, B.; Eiberg, H.; Kjer, P.; Rosenberg, T. Dominant optic atrophy mapped to chromosome 3q region. II. Clinical and epidemiological aspects. Acta Ophthalmol. Scand. 1996, 74, 3–7. [Google Scholar] [CrossRef]
- Votruba, M.; Moore, A.T.; Bhattacharya, S.S. Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. J. Med. Genet. 1998, 35, 793–800. [Google Scholar] [CrossRef]
- Votruba, M.; Fitzke, F.W.; Holder, G.E.; Carter, A.; Bhattacharya, S.S.; Moore, A.T. Clinical features in affected individuals from 21 pedigrees with dominant optic atrophy. Arch. Ophthalmol. 1998, 116, 351–358. [Google Scholar] [CrossRef]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Juschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye Res. 2021, 83, 100935. [Google Scholar] [CrossRef]
- Alavi, M.V.; Bette, S.; Schimpf, S.; Schuettauf, F.; Schraermeyer, U.; Wehrl, H.F.; Ruttiger, L.; Beck, S.C.; Tonagel, F.; Pichler, B.J.; et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007, 130, 1029–1042. [Google Scholar] [CrossRef]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Sarzi, E.; Seveno, M.; Piro-Megy, C.; Elziere, L.; Quiles, M.; Pequignot, M.; Muller, A.; Hamel, C.P.; Lenaers, G.; Delettre, C. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci. Rep. 2018, 8, 2468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wakabayashi, N.; Wakabayashi, J.; Tamura, Y.; Song, W.J.; Sereda, S.; Clerc, P.; Polster, B.M.; Aja, S.M.; Pletnikov, M.V.; et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol. Biol. Cell 2011, 22, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V.; Fuhrmann, N.; Nguyen, H.P.; Yu-Wai-Man, P.; Heiduschka, P.; Chinnery, P.F.; Wissinger, B. Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp. Neurol. 2009, 220, 404–409. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, I.; Reinhard, K.; Tolivia, J.; Wissinger, B.; Munch, T.A. Influence of Opa1 Mutation on Survival and Function of Retinal Ganglion Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4835–4845. [Google Scholar] [CrossRef]
- Heiduschka, P.; Schnichels, S.; Fuhrmann, N.; Hofmeister, S.; Schraermeyer, U.; Wissinger, B.; Alavi, M.V. Electrophysiological and histologic assessment of retinal ganglion cell fate in a mouse model for OPA1-associated autosomal dominant optic atrophy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1424–1431. [Google Scholar] [CrossRef]
- Juschke, C.; Klopstock, T.; Catarino, C.B.; Owczarek-Lipska, M.; Wissinger, B.; Neidhardt, J. Autosomal dominant optic atrophy: A novel treatment for OPA1 splice defects using U1 snRNA adaption. Mol. Ther. Nucleic Acids 2021, 26, 1186–1197. [Google Scholar] [CrossRef]
- Da Costa, R.; Roger, C.; Segelken, J.; Barben, M.; Grimm, C.; Neidhardt, J. A Novel Method Combining Vitreous Aspiration and Intravitreal AAV2/8 Injection Results in Retina-Wide Transduction in Adult Mice. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5326–5334. [Google Scholar] [CrossRef]
- Irigoyen, C.; Amenabar Alonso, A.; Sanchez-Molina, J.; Rodriguez-Hidalgo, M.; Lara-Lopez, A.; Ruiz-Ederra, J. Subretinal Injection Techniques for Retinal Disease: A Review. J. Clin. Med. 2022, 11, 4717. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Natkunarajah, M.; Trittibach, P.; McIntosh, J.; Duran, Y.; Barker, S.E.; Smith, A.J.; Nathwani, A.C.; Ali, R.R. Assessment of ocular transduction using single-stranded and self-complementary recombinant adeno-associated virus serotype 2/8. Gene Ther. 2008, 15, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Allocca, M.; Mussolino, C.; Garcia-Hoyos, M.; Sanges, D.; Iodice, C.; Petrillo, M.; Vandenberghe, L.H.; Wilson, J.M.; Marigo, V.; Surace, E.M.; et al. Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J. Virol. 2007, 81, 11372–11380. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Miyake, K.; Asakawa, N.; Miyake, N.; Shimada, T.; Takahashi, H. Direct comparison of administration routes for AAV8-mediated ocular gene therapy. Curr. Eye Res. 2013, 38, 569–577. [Google Scholar] [CrossRef]
- Lebherz, C.; Maguire, A.; Tang, W.; Bennett, J.; Wilson, J.M. Novel AAV serotypes for improved ocular gene transfer. J. Gene Med. 2008, 10, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Miyake, K.; Masuda, I.; Takahashi, H.; Shimada, T. Adeno-associated vector (type 8)-mediated expression of soluble Flt-1 efficiently inhibits neovascularization in a murine choroidal neovascularization model. Hum. Gene Ther. 2010, 21, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Cideciyan, A.V.; Schwartz, S.B.; Wang, L.; Conlon, T.J.; Boye, S.L.; Flotte, T.R.; Byrne, B.J.; et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Hum. Gene Ther. 2008, 19, 979–990. [Google Scholar] [CrossRef]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Nuzbrokh, Y.; Kassotis, A.S.; Ragi, S.D.; Jauregui, R.; Tsang, S.H. Treatment-Emergent Adverse Events in Gene Therapy Trials for Inherited Retinal Diseases: A Narrative Review. Ophthalmol. Ther. 2020, 9, 709–724. [Google Scholar] [CrossRef]
- Sun, S.; Erchova, I.; Sengpiel, F.; Votruba, M. Opa1 Deficiency Leads to Diminished Mitochondrial Bioenergetics With Compensatory Increased Mitochondrial Motility. Investig. Ophthalmol. Vis. Sci. 2020, 61, 42. [Google Scholar] [CrossRef] [PubMed]
- Pesch, U.E.; Leo-Kottler, B.; Mayer, S.; Jurklies, B.; Kellner, U.; Apfelstedt-Sylla, E.; Zrenner, E.; Alexander, C.; Wissinger, B. OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum. Mol. Genet. 2001, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Cohn, A.C.; Toomes, C.; Potter, C.; Towns, K.V.; Hewitt, A.W.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. Autosomal dominant optic atrophy: Penetrance and expressivity in patients with OPA1 mutations. Am. J. Ophthalmol. 2007, 143, 656–662. [Google Scholar] [CrossRef]
- Xu, X.; Wang, P.; Jia, X.; Sun, W.; Li, S.; Xiao, X.; Hejtmancik, J.F.; Zhang, Q. Pathogenicity evaluation and the genotype-phenotype analysis of OPA1 variants. Mol. Genet. Genom. 2021, 296, 845–862. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Lee, Y.M.; Lee, N.C. Gene therapy with modified U1 small nuclear RNA. Expert. Rev. Endocrinol. Metab. 2017, 12, 171–175. [Google Scholar] [CrossRef]
- Ast, G. How did alternative splicing evolve? Nat. Rev. Genet. 2004, 5, 773–782. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Akepati, V.R.; Muller, E.C.; Otto, A.; Strauss, H.M.; Portwich, M.; Alexander, C. Characterization of OPA1 isoforms isolated from mouse tissues. J. Neurochem. 2008, 106, 372–383. [Google Scholar] [CrossRef]
- Delettre, C.; Griffoin, J.M.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef]
- Del Dotto, V.; Mishra, P.; Vidoni, S.; Fogazza, M.; Maresca, A.; Caporali, L.; McCaffery, J.M.; Cappelletti, M.; Baruffini, E.; Lenaers, G.; et al. OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions. Cell Rep. 2017, 19, 2557–2571. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.M.; Chadderton, N.; Millington-Ward, S.; Palfi, A.; Shortall, C.; O’Byrne, J.J.; Cassidy, L.; Keegan, D.; Humphries, P.; Kenna, P.; et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Front. Neurosci. 2020, 14, 571479. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Cavallero, S.; Dominguez, E.; Barbe, P.; Simonutti, M.; Sahel, J.A.; Sennlaub, F.; Raoul, W.; Paques, M.; Bemelmans, A.P. Spectral-domain optical coherence tomography of the rodent eye: Highlighting layers of the outer retina using signal averaging and comparison with histology. PLoS ONE 2014, 9, e96494. [Google Scholar] [CrossRef]
- Dysli, C.; Enzmann, V.; Sznitman, R.; Zinkernagel, M.S. Quantitative Analysis of Mouse Retinal Layers Using Automated Segmentation of Spectral Domain Optical Coherence Tomography Images. Transl. Vis. Sci. Technol. 2015, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, M.; Ruitenberg, M.J.; Pollett, M.A.; Ehlert, E.M.; Twisk, J.; Verhaagen, J.; Harvey, A.R. Cellular tropism and transduction properties of seven adeno-associated viral vector serotypes in adult retina after intravitreal injection. Gene Ther. 2009, 16, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, D.; Kolstad, K.D.; Caporale, N.; Visel, M.; Klimczak, R.R.; Schaffer, D.V.; Flannery, J.G. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 2096–2102. [Google Scholar] [CrossRef]
- Reichel, F.F.; Peters, T.; Wilhelm, B.; Biel, M.; Ueffing, M.; Wissinger, B.; Bartz-Schmidt, K.U.; Klein, R.; Michalakis, S.; Fischer, M.D.; et al. Humoral Immune Response After Intravitreal But Not After Subretinal AAV8 in Primates and Patients. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1910–1915. [Google Scholar] [CrossRef]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; Lopez-Otin, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef]
- Suzuki, H.; Kumar, S.A.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; De Antonellis, P.; Cavalli, F.M.G.; Juraschka, K.; Farooq, H.; Shibahara, I.; et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Opa1+/+ ONL Thickness (µm) | Opa1enu/+ ONL Thickness (µm) | |||
|---|---|---|---|---|
| Proximal | Distal | Proximal | Distal | |
| non-injected | 56.05 ± 1.21 (n = 6) | 52.71 ± 2.08 (n = 6) | 57.99 ± 1.24 (n = 5) | 56.75 ± 1.10 (n = 5) |
| GFP | - | - | 56.08 ± 1.80 (n = 9) | 53.97 ± 2.12 (n = 9) |
| U1.wt | 56.77 ± 1.57 (n = 5) | 52.84 ± 2.35 (n = 5) | 56.28 ± 2.73 (n = 9) | 53.75 ± 2.72 (n = 9) |
| U1.ad | 54.77 ± 3.86 (n = 9) | 53.25 ± 3.07 (n = 9) | 56.08 ± 2.90 (n = 16) | 53.24 ± 2.29 (n = 16) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swirski, S.; May, O.; Ahlers, M.; Wissinger, B.; Greschner, M.; Jüschke, C.; Neidhardt, J. In Vivo Efficacy and Safety Evaluations of Therapeutic Splicing Correction Using U1 snRNA in the Mouse Retina. Cells 2023, 12, 955. https://doi.org/10.3390/cells12060955
Swirski S, May O, Ahlers M, Wissinger B, Greschner M, Jüschke C, Neidhardt J. In Vivo Efficacy and Safety Evaluations of Therapeutic Splicing Correction Using U1 snRNA in the Mouse Retina. Cells. 2023; 12(6):955. https://doi.org/10.3390/cells12060955
Chicago/Turabian StyleSwirski, Sebastian, Oliver May, Malte Ahlers, Bernd Wissinger, Martin Greschner, Christoph Jüschke, and John Neidhardt. 2023. "In Vivo Efficacy and Safety Evaluations of Therapeutic Splicing Correction Using U1 snRNA in the Mouse Retina" Cells 12, no. 6: 955. https://doi.org/10.3390/cells12060955
APA StyleSwirski, S., May, O., Ahlers, M., Wissinger, B., Greschner, M., Jüschke, C., & Neidhardt, J. (2023). In Vivo Efficacy and Safety Evaluations of Therapeutic Splicing Correction Using U1 snRNA in the Mouse Retina. Cells, 12(6), 955. https://doi.org/10.3390/cells12060955