

Myelinating Glia: Potential Therapeutic Targets in Polyglutamine Spinocerebellar Ataxias

Abstract
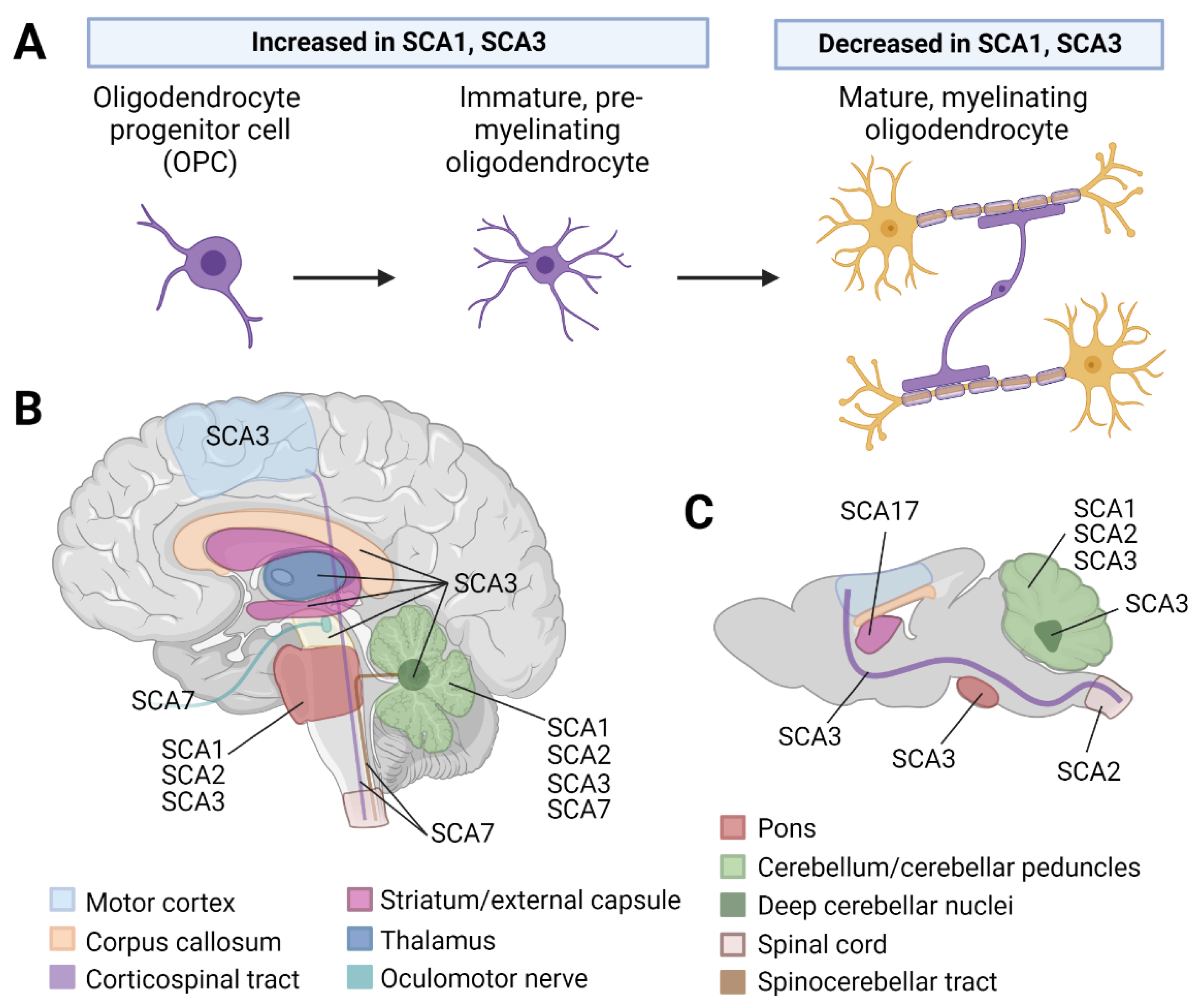
1. Introduction
2. Central Nervous System Myelinating Glia: Oligodendrocytes
2.1. White Matter Abnormalities Are Observed in Patients with SCA1, 2, 3, and 7
2.2. Oligodendrocyte Dysregulation Is Observed in SCA1 and 3 Rodent Models
3. Peripheral Nervous System Myelinating Glia: Schwann Cells
3.1. Peripheral Neuropathy Is Observed in Patients with SCA1, 2, 3, and 6
3.2. Schwann Cell Dysregulation Is Observed in SCA1, 3, and 7 Rodent Models
4. Therapeutic Considerations for Targeting Myelinating Glia
4.1. Targeting Oligodendrocytes
4.2. Targeting Schwann Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coarelli, G.; Brice, A.; Durr, A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Res 2018, 7, F1000 Faculty Rev-1781. [Google Scholar] [CrossRef]
- Paulson, H.L. The spinocerebellar ataxias. J. Neuroophthalmol. 2009, 29, 227–237. [Google Scholar] [CrossRef]
- Klockgether, T.; Mariotti, C.; Paulson, H. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef]
- Durr, A.; Stevanin, G.; Cancel, G.; Duyckaerts, C.; Abbas, N.; Didierjean, O.; Chneiweiss, H.; Benomar, A.; Lyon-Caen, O.; Julien, J.; et al. Spinocerebellar ataxia 3 and Machado-Joseph disease: Clinical, molecular, and neuropathological features. Ann. Neurol. 1996, 39, 490–499. [Google Scholar] [CrossRef]
- Rüb, U.; Schoels, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.W.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef]
- Corral-Juan, M.; Casquero, P.; Giraldo-Restrepo, N.; Laurie, S.; Martinez-Piñeiro, A.; Mateo-Montero, R.C.; Ispierto, L.; Vilas, D.; Tolosa, E.; Volpini, V.; et al. New spinocerebellar ataxia subtype caused by SAMD9L mutation triggering mitochondrial dysregulation (SCA49). Brain Commun. 2022, 4, fcac030. [Google Scholar] [CrossRef]
- Rafehi, H.; Read, J.; Szmulewicz, D.J.; Davies, K.C.; Snell, P.; Fearnley, L.G.; Scott, L.; Thomsen, M.; Gillies, G.; Pope, K.; et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA50/ATX-FGF14. Am. J. Hum. Genet. 2022, 110, 105–119. [Google Scholar] [CrossRef]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias–from genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013, 136 Pt 2, 471–482. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.S.; Gorospe, M.; et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Errea, O.; Rodriguez-Oroz, M. Oligodendrocytes, a New Player in the Etiology of Parkinson’s Disease. Mov. Disord. 2021, 36, 83. [Google Scholar] [CrossRef]
- Ettle, B.; Schlachetzki, J.; Winkler, J. Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef]
- Ferrari Bardile, C.; Garcia-Miralles, M.; Caron, N.S.; Rayan, N.A.; Langley, S.R.; Harmston, N.; Rondelli, A.M.; Teo, R.T.Y.; Waltl, S. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc. Natl. Acad. Sci. USA 2019, 116, 9622–9627. [Google Scholar] [CrossRef]
- Kenigsbuch, M.; Bost, P.; Halevi, S.; Chang, Y.; Chen, S.; Ma, Q.; Hajbi, R.; Schwikowski, B.; Bodenmiller, B.; Fu, H.; et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci. 2022, 25, 876–886. [Google Scholar] [CrossRef]
- Humphrey, J.; Venkatesh, S.; Hasan, R.; Herb, J.T.; de Paiva Lopes, K.; Küçükali, F.; Byrska-Bishop, M.; Evani, U.S.; Narzisi, G.; Fagegaltier, D. Integrative transcriptomic analysis of the amyotrophic lateral sclerosis spinal cord implicates glial activation and suggests new risk genes. Nat. Neurosci. 2022, 26, 150–162. [Google Scholar] [CrossRef]
- Suga, N.; Katsuno, M.; Koike, H.; Banno, H.; Suzuki, K.; Hashizume, A.; Mano, T.; Iijima, M.; Kawagashira, Y.; Hirayama, M.; et al. Schwann cell involvement in the peripheral neuropathy of spinocerebellar ataxia type 3. Neuropathol. Appl. Neurobiol. 2014, 40, 628–639. [Google Scholar] [CrossRef]
- Velázquez-Perez, L.; Rodriguez-Labrada, R.; Canales-Ochoa, N.; Sanchez-Cruz, G.; Fernandez-Ruiz, J.; Montero, J.M.; Aguilera-Rodríguez, R.; Diaz, R.; Almaguer-Mederos, L.E.; Truitz, A.P. Progression markers of Spinocerebellar ataxia 2. A twenty years neurophysiological follow up study. J. Neurol. Sci. 2010, 290, 22–26. [Google Scholar] [CrossRef]
- Linnemann, C.; du Montcel, S.T.; Rakowicz, M.; Schmitz-Hübsch, T.T.; Szymanski, S.S.; Berciano, J.J.; Van De Warrenburg, B.P.C.B.; Pedersen, K.; Depondt, C.; Rola, R.R.; et al. Peripheral Neuropathy in Spinocerebellar Ataxia Type 1, 2, 3, and 6. Cerebellum 2016, 15, 165–173. [Google Scholar] [CrossRef]
- Dugas, J.C.; Tai, Y.C.; Speed, T.P.; Ngai, J.; Barres, B.A. Functional genomic analysis of oligodendrocyte differentiation. J. Neurosci. 2006, 26, 10967–10983. [Google Scholar] [CrossRef]
- Warrington, A.E.; Pfeiffer, S.E. Proliferation and differentiation of O4+ oligodendrocytes in postnatal rat cerebellum: Analysis in unfixed tissue slices using anti-glycolipid antibodies. J. Neurosci. Res. 1992, 33, 338–353. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta. Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells. 2019, 8, 1424. [Google Scholar] [CrossRef]
- Williams, K.A.; Deber, C.M.; Klrschner, O.A. The structure and function of central nervous system myelin. Crit. Rev. Clin. Lab. Sci. 1993, 30, 29–64. [Google Scholar] [CrossRef]
- Norton, W.T.; Cammer, W. Isolation and Characterization of Myelin. In Myelin; Morell, P., Ed.; Springer: Boston, MA, USA, 1984; pp. 147–195. [Google Scholar]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin Fat Facts: An Overview of Lipids and Fatty Acid Metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef]
- Marcus, J.; Popko, B. Galactolipids are molecular determinants of myelin development and axo-glial organization. Biochim. Biophys. Acta 2002, 1573, 406–413. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Dai, X.; Lercher, L.D.; Clinton, P.M.; Du, Y.; Livingston, D.L.; Vieira, C.; Yang, L.; Shen, M.M.; Dreyfus, C.F. The trophic role of oligodendrocytes in the basal forebrain. J. Neurosci. 2003, 23, 5846–5853. [Google Scholar] [CrossRef]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef]
- Park, Y.W.; Joers, J.M.; Guo, B.; Hutter, D.; Bushara, K.; Adanyeguh, I.M.; Eberly, L.; Oz, G.; Lenglet, C. Assessment of Cerebral and Cerebellar White Matter Microstructure in Spinocerebellar Ataxias 1, 2, 3, and 6 Using Diffusion MRI. Front. Neurol. 2020, 11, 411. [Google Scholar] [CrossRef]
- Soares, J.M.; Marques, P.; Alves, V.; Sousa, N. A hitchhiker’s guide to diffusion tensor imaging. Front. Neurosci. 2013, 7, 31. [Google Scholar] [CrossRef]
- Salat, D.H. Chapter 12–Diffusion Tensor Imaging in the Study of Aging and Age-Associated Neural Disease. In Diffusion MRI, 2nd ed.; Diffusion, M.R.I., Ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Solodkin, A.; Peri, E.; Chen, E.E.; Ben-Jacob, E.; Gomez, C.M. Loss of intrinsic organization of cerebellar networks in spinocerebellar ataxia type 1: Correlates with disease severity and duration. Cerebellum 2011, 10, 218–232. [Google Scholar] [CrossRef]
- Chandrasekaran, J.; Petit, E.; Park, Y.W.; Tezenas du Montcel, S.; Joers, J.M.; Deelchand, D.K.; Považan, M.; Banan, G.; Valabregue, R.; Ehses, P.; et al. Clinically meaningful MR endpoints sensitive to preataxic SCA1 and SCA3. Ann. Neurol. 2022. [Google Scholar] [CrossRef]
- Han, Q.; Yang, J.; Xiong, H.; Shang, H. Voxel-based meta-analysis of gray and white matter volume abnormalities in spinocerebellar ataxia type 2. Brain Behav. 2018, 8, e01099. [Google Scholar] [CrossRef]
- Stezin, A.; Bhardwaj, S.; Khokhar, S.; Hegde, S.; Jain, S.; Bharath, R.D.; Saini, J.; Pal, P.K. In vivo microstructural white matter changes in early spinocerebellar ataxia 2. Acta Neurol. Scand. 2021, 143, 326–332. [Google Scholar] [CrossRef]
- Chen, H.-C.; Lirng, J.-F.; Soong, B.-W.; Guo, W.Y.; Wu, H.-M.; Chen, C.C.-C.; Chang, C.-Y. The merit of proton magnetic resonance spectroscopy in the longitudinal assessment of spinocerebellar ataxias and multiple system atrophy-cerebellar type. Cerebellum Ataxias 2014, 1, 17. [Google Scholar] [CrossRef]
- Sen, N.-E.; Arsovic, A.; Meierhofer, D.; Brodesser, S.; Oberschmidt, C.; Canet-Pons, J.; Kaya, Z.-E.; Halbach, M.-V.; Gispert, S.; Sandhoff, K.; et al. In Human and Mouse Spino-Cerebellar Tissue, Ataxin-2 Expansion Affects Ceramide-Sphingomyelin Metabolism. Int. J. Mol. Sci. 2019, 20, 5854. [Google Scholar] [CrossRef]
- Lukas, C.; Schöls, L.; Bellenberg, B.; Rüb, U.; Przuntek, H.; Schmid, G.; Köster, O.; Suchan, B. Dissociation of grey and white matter reduction in spinocerebellar ataxia type 3 and 6: A voxel-based morphometry study. Neurosci. Lett. 2006, 408, 230–235. [Google Scholar] [CrossRef]
- Kang, J.-S.; Klein, J.C.; Baudrexel, S.; Deichmann, R.; Nolte, D.; Hilker, R. White matter damage is related to ataxia severity in SCA3. J. Neurol. 2014, 261, 291–299. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Z.; Lin, W.; Li, M.; Ye, Z.; Qiu, Y.; Xia, X.; Chen, N.; Hu, J.; Gan, S.; et al. Altered brain white matter structural motor network in spinocerebellar ataxia type 3. Ann. Clin. Transl. Neurol. 2022. [Google Scholar] [CrossRef]
- Joers, J.M.; Deelchand, D.K.; Lyu, T.; Emir, U.E.; Hutter, D.; Mn, C.M.G.; Bushara, K.O.; Eberly, L.E.; Öz, G. Neurochemical abnormalities in premanifest and early spinocerebellar ataxias. Ann. Neurol. 2018, 83, 816–829. [Google Scholar] [CrossRef]
- Miranda, C.O.; Nobre, R.J.; Paiva, V.H.; Duarte, J.V.; Castelhano, J.; Petrella, L.I.; Sereno, J.; Santana, M.; Afonso, S.; Januário, C.; et al. Cerebellar morphometric and spectroscopic biomarkers for Machado-Joseph Disease. Acta Neuropathol. Commun. 2022, 10, 37. [Google Scholar] [CrossRef]
- Lirng, J.-F.; Wang, P.-S.; Chen, H.-C.; Soong, B.-W.; Guo, W.Y.; Wu, H.-M.; Chang, C.-Y. Differences between spinocerebellar ataxias and multiple system atrophy-cerebellar type on proton magnetic resonance spectroscopy. PLoS ONE 2012, 7, e47925. [Google Scholar] [CrossRef]
- Lei, L.; Liao, Y.; Liao, W.; Zhou, J.; Yuan, Y.; Wang, J.; Jiang, H.; Shen, L.; Tang, T. Magnetic resonance spectroscopy of the cerebellum in patients with spinocerebellar ataxia type 3/Machado-Joseph disease. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2011, 36, 511–519. [Google Scholar]
- Chen, M.-L.; Lin, C.-C.; Rosenthal, L.S.; Opal, P.; Kuo, S.-H. Rating scales and biomarkers for CAG-repeat spinocerebellar ataxias: Implications for therapy development. J. Neurol. Sci. 2021, 424, 117417. [Google Scholar] [CrossRef]
- Costa, M.D.C.; Radzwion, M.; McLoughlin, H.S.; Ashraf, N.S.; Fischer, S.; Shakkottai, V.G.; Maciel, P.; Paulson, H.L.; Öz, G. In Vivo Molecular Signatures of Cerebellar Pathology in Spinocerebellar Ataxia Type 3. Mov. Disord. 2020, 35, 1744–1786. [Google Scholar] [CrossRef]
- Schuster, K.H.; Putka, A.F.; McLoughlin, H.S. Disease-associated oligodendrocyte signatures are spatiotemporally dysregulated in Spinocerebellar Ataxia Type 3. Front. Neurosci. 2023, 17, 149. [Google Scholar]
- Falcon, M.; Gomez, C.; Chen, E.; Shereen, A.; Solodkin, A. Early Cerebellar Network Shifting in Spinocerebellar Ataxia Type 6. Cereb. Cortex 2016, 26, 3205–3218. [Google Scholar] [CrossRef]
- Alcauter, S.; Barrios, F.A.; Díaz, R.; Fernández-Ruiz, J. Gray and white matter alterations in spinocerebellar ataxia type 7: An in vivo DTI and VBM study. Neuroimage 2011, 55, 1–7. [Google Scholar] [CrossRef]
- Masciullo, M.; Modoni, A.; Pomponi, M.G.; Tartaglione, T.; Falsini, B.; Tonali, P.; Silvestri, G. Evidence of white matter involvement in SCA 7. J. Neurol. 2007, 254, 536–538. [Google Scholar] [CrossRef]
- Hernandez-Castillo, C.; Vaca-Palomares, I.; Barrios, F.; Martinez, L.; Boll, M.-C.; Fernandez-Ruiz, J. Ataxia Severity Correlates with White Matter Degeneration in Spinocerebellar Ataxia Type 7. AJNR Am. J. Neuroradiol. 2016, 37, 2050–2054. [Google Scholar] [CrossRef]
- Parker, J.A.; Merchant, S.H.; Attaripour-Isfahani, S.; Cho, H.J.; McGurrin, P.; Brooks, B.P.; La Spada, A.R.; Hallett, M.; Huryn, L.A.; Horovitz, S.G. In vivo assessment of neurodegeneration in Spinocerebellar Ataxia type 7. Neuroimage Clin. 2021, 29, 102561. [Google Scholar] [CrossRef]
- Rüb, U.; Brunt, E.; Gierga, K.; Seidel, K.; Schultz, C.; Schöls, L.; Auburger, G.; Heinsen, H.; Ippel, P.; Glimmerveen, W.; et al. Spinocerebellar ataxia type 7 (SCA7): First report of a systematic neuropathological study of the brain of a patient with a very short expanded CAG-repeat. Brain Pathol. 2005, 15, 287–295. [Google Scholar] [CrossRef]
- Rüb, U.; Brunt, E.R.; Seidel, K.; Gierga, K.; Mooy, C.M.; Kettner, M.; Van Broeckhoven, C.; Bechmann, I.; La Spada, A.R.; Schöls, L.; et al. Spinocerebellar ataxia type 7 (SCA7): Widespread brain damage in an adult-onset patient with progressive visual impairments in comparison with an adult-onset patient without visual impairments. Neuropathol. Appl. Neurobiol. 2008, 34, 155–168. [Google Scholar] [CrossRef]
- Gierga, K.; Bürk, K.; Bauer, M.; Diaz, G.O.; Auburger, G.; Schultz, C.; Vuksic, M.; Schöls, L.; De Vos, R.A.I.; Braak, H.; et al. Involvement of the cranial nerves and their nuclei in spinocerebellar ataxia type 2 (SCA2). Acta Neuropathol. 2005, 109, 617–631. [Google Scholar] [CrossRef]
- Guimaraes, R.P.; D'Abreu, A.; Yasuda, C.L.; França, M.C.; Silva, B.H.B.; Cappabianco, F.A.M.; Bergo, F.P.; Lopes-Cendes, I.T.; Cendes, F. A multimodal evaluation of microstructural white matter damage in spinocerebellar ataxia type 3. Mov. Disord. 2013, 28, 1125–1132. [Google Scholar] [CrossRef]
- Rezende, T.J.R.; De Paiva, J.L.R.; Martinez, A.R.M.; Lopes-Cendes, I.; Pedroso, J.L.; Barsottini, O.G.P.; Cendes, F.; França, M.C., Jr. Structural signature of SCA3: From presymptomatic to late disease stages. Ann. Neurol. 2018, 84, 401–408. [Google Scholar] [CrossRef]
- Assaf, Y.; Pasternak, O. Diffusion tensor imaging (DTI)-based white matter mapping in brain research: A review. J. Mol. Neurosci. 2008, 34, 51–61. [Google Scholar] [CrossRef]
- Albi, A.; Pasternak, O.; Minati, L.; Marizzoni, M.; Bartrés-Faz, D.; Bargalló, N.; Bosch, B.; Rossini, P.M.; Marra, C.; Müller, B.; et al. Free water elimination improves test-retest reproducibility of diffusion tensor imaging indices in the brain: A longitudinal multisite study of healthy elderly subjects. Hum. Brain Mapp. 2017, 38, 12–26. [Google Scholar] [CrossRef]
- Brockmann, K.; Reimold, M.; Globas, C.; Hauser, T.K.; Walter, U.; Machulla, H.-J.; Rolfs, A.; Schöls, L. PET and MRI reveal early evidence of neurodegeneration in spinocerebellar ataxia type 17. J. Nucl. Med. 2012, 53, 1074–1080. [Google Scholar] [CrossRef]
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte Bioenergetics in Health and Disease. Neuroscientist 2019, 25, 334–343. [Google Scholar] [CrossRef]
- Yan, H.; Rivkees, S.A. Hypoglycemia influences oligodendrocyte development and myelin formation. Neuroreport 2006, 17, 55–59. [Google Scholar] [CrossRef]
- Tejwani, L.; Ravindra, N.G.; Nguyen, B.; Luttik, K.; Lee, C.; Gionco, J.; Kim, K.; Yoon, J.; Haidery, F.; Ro, H.; et al. Longitudinal single-cell transcriptional dynamics throughout neurodegeneration in SCA1. bioRxiv 2021. [Google Scholar] [CrossRef]
- Canet-Pons, J.; Sen, N.-E.; Arsović, A.; Almaguer-Mederos, L.-E.; Halbach, M.V.; Key, J.; Döring, C.; Kerksiek, A.; Picchiarelli, G.; Cassel, R.; et al. Atxn2-CAG100-KnockIn mouse spinal cord shows progressive TDP43 pathology associated with cholesterol biosynthesis suppression. Neurobiol. Dis. 2021, 152, 105289. [Google Scholar] [CrossRef]
- Sen, N.-E.; Canet-Pons, J.; Halbach, M.V.; Arsovic, A.; Pilatus, U.; Chae, W.-H.; Kaya, Z.-E.; Seidel, K.; Rollmann, E.; Mittelbronn, M.; et al. Generation of an Atxn2-CAG100 knock-in mouse reveals N-acetylaspartate production deficit due to early Nat8l dysregulation. Neurobiol. Dis. 2019, 132, 104559. [Google Scholar] [CrossRef]
- Schuster, K.H.; Zalon, A.J.; Zhang, H.; DiFranco, D.M.; Stec, N.R.; Haque, Z.; Blumenstein, K.G.; Pierce, A.M.; Guan, Y.; Paulson, H.L.; et al. Impaired oligodendrocyte maturation is an early feature in SCA3 disease pathogenesis. J. Neurosci. 2022, 42, 1604–1617. [Google Scholar] [CrossRef]
- Schuster, K.H.; Putka, A.F.; McLoughlin, H.S. Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture. Cells 2022, 11, 2615. [Google Scholar] [CrossRef]
- Pessentheiner, A.R.; Pelzmann, H.J.; Walenta, E.; Schweiger, M.; Groschner, L.N.; Graier, W.F.; Kolb, D.; Uno, K.; Miyazaki, T.; Nitta, A.; et al. NAT8L (N-acetyltransferase 8-like) accelerates lipid turnover and increases energy expenditure in brown adipocytes. J. Biol. Chem. 2013, 288, 36040–36051. [Google Scholar] [CrossRef]
- Chakraborty, G.; Mekala, P.; Yahya, D.; Wu, G.; Ledeen, R.W. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: Evidence for myelin-associated aspartoacylase. J. Neurochem. 2001, 78, 736–745. [Google Scholar] [CrossRef]
- Wiame, E.; Tyteca, D.; Pierrot, N.; Collard, F.; Amyere, M.; Noel, G.; Desmedt, J.; Nassogne, M.C.; Vikkula, M.; Octave, J.N.; et al. Molecular identification of aspartate N-acetyltransferase and its mutation in hypoacetylaspartia. Biochem. J. 2009, 425, 127–136. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Wang, M.-D.; Gomes, J.; Cashman, N.R.; Little, J.; Krewski, D. Intermediate CAG repeat expansion in the ATXN2 gene is a unique genetic risk factor for ALS--a systematic review and meta-analysis of observational studies. PLoS ONE 2014, 9, e105534. [Google Scholar] [CrossRef]
- Ramani, B.; Panwar, B.; Moore, L.R.; Wang, B.; Huang, R.; Guan, Y.; Paulson, H.L. Comparison of spinocerebellar ataxia type 3 mouse models identifies early gain-of-function, cell-autonomous transcriptional changes in oligodendrocytes. Hum. Mol. Genet. 2017, 26, 3362–3374. [Google Scholar] [CrossRef]
- Haas, E.; Incebacak, R.D.; Hentrich, T.; Huridou, C.; Schmidt, T.; Casadei, N.; Maringer, Y.; Bahl, C.; Zimmermann, F.; Mills, J.D.; et al. A Novel SCA3 Knock-in Mouse Model Mimics the Human SCA3 Disease Phenotype Including Neuropathological, Behavioral, and Transcriptional Abnormalities Especially in Oligodendrocytes. Mol. Neurobiol. 2022, 59, 495–522. [Google Scholar] [CrossRef]
- Spitzer, S.O.; Sitnikov, S.; Kamen, Y.; Evans, K.A.; Kronenberg-Versteeg, D.; Dietmann, S.; de Faria, O.; Agathou, S.; Káradóttir, R.T. Oligodendrocyte Progenitor Cells Become Regionally Diverse and Heterogeneous with Age. Neuron 2019, 101, 459–471.e5. [Google Scholar] [CrossRef]
- Louis, E.D.; Kuo, S.-H.; Vonsattel, J.-P.G.; Faust, P.L. Torpedo formation and Purkinje cell loss: Modeling their relationship in cerebellar disease. Cerebellum 2014, 13, 433–439. [Google Scholar] [CrossRef]
- Ljungberg, L.; Lang-Ouellette, D.; Yang, A.; Jayabal, S.; Quilez, S.; Watt, A.J. Transient Developmental Purkinje Cell Axonal Torpedoes in Healthy and Ataxic Mouse Cerebellum. Front. Cell. Neurosci. 2016, 10, 248. [Google Scholar] [CrossRef]
- Takahashi, N.; Iwatsubo, T.; Nakano, I.; Machinami, R. Focal appearance of cerebellar torpedoes associated with discrete lesions in the cerebellar white matter. Acta Neuropathol. 1992, 84, 153–156. [Google Scholar] [CrossRef]
- Grimaldi, G.; Manto, M. Is essential tremor a Purkinjopathy? The role of the cerebellar cortex in its pathogenesis. Mov. Disord. 2013, 28, 1759–1761. [Google Scholar] [CrossRef]
- Yang, Q.; Hashizume, Y.; Yoshida, M.; Wang, Y.; Goto, Y.; Mitsuma, N.; Ishikawa, K.; Mizusawa, H. Morphological Purkinje cell changes in spinocerebellar ataxia type 6. Acta Neuropathol. 2000, 100, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Kelp, A.; Koeppen, A.H.; Petrasch-Parwez, E.; Calaminus, C.; Bauer, C.; Portal, E.; Yu-Taeger, L.; Pichler, B.; Bauer, P.; Riess, O.; et al. A novel transgenic rat model for spinocerebellar ataxia type 17 recapitulates neuropathological changes and supplies in vivo imaging biomarkers. J. Neurosci. 2013, 33, 9068–9081. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Doussau, F.; Perot, J.-B.; Roux, M.J.; Keime, C.; Hache, A.; Piguet, F.; Novati, A.; Weber, C.; Yalcin, B.; et al. SCA7 Mouse Cerebellar Pathology Reveals Preferential Downregulation of Key Purkinje Cell-Identity Genes and Shared Disease Signature with SCA1 and SCA2. J. Neurosci. 2021, 41, 4910–4936. [Google Scholar] [CrossRef]
- Nocera, G.; Jacob, C. Mechanisms of Schwann cell plasticity involved in peripheral nerve repair after injury. Cell. Mol. Life Sci. 2020, 77, 3977–3989. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef]
- Madduri, S.; Gander, B. Schwann cell delivery of neurotrophic factors for peripheral nerve regeneration. J. Peripher. Nerv. Syst. 2010, 15, 93–103. [Google Scholar] [CrossRef]
- Hicks, C.W.; Wang, D.; Windham, B.G.; Matsushita, K.; Selvin, E. Prevalence of peripheral neuropathy defined by monofilament insensitivity in middle-aged and older adults in two US cohorts. Sci. Rep. 2021, 11, 19159. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, M.L.E.; Pedroso, J.L.; Braga-Neto, P.; Abrahao, A.; de Albuquerque, M.V.C.; Borges, F.R.P.; Saraiva-Pereira, M.L.; Jardim, L.B.; Braga, N.I.D.O.; Manzano, G.M.; et al. Pattern of Peripheral Nerve Involvement in Spinocerebellar Ataxia Type 2: A Neurophysiological Assessment. Cerebellum 2016, 15, 767–773. [Google Scholar] [CrossRef]
- Dong, S.; Zhu, D.; Yang, W.; Li, J.; Chen, X. Prominent lower motor neuron involvement in patients with intermediate-length CAG repeats in ATXN3 gene. Neurol. Sci. 2022, 43, 6993–6995. [Google Scholar] [CrossRef]
- Takechi, Y.; Mieda, T.; Iizuka, A.; Toya, S.; Suto, N.; Takagishi, K.; Nakazato, Y.; Nakamura, K.; Hirai, H. Impairment of spinal motor neurons in spinocerebellar ataxia type 1-knock-in mice. Neurosci. Lett. 2013, 535, 67–72. [Google Scholar] [CrossRef]
- Mieda, T.; Suto, N.; Iizuka, A.; Matsuura, S.; Iizuka, H.; Takagishi, K.; Nakamura, K.; Hirai, H. Mesenchymal stem cells attenuate peripheral neuronal degeneration in spinocerebellar ataxia type 1 knockin mice. J. Neurosci. Res. 2016, 94, 246–252. [Google Scholar] [CrossRef]
- Cemal, C.K.; Carroll, C.; Lawrence, L.; Lowrie, M.B.; Ruddle, P.; Al-Mahdawi, S.; King, R.H.; Pook, M.A.; Huxley, C.; Chamberlain, S. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum. Mol. Genet. 2002, 11, 1075–1094. [Google Scholar] [CrossRef]
- França, M.C.; D’Abreu, A.; Friedman, J.H.; Nucci, A.; Lopes-Cendes, I. Chronic pain in Machado-Joseph disease: A frequent and disabling symptom. Arch. Neurol. 2007, 64, 1767–1770. [Google Scholar] [CrossRef]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.L.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.; Rainwater, O.; Hofstra, B.; Benneyworth, M.; et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018, 3, e123193. [Google Scholar] [CrossRef]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef]
- Moore, L.R.; Rajpal, G.; Dillingham, I.T.; Qutob, M.; Blumenstein, K.G.; Gattis, D.; Hung, G.; Kordasiewicz, H.B.; Paulson, H.L.; McLoughlin, H.S. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol. Ther. Nucleic Acids 2017, 7, 200–210. [Google Scholar] [CrossRef]
- Schuster, K.H.; Zalon, A.J.; DiFranco, D.M.; Putka, A.F.; Stec, N.; Jarrah, S.; Naeem, A.; Haque, Z.; Zhang, H.; Guan, Y.; et al. ASOs are an effective treatment for disease-associated oligodendrocyte signatures in premanifest and symptomatic SCA3 mice. bioRxiv 2022. [Google Scholar] [CrossRef]
- Nóbrega, C.; Mendonça, L.; Marcelo, A.; Lamazière, A.; Tomé, S.; Despres, G.; Matos, C.A.; Mechmet, F.; Langui, D.; Dunnen, W.D.; et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol. 2019, 138, 837–858. [Google Scholar] [CrossRef]
- Morell, P.; Jurevics, H. Origin of cholesterol in myelin. Neurochem. Res. 1996, 21, 463–470. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid. Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Muse, E.D.; Jurevics, H.; Toews, A.D.; Matsushima, G.K.; Morell, P. Parameters related to lipid metabolism as markers of myelination in mouse brain. J. Neurochem. 2001, 76, 77–86. [Google Scholar] [CrossRef]
- Mathews, E.S.; Appel, B. Cholesterol Biosynthesis Supports Myelin Gene Expression and Axon Ensheathment through Modulation of P13K/Akt/mTor Signaling. J. Neurosci. 2016, 36, 7628–7639. [Google Scholar] [CrossRef]
- Dietz, K.C.; Polanco, J.J.; Pol, S.U.; Sim, F.J. Targeting human oligodendrocyte progenitors for myelin repair. Exp. Neurol. 2016, 283 Pt B, 489–500. [Google Scholar] [CrossRef]
- Seeker, L.A.; Williams, A. Oligodendroglia heterogeneity in the human central nervous system. Acta Neuropathol. 2022, 143, 143–157. [Google Scholar] [CrossRef]
- Nistor, G.I.; Totoiu, M.O.; Haque, N.; Carpenter, M.K.; Keirstead, H.S. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia 2005, 49, 385–396. [Google Scholar] [CrossRef]
- Izrael, M.; Zhang, P.; Kaufman, R.; Shinder, V.; Ella, R.; Amit, M.; Itskovitz-Eldor, J.; Chebath, J.; Revel, M. Human oligodendrocytes derived from embryonic stem cells: Effect of noggin on phenotypic differentiation in vitro and on myelination in vivo. Mol. Cell. Neurosci. 2007, 34, 310–323. [Google Scholar] [CrossRef]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.-L.; Cui, Q.-L.; Schambach, A.; Kim, K.-P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef]
- Ng, A.H.M.; Khoshakhlagh, P.; Arias, J.E.R.; Pasquini, G.; Wang, K.; Swiersy, A.; Shipman, S.L.; Appleton, E.; Kiaee, K.; Kohman, R.E.; et al. A comprehensive library of human transcription factors for cell fate engineering. Nat. Biotechnol. 2021, 39, 510–519. [Google Scholar] [CrossRef]
- Huang, B.; Wei, W.; Wang, G.; Gaertig, M.A.; Feng, Y.; Wang, W.; Li, X.-J.; Li, S. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 2015, 85, 1212–1226. [Google Scholar] [CrossRef]
- Osipovitch, M.; Martinez, A.A.; Mariani, J.N.; Cornwell, A.; Dhaliwal, S.; Zou, L.; Chandler-Militello, D.; Wang, S.; Li, X.; Benraiss, S.-J.; et al. Human ESC-Derived Chimeric Mouse Models of Huntington’s Disease Reveal Cell-Intrinsic Defects in Glial Progenitor Cell Differentiation. Cell Stem Cell 2019, 24, 107–122.e7. [Google Scholar] [CrossRef]
- Huang, Z.; Powell, R.; Phillips, J.B.; Haastert-Talini, K. Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering. Cells 2020, 9, 2497. [Google Scholar] [CrossRef]
- Kim, H.-S.; Lee, J.; Lee, D.Y.; Kim, Y.-D.; Kim, J.Y.; Lim, H.J.; Lim, S.; Cho, Y.S. Schwann Cell Precursors from Human Pluripotent Stem Cells as a Potential Therapeutic Target for Myelin Repair. Stem Cell Rep. 2017, 8, 1714–1726. [Google Scholar] [CrossRef]
- Sakaue, M.; Sieber-Blum, M. Human epidermal neural crest stem cells as a source of Schwann cells. Development 2015, 142, 3188–3197. [Google Scholar]
- Verovskaya, E.; Broekhuis, M.J.C.; Zwart, E.; Ritsema, M.; Van Os, R.; de Haan, G.; Bystrykh, L.V. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood 2013, 122, 523–532. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Belfiore, L.; Chu, T.-H.; Fleming, T.; Midha, R.; Biernaskie, J.; Schuurmans, C. Insights Into the Role and Potential of Schwann Cells for Peripheral Nerve Repair From Studies of Development and Injury. Front. Mol. Neurosci. 2020, 13, 608442. [Google Scholar] [CrossRef]
- Zhao, H.T.; Damle, S.; Ikeda-Lee, K.; Kuntz, S.; Karli, I.-L.; Mohan, A.; Kim, A.; Hung, G.; Scheideler, M.A.; Scherer, S.S.; et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J. Clin. Investig. 2018, 128, 359–368. [Google Scholar] [CrossRef]
- Stavrou, M.; Kagiava, A.; Choudury, S.G.; Jennings, M.J.; Wallace, L.M.; Fowler, A.M.; Heslegrave, A.; Richter, J.; Tryfonos, C.; Christodoulou, C.; et al. A translatable RNAi-driven gene therapy silences PMP22/Pmp22 genes and improves neuropathy in CMT1A mice. J. Clin. Investig. 2022, 132, e159814. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Method | Findings * | Ref |
|---|---|---|---|
| SCA1 | DTI | Increased axial diffusivity in the middle cerebellar peduncle | [32] |
| Fractional anisotropy values in the superior cerebellar peduncle are positively correlated with duration of illness and negatively correlated with SARA score | [35] | ||
| MRS | Higher tCr and lower tNAA in preataxic patients | [36] | |
| SCA2 | DTI | White matter microstructural changes in the pons and cerebellar peduncles, hemispheres, and vermis | [37] |
| Axial diffusivity in the right corticospinal tract and right superior cerebellar peduncle is negatively correlated with SARA scores | [38] | ||
| MRS | Decreased NAA to Cr ratio in the cerebellar hemisphere | [39] | |
| Other | Reduced sulfatide and galactosylceramide in one postmortem patient cerebellum | [40] | |
| SCA3 | DTI | White matter alterations in disease-vulnerable brain regions (cerebellar peduncles, dentate nucleus, pons, midbrain, and thalamus) | [41,42] |
| Direct relationship between disease duration and fractional anisotropy values | [43] | ||
| Motor network white matter changes correlate with disease severity and occur prior to onset of ataxia symptoms | [43] | ||
| MRS | Reduced tNAA in cerebellar vermis and white matter | [44] | |
| Inverse relationship between tNAA levels and disease severity | [45] | ||
| Reduced NAA to Cr ratio in cerebellar vermis, hemispheres, and dentate nucleus | [46,47,48] | ||
| Other | Decreased myelin basic protein and myelin staining in patient postmortem cerebellar tissue | [49,50] | |
| SCA6 | DTI | Limited but significant damage to white matter microstructure | [32] |
| Increased fractional anisotropy and decreased radial diffusivity and in the superior cerebellar peduncle of preataxic patients | [51] | ||
| Decreased fractional anisotropy and increased radial diffusivity in the superior cerebellar peduncle of moderate to severe symptomatic patients | [51] | ||
| SCA7 | DTI | DTI and MRI reveal loss of myelinated axons in the spinocerebellar tract, oculomotor nerve, cerebellar white matter, and corpus callosum | [52,53] |
| Increased mean diffusivity and decreased fractional anisotropy of the cerebellar peduncles and corticospinal tract | [54,55] | ||
| Significant correlation between mean diffusivity and SARA score in anterior cerebellar white matter, superior cerebellar peduncles, and middle occipital gyrus | [54] | ||
| Inverse relationship between whole-brain parenchymal fractional anisotropy/cerebellar parenchymal tissue volume and SARA score | [55] | ||
| Other | Myelin abnormalities in central nervous fiber tracts outside the optic tract in two adult-onset patients | [56,57] |
| Disease | Mouse Model | Findings * | Ref |
|---|---|---|---|
| SCA1 | Knock-in Atxn1154Q/+ | Dysregulation of mature oligodendrocyte transcripts and protein in mouse cerebellar tissue. Cerebellar TEM analysis showed age-dependent reductions in myelination. | [66] |
| SCA2 | Atxn2-CAG100 knock-in | Transcriptional dysregulation of myelin and lipid synthesis (including Nat8l) identified by RNAseq of mouse spinal cord. Decreased levels of cholesterol biosynthesis pathway intermediates identified by gas chromatography–mass spectrometry of mouse spinal cord. | [67] |
| Decreased NAT8L in the cerebellum by Western blot analysis. | [68] | ||
| Perturbations in the levels of myelin-enriched lipids in the cerebellum and spinal cord by liquid chromatography–mass spectrometry. | [40] | ||
| SCA3 | YACQ84 transgenic | Decreased mature oligodendrocyte transcript and protein expression in the brainstem and cerebellum by RNAseq and Western blot analysis, respectively. This is paralleled by a reduced number of mature oligodendrocyte cell counts (immunohistochemistry) and thinner myelination (TEM analysis) in disease-vulnerable brain regions. | [69] |
| Onset of mature oligodendrocyte transcriptional demise parallels onset of motor deficits. | [50] | ||
| The maturation deficit is cell autonomous and due to a toxic gain of function by primary oligodendrocyte culture. | [69,70] | ||
| Atxn3 Q82 and Q300 knock-in | PolyQ repeat-dependent spatiotemporal dysregulation of mature oligodendrocyte transcripts; Q300 mice show dysfunction earlier than Q82 mice. | [50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putka, A.F.; Mato, J.P.; McLoughlin, H.S. Myelinating Glia: Potential Therapeutic Targets in Polyglutamine Spinocerebellar Ataxias. Cells 2023, 12, 601. https://doi.org/10.3390/cells12040601
Putka AF, Mato JP, McLoughlin HS. Myelinating Glia: Potential Therapeutic Targets in Polyglutamine Spinocerebellar Ataxias. Cells. 2023; 12(4):601. https://doi.org/10.3390/cells12040601
Chicago/Turabian StylePutka, Alexandra F., Juan P. Mato, and Hayley S. McLoughlin. 2023. "Myelinating Glia: Potential Therapeutic Targets in Polyglutamine Spinocerebellar Ataxias" Cells 12, no. 4: 601. https://doi.org/10.3390/cells12040601
APA StylePutka, A. F., Mato, J. P., & McLoughlin, H. S. (2023). Myelinating Glia: Potential Therapeutic Targets in Polyglutamine Spinocerebellar Ataxias. Cells, 12(4), 601. https://doi.org/10.3390/cells12040601