

Differential Occupancy and Regulatory Interactions of KDM6A in Bladder Cell Lines

 , , , and
, , , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Procedures
2.1.1. Cell Culture
2.1.2. Chromatin Immunoprecipitation
2.1.3. ChIP-Seq
2.1.4. RT-qPCR
2.1.5. ChIP-qPCR
2.1.6. Primer Design
2.1.7. Western Blotting
2.1.8. Identification of KDM6A Mutation
2.1.9. Plasmid Constructs and Transfection
2.1.10. Co-IP
2.2. Data Analysis and Structural Modeling Procedures
2.2.1. Alignment and Processing of Next-Generation Sequencing Data
2.2.2. Peak Calling and Peak Overlap
2.2.3. Clustering of KDM6A Peaks
2.2.4. Analysis of T24 KDM6A Peaks
2.2.5. Transcription Factor Motif Finding
2.2.6. Gene Ontology Analysis
2.2.7. Data Visualization
2.2.8. String Protein Interaction Data Visualization
2.2.9. HHEX and HES1 Gene Expression-Related Associations
2.2.10. Survival Analysis
2.2.11. Comparison of T24 KDM6A Peaks with Published Data
2.2.12. Structural Modeling
KDM6A:TLE1 Interaction Modeling
TLE1:HES1 Interaction Modeling
TLE1:RUNX1/2 Interaction Modeling
TLE1:HHEX Modeling
2.2.13. Statistical Analysis
3. Results
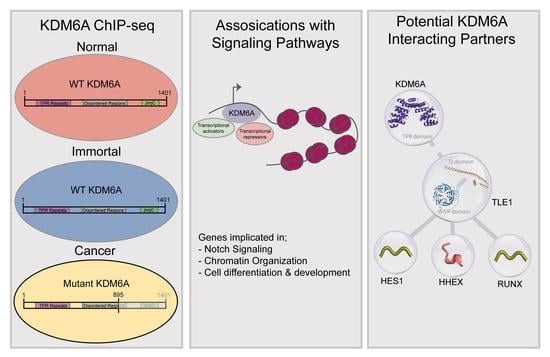
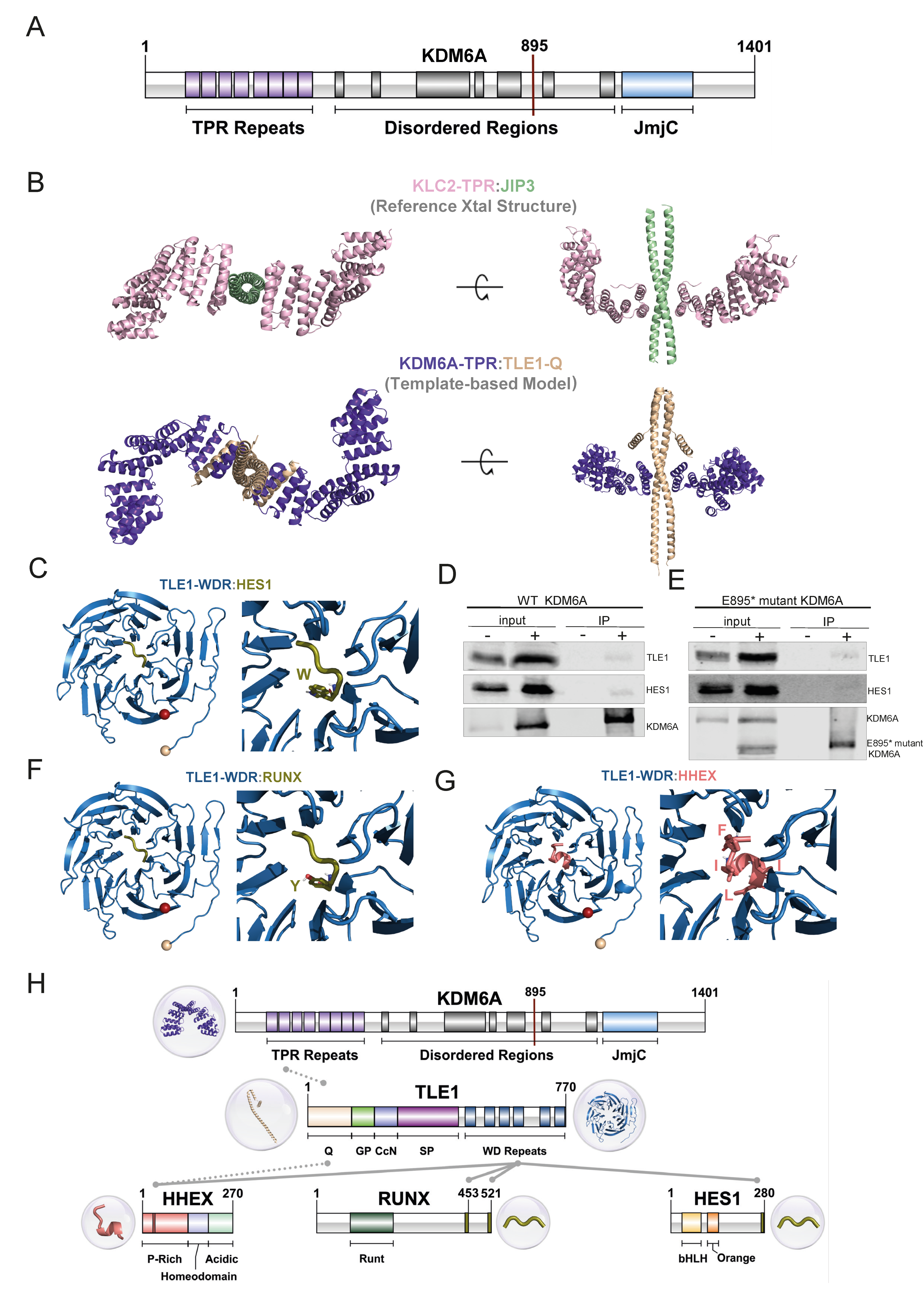
3.1. KDM6A Mutation Status, Identification, and Classification of KDM6A Peaks in Bladder Cell Lines
3.2. Characterization of Genomic Loci with Differential KDM6A Signals
3.3. KDM6A Occupies the Genes Involved in Notch Signaling
3.4. Truncated KDM6A Associates with Genes Deregulated in Cancer
3.5. Structural Modeling and Co-IP Illustrate Previously Uncovered KDM6A-TLE-HES1 Interactions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bennett, R.L.; Licht, J.D. Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.G.; Oh, W.K.; Galsky, M.D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J. Clin. 2020, 70, 404–423. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Sinha, R.J.; Bhaskar, V.; Aeron, R.; Sharma, A.; Singh, V. Role of gemcitabine and cisplatin as neoadjuvant chemotherapy in muscle invasive bladder cancer: Experience over the last decade. Asian J. Urol. 2019, 6, 222–229. [Google Scholar] [CrossRef]
- Koch, J.; Lang, A.; Whongsiri, P.; Schulz, W.A.; Hoffmann, M.J.; Greife, A. KDM6A mutations promote acute cytoplasmic DNA release, DNA damage response and mitosis defects. BMC Mol. Cell Biol. 2021, 22, 54. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Hong, S.; Cho, Y.-W.; Yu, L.-R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef]
- Der Meulen, V.; Speleman, F.; Van Vlierberghe, P. The H3K27me3 demethylase UTX in normal development and disease. Epigenetics 2014, 9, 658–668. [Google Scholar] [CrossRef]
- Welstead, G.G.; Creyghton, M.P.; Bilodeau, S.; Cheng, A.W.; Markoulaki, S.; Young, R.A.; Jaenisch, R. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc. Natl. Acad. Sci. USA 2012, 109, 13004–13009. [Google Scholar] [CrossRef]
- Dhar, S.S.; Lee, S.-H.; Chen, K.; Zhu, G.; Oh, W.; Allton, K.; Gafni, O.; Kim, Y.Z.; Tomoiga, A.S.; Barton, M.; et al. An essential role for UTX in resolution and activation of bivalent promoters. Nucleic Acids Res. 2016, 44, 3659–3674. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.W.; Lee, S.K. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev. Cell 2012, 22, 25–37. [Google Scholar] [CrossRef]
- Thieme, S.; Gyárfás, T.; Richter, C.; Özhan, G.; Fu, J.; Alexopoulou, D.; Muders, M.H.; Michalk, I.; Jakob, C.; Dahl, A.; et al. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 2013, 121, 2462–2473. [Google Scholar] [CrossRef]
- Kim, J.-H.; Sharma, A.; Dhar, S.S.; Lee, S.-H.; Gu, B.; Chan, C.-H.; Lin, H.-K.; Lee, M.G. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014, 74, 1705–1717. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Sengoku, T.; Yokoyama, S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011, 25, 2266–2277. [Google Scholar] [CrossRef]
- Esposito, C.; Wiedmer, L.; Caflisch, A. In Silico Identification of JMJD3 Demethylase Inhibitors. J. Chem. Inf. Model 2018, 58, 2151–2163. [Google Scholar] [CrossRef]
- Pickles, L.M.; Roe, M.; Hemingway, E.J.; Stifani, S.; Pearl, L.H. Crystal structure of the C-terminal WD40 repeat domain of the human Groucho/TLE1 transcriptional corepressor. Structure 2002, 10, 751–761. [Google Scholar] [CrossRef]
- Jennings, B.; Pickles, L.M.; Wainwright, S.M.; Roe, M.; Pearl, L.H.; Ish-Horowicz, D. Molecular recognition of transcriptional repressor motifs by the WD domain of the Groucho/TLE corepressor. Mol. Cell. 2006, 22, 645–655. [Google Scholar] [CrossRef]
- Chodaparambil, J.; Pate, K.T.; Hepler, M.R.D.; Tsai, B.P.; Muthurajan, U.M.; Luger, K.; Waterman, M.L.; Weis, W. Molecular functions of the TLE tetramerization domain in Wnt target gene repression. Embo J. 2014, 33, 719–731. [Google Scholar] [CrossRef]
- McGrath, S.; Tortorici, M.; Drouin, L.; Solanki, S.; Vidler, L.; Westwood, I.; Gimeson, P.; Van Montfort, R.; Hoelder, S. Structure-Enabled Discovery of a Stapled Peptide Inhibitor to Target the Oncogenic Transcriptional Repressor TLE1. Chemistry 2017, 23, 9577–9584. [Google Scholar] [CrossRef]
- Cockburn, J.J.; Hesketh, S.J.; Mulhair, P.; Thomsen, M.; O’Connell, M.J.; Way, M. Insights into Kinesin-1 Activation from the Crystal Structure of KLC2 Bound to JIP3. Structure 2018, 26, 1486–1498.e6. [Google Scholar] [CrossRef]
- Ye, Y.; Godzik, A. FATCAT: A web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004, 32, W582–W585. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jimenez-Garcia, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M.J.J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Bonvin, A.M.J.J.; Karaca, E.; Kastritis, P.L.; Rodrigues, J.P.G.L.M. Defining distance restraints in HADDOCK. Nat. Protoc. 2018, 13, 1503. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lang, A.; Yilmaz, M.; Hader, C.; Murday, S.; Kunz, X.; Wagner, N.; Wiek, C.; Petzsch, P.; Köhrer, K.; Koch, J.; et al. Contingencies of UTX/KDM6A Action in Urothelial Carcinoma. Cancers 2019, 11, 481. [Google Scholar] [CrossRef]
- Ghule, P.N.; Xie, R.L.; Colby, J.L.; Jones, S.N.; Lian, J.B.; Van Wijnen, A.J.; Stein, J.L.; Stein, G.S. p53 checkpoint ablation exacerbates the phenotype of Hinfp dependent histone H4 deficiency. Cell Cycle 2015, 14, 2501–2508. [Google Scholar] [CrossRef]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef]
- Guneri-Sozeri, P.Y.; Özden-Yılmaz, G.; Kisim, A.; Cakiroglu, E.; Eray, A.; Uzuner, H.; Karakülah, G.; Pesen-Okvur, D.; Senturk, S.; Erkek-Ozhan, S. FLI1 and FRA1 transcription factors drive the transcriptional regulatory networks characterizing muscle invasive bladder cancer. Commun. Biol. 2023, 6, 199. [Google Scholar] [CrossRef]
- Maraver, A.; Fernandez-Marcos, P.J.; Cash, T.P.; Mendez-Pertuz, M.; Dueñas, M.; Maietta, P.; Martinelli, P.; Muñoz-Martin, M.; Martínez-Fernández, M.; Cañamero, M.; et al. NOTCH pathway inactivation promotes bladder cancer progression. J. Clin. Investig. 2015, 125, 824–830. [Google Scholar] [CrossRef]
- Paraskevopoulou, V.; Bonis, V.; Dionellis, V.S.; Paschalidis, N.; Melissa, P.; Chavdoula, E.; Vasilaki, E.; Pateras, I.S.; Klinakis, A. Notch controls urothelial integrity in the mouse bladder. JCI Insight 2020, 5, e133232. [Google Scholar] [CrossRef]
- Sang, L.; Roberts, J.; Coller, H. Hijacking HES1: How tumors co-opt the anti-differentiation strategies of quiescent cells. Trends Mol. Med. 2010, 16, 17–26. [Google Scholar] [CrossRef]
- Choi, K.; Ahn, Y.-H.; Gibbons, D.L.; Tran, H.T.; Creighton, C.J.; Girard, L.; Minna, J.D.; Qin, F.X.-F.; Kurie, J.M. Distinct biological roles for the notch ligands Jagged-1 and Jagged-2. J. Biol. Chem. 2009, 284, 17766–17774. [Google Scholar] [CrossRef]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef]
- Seenundun, S.; Rampalli, S.; Liu, Q.-C.; Aziz, A.; Palii, C.; Hong, S.; Blais, A.; Brand, M.; Ge, K.; Dilworth, F.J. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010, 29, 1401–1411. [Google Scholar] [CrossRef]
- Supernat, A.; Łapińska-Szumczyk, S.; Sawicki, S.; Wydra, D.; Biernat, W.; Żaczek, A.J. Deregulation of RAD21 and RUNX1 expression in endometrial cancer. Oncol. Lett. 2012, 4, 727–732. [Google Scholar] [CrossRef]
- Jankovic, D.; Gorello, P.; Liu, T.; Ehret, S.; La Starza, R.; Desjobert, C.; Baty, F.; Brutsche, M.; Jayaraman, P.S.; Santoro, A.; et al. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood 2008, 111, 5672–5682. [Google Scholar] [CrossRef]
- Minuti, G.; Landi, L. MET deregulation in breast cancer. Ann. Transl. Med. 2015, 3, 181. [Google Scholar]
- Barrows, D.; Feng, L.; Carroll, T.S.; Allis, C.D. Loss of UTX/KDM6A and the activation of FGFR3 converge to regulate differentiation gene-expression programs in bladder cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 25732–25741. [Google Scholar] [CrossRef]
- Gaston, K.; Tsitsilianos, M.-A.; Wadey, K.; Jayaraman, P.-S. Misregulation of the proline rich homeodomain (PRH/HHEX) protein in cancer cells and its consequences for tumour growth and invasion. Cell Biosci. 2016, 6, 12. [Google Scholar] [CrossRef]
- Matsumori, T.; Kodama, Y.; Takai, A.; Shiokawa, M.; Nishikawa, Y.; Matsumoto, T.; Takeda, H.; Marui, S.; Okada, H.; Hirano, T.; et al. Hes1 Is Essential in Proliferating Ductal Cell-Mediated Development of Intrahepatic Cholangiocarcinoma. Cancer Res. 2020, 80, 5305–5316. [Google Scholar] [CrossRef]
- Kuriyama, K.; Kodama, Y.; Shiokawa, M.; Nishikawa, Y.; Marui, S.; Kuwada, T.; Sogabe, Y.; Kakiuchi, N.; Tomono, T.; Matsumori, T.; et al. Essential role of Notch/Hes1 signaling in postnatal pancreatic exocrine development. J. Gastroenterol. 2021, 56, 673–687. [Google Scholar] [CrossRef]
- Villasenor, A.; Gauvrit, S.; Collins, M.M.; Maischein, H.-M.; Stainier, D.Y. Hhex regulates the specification and growth of the hepatopancreatic ductal system. Dev. Biol. 2020, 458, 228–236. [Google Scholar] [CrossRef]
- Evseeva, M.N.; Dyikanov, D.T.; Karagyaur, M.N.; Prikazchikova, T.A.; Sheptulina, A.F.; Balashova, M.S.; Zatsepin, T.S.; Rubtsov, Y.P.; Kulebyakin, K.Y. Hematopoietically-expressed homeobox protein HHEX regulates adipogenesis in preadipocytes. Biochimie 2021, 185, 68–77. [Google Scholar] [CrossRef]
- Boglev, Y.; Wilanowski, T.; Caddy, J.; Parekh, V.; Auden, A.; Darido, C.; Hislop, N.R.; Cangkrama, M.; Ting, S.B.; Jane, S.M. The unique and cooperative roles of the Grainy head-like transcription factors in epidermal development reflect unexpected target gene specificity. Dev. Biol. 2011, 349, 512–522. [Google Scholar] [CrossRef]
- Cheng, Z.-Y.; He, T.-T.; Gao, X.-M.; Zhao, Y.; Wang, J. ZBTB Transcription Factors: Key Regulators of the Development, Differentiation and Effector Function of T Cells. Front. Immunol. 2021, 12, 713294. [Google Scholar] [CrossRef]
- Wehn, A.K.; Farkas, D.R.; Sedlock, C.E.; Subedi, D.; Chapman, D.L. Functionally distinct roles for T and Tbx6 during mouse development. Biol. Open 2020, 9, bio054692. [Google Scholar] [CrossRef]
- Tran, N.; Broun, A.; Ge, K. Lysine Demethylase KDM6A in Differentiation, Development, and Cancer. Mol. Cell Biol. 2020, 40, e00341-20. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Agarwal, M.; Kumar, P.; Mathew, S. The Groucho/Transducin-like enhancer of split protein family in animal development. IUBMB Life 2015, 67, 472–481. [Google Scholar] [CrossRef]
- Grbavec, D.; Lo, R.; Liu, Y.; Greenfield, A.; Stifani, S. Groucho/transducin-like enhancer of split (TLE) family members interact with the yeast transcriptional co-repressor SSN6 and mammalian SSN6-related proteins: Implications for evolutionary conservation of transcription repression mechanisms. Biochem. J. 1999, 337 Pt 1, 13–17. [Google Scholar] [CrossRef]
- Chen, G.; Nguyen, P.; Courey, A. A role for Groucho tetramerization in transcriptional repression. Mol. Cell Biol. 1998, 18, 7259–7268. [Google Scholar] [CrossRef]
- Gasperowicz, M.; Otto, F. Mammalian Groucho homologs: Redundancy or specificity? J. Cell Biochem. 2005, 95, 670–687. [Google Scholar] [CrossRef]
- McLarren, K.W.; Theriault, F.M.; Stifani, S. Association with the nuclear matrix and interaction with Groucho and RUNX proteins regulate the transcription repression activity of the basic helix loop helix factor Hes1. J. Biol. Chem. 2001, 276, 1578–1584. [Google Scholar] [CrossRef]
- Ciarapica, R.; Methot, L.; Tang, Y.; Lo, R.; Dali, R.; Buscarlet, M.; Locatelli, F.; Del Sal, G.; Rota, R.; Stifani, S. Prolyl isomerase Pin1 and protein kinase HIPK2 cooperate to promote cortical neurogenesis by suppressing Groucho/TLE:Hes1-mediated inhibition of neuronal differentiation. Cell Death Differ. 2014, 21, 321–332. [Google Scholar] [CrossRef]
- Swingler, T.E.; Bess, K.L.; Yao, J.; Stifani, S.; Jayaraman, P.-S. The proline-rich homeodomain protein recruits members of the Groucho/Transducin-like enhancer of split protein family to co-repress transcription in hematopoietic cells. J. Biol. Chem. 2004, 279, 34938–34947. [Google Scholar] [CrossRef]
- Soufi, A.; Jayaraman, P. PRH/Hex: An oligomeric transcription factor and multifunctional regulator of cell fate. Biochem. J. 2008, 412, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Greife, A.; Jankowiak, S.; Steinbring, J.; Nikpour, P.; Niegisch, G.; Hoffmann, M.J.; A Schulz, W. Canonical Notch signalling is inactive in urothelial carcinoma. BMC Cancer 2014, 14, 628. [Google Scholar] [CrossRef] [PubMed]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef] [PubMed]
- Seelk, S.; Adrian-Kalchhauser, I.; Hargitai, B.; Hajduskova, M.; Gutnik, S.; Tursun, B.; Ciosk, R. Increasing Notch signaling antagonizes PRC2-mediated silencing to promote reprograming of germ cells into neurons. eLife 2016, 5, e15477. [Google Scholar] [CrossRef]
- Zhang, C.; Berndt-Paetz, M.; Neuhaus, J. A Comprehensive Bioinformatics Analysis of Notch Pathways in Bladder Cancer. Cancers 2021, 13, 3089. [Google Scholar] [CrossRef]
- Marfil, V.; Blazquez, M.; Serrano, F.; Castell, J.V.; Bort, R. Growth-promoting and tumourigenic activity of c-Myc is suppressed by Hhex. Oncogene 2015, 34, 3011–3022. [Google Scholar] [CrossRef]
- Shi, B.; Li, W.; Song, Y.; Wang, Z.; Ju, R.; Ulman, A.; Hu, J.; Palomba, F.; Zhao, Y.; Le, J.P.; et al. UTX condensation underlies its tumour-suppressive activity. Nature 2021, 597, 726–731. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özden-Yılmaz, G.; Savas, B.; Bursalı, A.; Eray, A.; Arıbaş, A.; Senturk, S.; Karaca, E.; Karakülah, G.; Erkek-Ozhan, S. Differential Occupancy and Regulatory Interactions of KDM6A in Bladder Cell Lines. Cells 2023, 12, 836. https://doi.org/10.3390/cells12060836
Özden-Yılmaz G, Savas B, Bursalı A, Eray A, Arıbaş A, Senturk S, Karaca E, Karakülah G, Erkek-Ozhan S. Differential Occupancy and Regulatory Interactions of KDM6A in Bladder Cell Lines. Cells. 2023; 12(6):836. https://doi.org/10.3390/cells12060836
Chicago/Turabian StyleÖzden-Yılmaz, Gülden, Busra Savas, Ahmet Bursalı, Aleyna Eray, Alirıza Arıbaş, Serif Senturk, Ezgi Karaca, Gökhan Karakülah, and Serap Erkek-Ozhan. 2023. "Differential Occupancy and Regulatory Interactions of KDM6A in Bladder Cell Lines" Cells 12, no. 6: 836. https://doi.org/10.3390/cells12060836
APA StyleÖzden-Yılmaz, G., Savas, B., Bursalı, A., Eray, A., Arıbaş, A., Senturk, S., Karaca, E., Karakülah, G., & Erkek-Ozhan, S. (2023). Differential Occupancy and Regulatory Interactions of KDM6A in Bladder Cell Lines. Cells, 12(6), 836. https://doi.org/10.3390/cells12060836